Optimization of Aminoimidazole Derivatives as Src Family Kinase Inhibitors

 , and
, and
Abstract
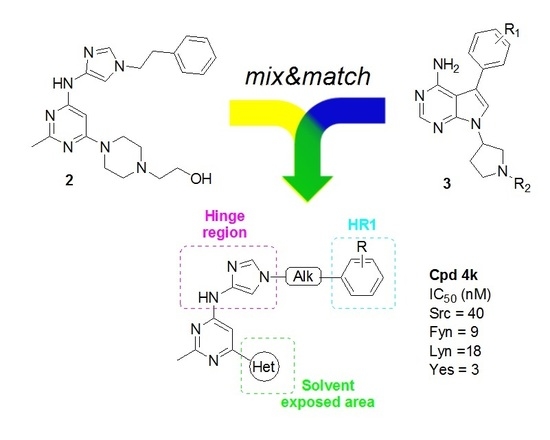
:
1. Introduction
2. Results and Discussion
2.1. Docking Studies
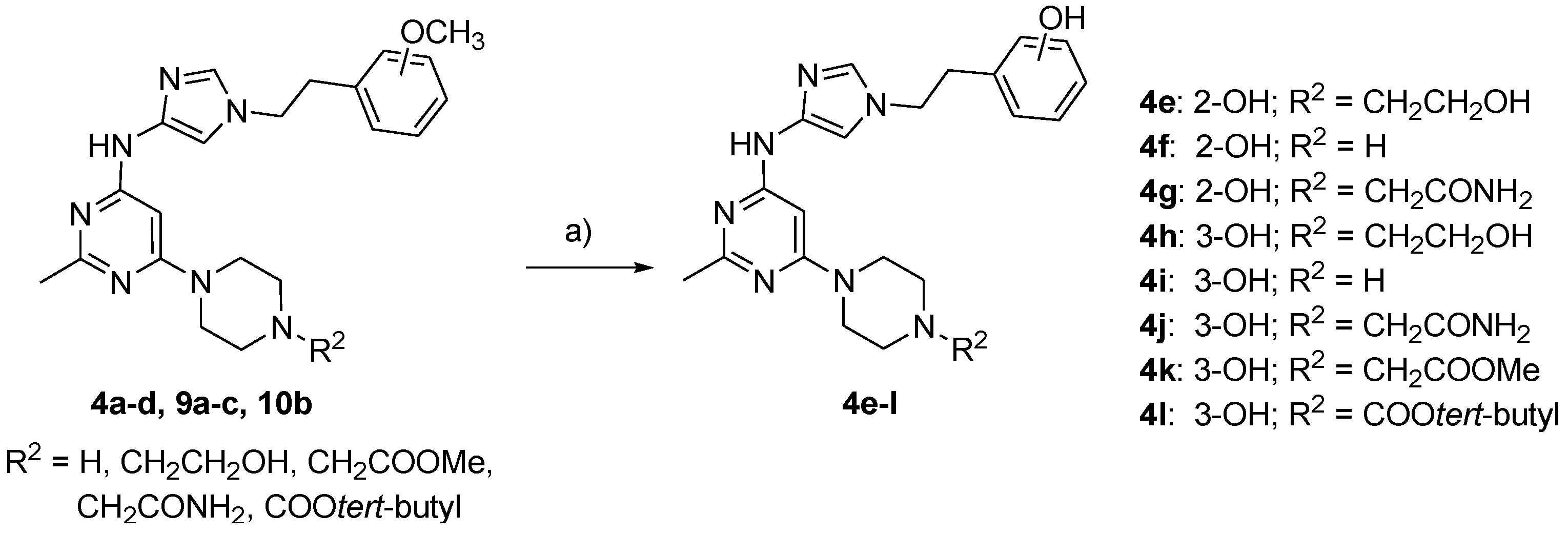
2.2. Chemistry
2.3. Enzymatic Assays
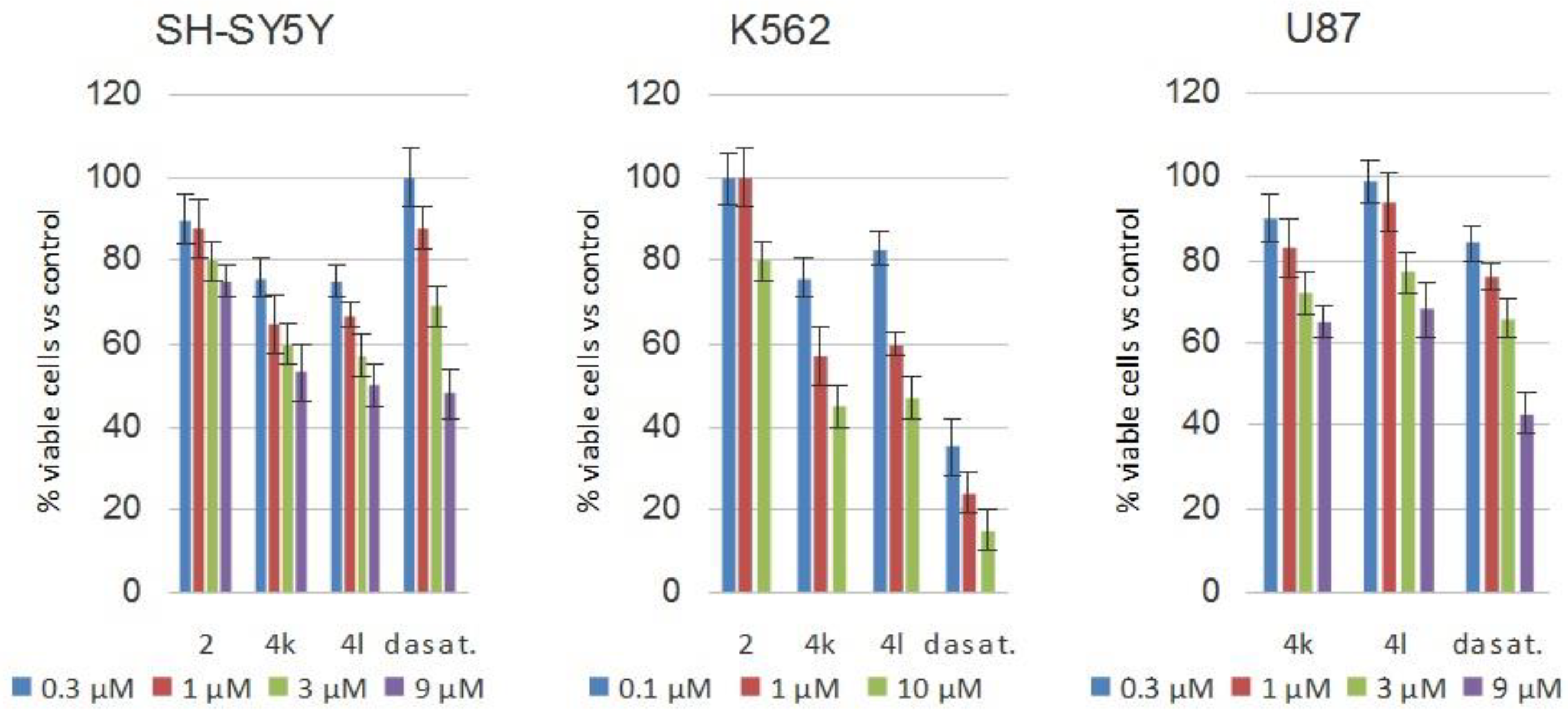
2.4. Cellular Assays
3. Materials and Methods
3.1. Computational Studies
3.1.1. Protein Preparation
3.1.2. Ligands Preparation
3.1.3. Molecular Docking
3.2. Chemistry
3.2.1. General Information
3.2.2. General Procedure for the Synthesis of 1-(2-Phenethyl)-4-nitro-1H-imidazole Derivatives 6a,b
3.2.3. General Procedure for the Synthesis of 1-(2-Phenylethyl)-1H-imidazol-4-amines 7a,b
3.2.4. General Procedure for the Synthesis of 6-Chloro-2-methyl-N-[1-(2-phenylethyl)-1H-imidazol-4-yl]pyrimidin-4-amines 8a,b
3.2.5. General Procedure for the Synthesis of 2-Methyl-N-[1-(2-phenylethyl)-1H-imidazol-4-yl]-6-piperazin-1-ylpyrimidin-4-amines 4a,c and 9a–c
3.2.6. General Procedure for the Synthesis of Methyl [4-(2-methyl-6-{[1-(2-phenylethyl)-1H-imidazol-4-yl]amino}pyrimidin-4-yl)piperazin-1-yl]acetate Derivatives 10a,b
3.2.7. General Procedure for the Synthesis of 2-[4-(2-Methyl-6-{[1-(2-phenylethyl)-1H-imidazol-4-yl]amino}pyrimidin-4-yl)piperazin-1-yl]acetamide Derivatives 4b,d
3.2.8. General Procedure for the Synthesis of (2-{4-[(2-Methyl-6-piperazin-1-ylpyrimidin-4-yl)amino]-1H-imidazol-1-yl}ethyl)phenol Derivatives 4e–l
3.3. Enzymatic Assays
3.4. Cellular Assays
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABCB1 | ATP-binding cassette sub-family B member 1 |
| ALL | acute lymphoblastic leukemia |
| Bcr-Abl | breakpoint cluster region-Abelson |
| Btk | Bruton’s tyrosine kinase |
| CML | chronic myeloid leukemia |
| c-Src | cellular-sarcoma tyrosine kinase |
| DIPEA | N,N-diisopropylethylamine |
| EMA | European Medicines Agency |
| FDA | Food and Drug Admnistration |
| FRET | fluorescence resonance energy transfer |
| GBM | glioblastoma multiforme |
| HRI | hydrophobic region I |
| HTS | high-throughput screening |
| NB | neuroblastoma |
| NCEs | new chemical entities |
| PDGFR | platelet derived growth factor receptor |
| Ph+ | Philadelphia chromosome–positive |
| RET | REarranged during Transfection |
| SFK | Src family kinase |
| SH | Src homology |
| STK | serine/threonine kinase |
| TK | tyrosine kinase |
| VEGFR | vascular endothelial growth factor |
References
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. Protein kinases—The major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 2002, 1, 309–315. [Google Scholar] [CrossRef] [PubMed]
- From the Blue Ridge Institute for Medical Research in Horse Shoe, North Carolina USA. FDA-Approved Protein Kinase Inhibitors Compiled by Robert Roskoski Jr. Available online: http://www.brimr.org/PKI/PKIs.htm (accessed on 23 August 2018).
- Taylor, S.S.; Radzio-Andzelm, E.; Hunter, T. How do protein kinases discriminate between serine/threonine and tyrosine? Structural insights from the insulin receptor protein-tyrosine kinase. FASEB J. 1995, 9, 1255–1266. [Google Scholar] [CrossRef] [PubMed]
- Boggon, T.J.; Eck, M.J. Structure and regulation of Src family kinases. Oncogene 2004, 23, 7918–7927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowan-Jacob, S.W.; Fendrich, G.; Manley, P.W.; Jahnke, W.; Fabbro, D.; Liebetanz, J.; Meyer, T. The crystal structure of a c-Src complex in an active conformation suggests possible steps in c-Src activation. Structure 2005, 13, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Huse, M.; Kuriyan, J. The conformational plasticity of protein kinases. Cell 2002, 109, 275–282. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 2015, 94, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, F.; Schenone, S.; Brullo, C.; Botta, M. An update on dual Src/Abl inhibitors. Future Med. Chem. 2012, 4, 799–822. [Google Scholar] [CrossRef] [PubMed]
- Um, J.W.; Nygaard, H.B.; Heiss, J.K.; Kostylev, M.A.; Stagi, M.; Vortmeyer, A.; Wisniewski, T.; Gunther, E.C.; Strittmatter, S.M. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 2012, 15, 1227–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musumeci, F.; Schenone, S.; Brullo, C.; Desogus, A.; Botta, L.; Tintori, C. Hck inhibitors as potential therapeutic agents in cancer and HIV infection. Curr. Med. Chem. 2015, 22, 1540–1564. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Kantarjian, H. Chronic myeloid leukemia: 2014 update on diagnosis, monitoring, and management. Am. J. Hematol. 2014, 89, 547–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, N.P.; Tran, C.; Lee, F.Y.; Chen, P.; Morris, D.; Sawyers, C.L. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 2004, 305, 399–401. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Cierci, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Hantschel, O.; Rix, U.; Schmidt, U.; Bürckstümmer, T.; Kneidinger, M.; Schütze, G.; Colinge, J.; Bennett, K.L.; Ellmeier, W.; Valent, P.; et al. The Btk tyrosine kinase is a major target of the Bcr-Abl inhibitor dasatinib. Proc. Natl. Acad. Sci. USA 2007, 104, 13283–13288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ClinicalTrials.gov. 3 Studies Found for Dasatinib|Neuroblastoma. Available online: https://www.clinicaltrials.gov/ct2/results?cond=Neuroblastoma&term=dasatinib&cntry=&state=&city=&dist= (accessed on 4 July 2018).
- FDA Grants Accelerated Approval to Bosutinib for Treatment of Newly-Diagnosed PH+ CML. Available online: https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm589856.htm (accessed on 23 August 2018).
- Highlights of Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/203469lbl.pdf (accessed on 23 August 2018).
- Hassan, A.Q.; Sharma, S.V.; Warmuth, M. Allosteric inhibition of BCR-ABL. Cell Cycle 2010, 9, 3710–3714. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Alessi, D.R. Kinase drug discovery—What’s next in the field? ACS Chem. Biol. 2013, 8, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.C.; Shokat, K.M. The evolution of protein kinase inhibitors from antagonists to agonists of cellular signaling. Annu. Rev. Biochem. 2011, 80, 769–795. [Google Scholar] [CrossRef] [PubMed]
- Noble, M.E.; Endicott, J.A.; Johnosn, L.N. Protein kinase inhibitors: Insights into drug design from structure. Science 2004, 303, 1800–1805. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Ioannidis, S.; Chuaqui, C.; Almeida, L.; Alimzhanov, M.; Bebernitz, G.; Bell, K.; Block, M.; Howard, T.; Huang, S.; et al. Discovery of 1-methyl-1H-imidazole derivatives as potent Jak2 inhibitors. J. Med. Chem. 2014, 57, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Francini, C.M.; Fallacara, A.L.; Artusi, R.; Mennuni, L.; Calgani, A.; Angelucci, A.; Schenone, S.; Botta, M. Identification of aminoimidazole and aminothiazole derivatives as Src family kinase inhibitors. ChemMedchem 2015, 10, 2027–2041. [Google Scholar] [CrossRef] [PubMed]
- Altmann, E.; Missbach, M.; Green, J.; Susa, M.; Wagenknecht, H.A.; Widler, L. 7-Pyrrolidinyl- and 7-piperidinyl-5-aryl-pyrrolo[2,3-d]pyrimidines-potent inhibitors of the tyrosine kinase c-Src. Bioorg. Med. Chem. Lett. 2001, 11, 853–856. [Google Scholar] [CrossRef]
- Getlik, M.; Grutter, C.; Simard, J.R.; Kluter, S.; Rabiller, M.; Rode, H.B.; Robubi, A.; Rauh, D. Hybrid compound design to overcome the gatekeeper T338M mutation in cSrc. J. Med. Chem. 2009, 52, 3915–3926. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shaw, D.E.; Shelley, M.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Breitenlechner, C.B.; Kairies, N.A.; Honold, K.; Scheiblich, S.; Koll, H.; Greiter, E.; Koch, S.; Schaefer, W.; Huber, R.; Engh, R.A. Crystal structures of active SRC kinase domain complexes. J. Mol. Biol. 2005, 353, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Almeida, L.; Chuaqui, C.E.; Ioannidis, S.; Peng, B.; Su, M. Tricyclic 2,4-Diamino-l,3,5-Triazine Derivatives Useful for the Treatment of Cancer and Myeloproliferative Disorders. WO2009150462, 17 December 2009. [Google Scholar]
- Bolen, J.B.; Rosen, N.; Israel, M.A. Increased pp60c-src tyrosyl kinase activity in human neuroblastoma is associated with amino-terminal tyrosine phosphorylation of the src gene product. Proc. Natl. Acad. Sci. USA 1985, 82, 7275–7279. [Google Scholar] [CrossRef] [PubMed]
- Finlay, D.; Vuori, K. Novel noncatalytic role for caspase-8 in promoting SRC-mediated adhesion and Erk signaling in neuroblastoma cells. Cancer Res. 2007, 67, 11704–11711. [Google Scholar] [CrossRef] [PubMed]
- Palacios-Moreno, J.; Foltz, L.; Guo, A.; Stokes, M.P.; Kuehn, E.D.; George, L.; Comb, M.; Grimes, M.L. Neuroblastoma tyrosine kinase signaling networks involve FYN and LYN in endosomes and lipid rafts. PLoS Comput. Biol. 2015, 11, e1004130. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Bernasconi, P.; Clauser, K.R.; Mani, D.R.; Finn, S.P.; Beroukhim, R.; Burns, M.; Julian, B.; Peng, X.P.; Hieronymus, H.; et al. Bead-based profiling of tyrosine kinase phosphorylation identifies SRC as a potential target for glioblastoma therapy. Nat. Biotechnol. 2009, 27, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Eramo, A.; Ricci-Vitiani, L.; Zeuner, A.; Pallini, R.; Lotti, F.; Sette, G.; Pilozzi, E.; Larocca, L.M.; Peschle, C.; De Maria, R. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ. 2006, 13, 1238–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aid. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Cmpd | R1 | R2 | Src Docking Score (kcal/mol) | Src IC50 (nM) a | Fyn IC50 (nM) a | Lyn IC50 (nM) a | Yes IC50 (nM) a |
|---|---|---|---|---|---|---|---|
| dasat. | <4 b | <9 b | <0.9 b | <3 b | |||
| 2 | H | -CH2CH2OH | −10.02 | 220 ±0.03 | 689 ± 0.10 | 1300 ± 0.02 | 167 ± 0.03 |
| 4a | -3OMe | -CH2CH2OH | −10.10 | 260 ± 23 | 146 ± 30 | 451 ± 13 | 560 ± 40 |
| 4b | -3OMe | -CH2CONH2 | −7.26 | 1476 ± 185 | 1454 ± 768 | 1808 ± 307 | 1843 ± 812 |
| 4c | -2OMe | -CH2CH2OH | −10.58 | 225 ± 8 | 439 ± 45 | 145 ± 31 | 55 ± 9 |
| 4d | -2OMe | -CH2CONH2 | −8.33 | 1533 ± 44 | 1387 ± 93 | 1205 ± 189 | NA |
| 4e | -2OH | -CH2CH2OH | −10.97 | 59 ± 5 | 73 ± 5 | 17 ± 0.54 | 11 ± 0.6 |
| 4f | -2OH | -H | −10.98 | 56 ± 5 | 50 ± 4 | 51 ± 3 | 3 ± 1 |
| 4g | -2OH | -CH2CONH2 | −11.23 | 40 ± 5 | 40 ± 5 | 19 ± 2 | 15 ± 4 |
| 4h | -3OH | -CH2CH2OH | −10.80 | 93 ± 19 | 10 ± 0.4 | 20 ± 0.8 | 3 ± 1 |
| 4i | -3OH | -H | −10.68 | 83 ± 18 | 10 ± 0.4 | 27 ± 1 | 3 ± 0.8 |
| 4j | -3OH | -CH2CONH2 | −11.33 | 40 ± 3 | 12 ± 2 | 10 ± 1.2 | 3 ± 0.7 |
| 4k | -3OH | -CH2COOMe | −10.99 | 40 ± 2 | 9 ± 1 | 18 ± 1 | 3 ± 1 |
| 4l | -3OH | -COOtert-butyl | −10.99 | 50 ± 3 | 14 ± 1.5 | 26 ± 3 | 7 ± 0.35 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Francini, C.M.; Musumeci, F.; Fallacara, A.L.; Botta, L.; Molinari, A.; Artusi, R.; Mennuni, L.; Angelucci, A.; Schenone, S. Optimization of Aminoimidazole Derivatives as Src Family Kinase Inhibitors. Molecules 2018, 23, 2369. https://doi.org/10.3390/molecules23092369
Francini CM, Musumeci F, Fallacara AL, Botta L, Molinari A, Artusi R, Mennuni L, Angelucci A, Schenone S. Optimization of Aminoimidazole Derivatives as Src Family Kinase Inhibitors. Molecules. 2018; 23(9):2369. https://doi.org/10.3390/molecules23092369
Chicago/Turabian StyleFrancini, Cinzia Maria, Francesca Musumeci, Anna Lucia Fallacara, Lorenzo Botta, Alessio Molinari, Roberto Artusi, Laura Mennuni, Adriano Angelucci, and Silvia Schenone. 2018. "Optimization of Aminoimidazole Derivatives as Src Family Kinase Inhibitors" Molecules 23, no. 9: 2369. https://doi.org/10.3390/molecules23092369