d-Amino Acid Pseudopeptides as Potential Amyloid-Beta Aggregation Inhibitors

Abstract
:
1. Introduction
2. Methods
2.1. The Interaction of SGB1 and SGD1 with R (Aβ13–23)
2.1.1. Molecular Dynamics Simulation
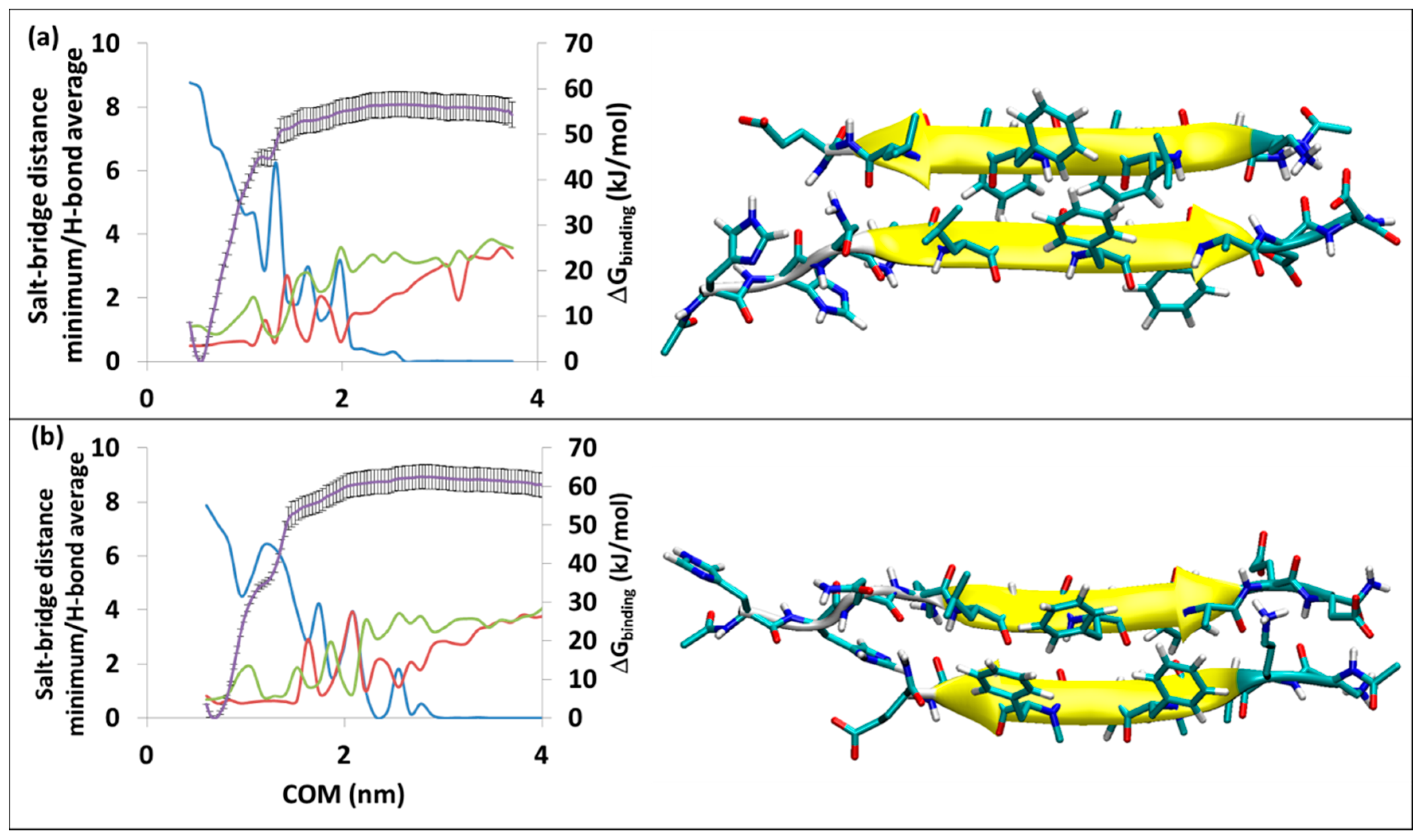
2.1.2. Steered MD (MD-SMD) Calculations and Umbrella Sampling Calculation (MD-US)
2.2. The Interaction of SGB1 and SGD1 with Aβ42
2.2.1. Molecular Dynamics Simulation
2.2.2. Relative Energy Determination
3. Results for SGB1, SGD1, SGB1-R and SGD1-R
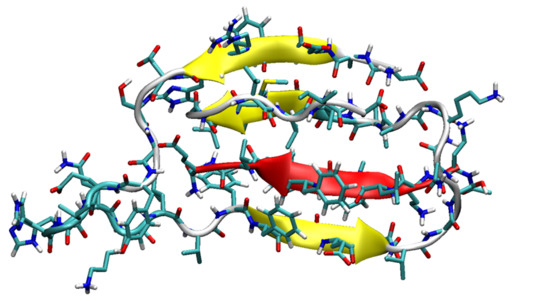
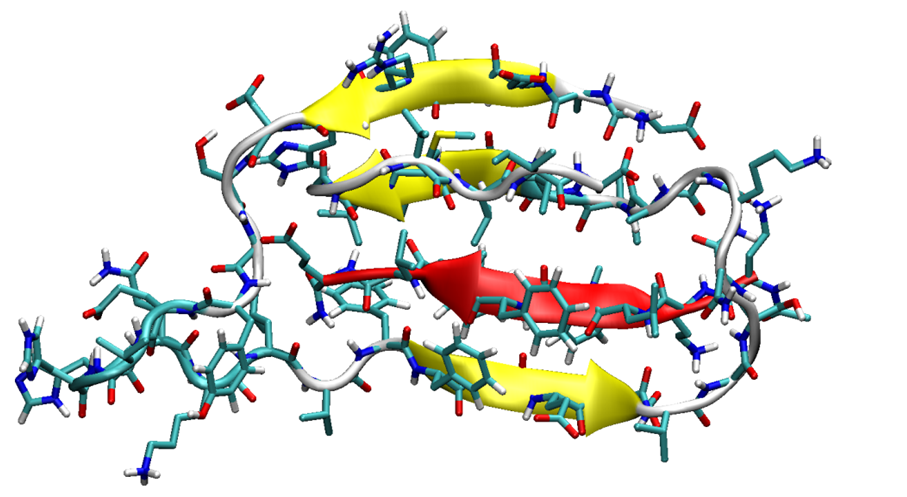
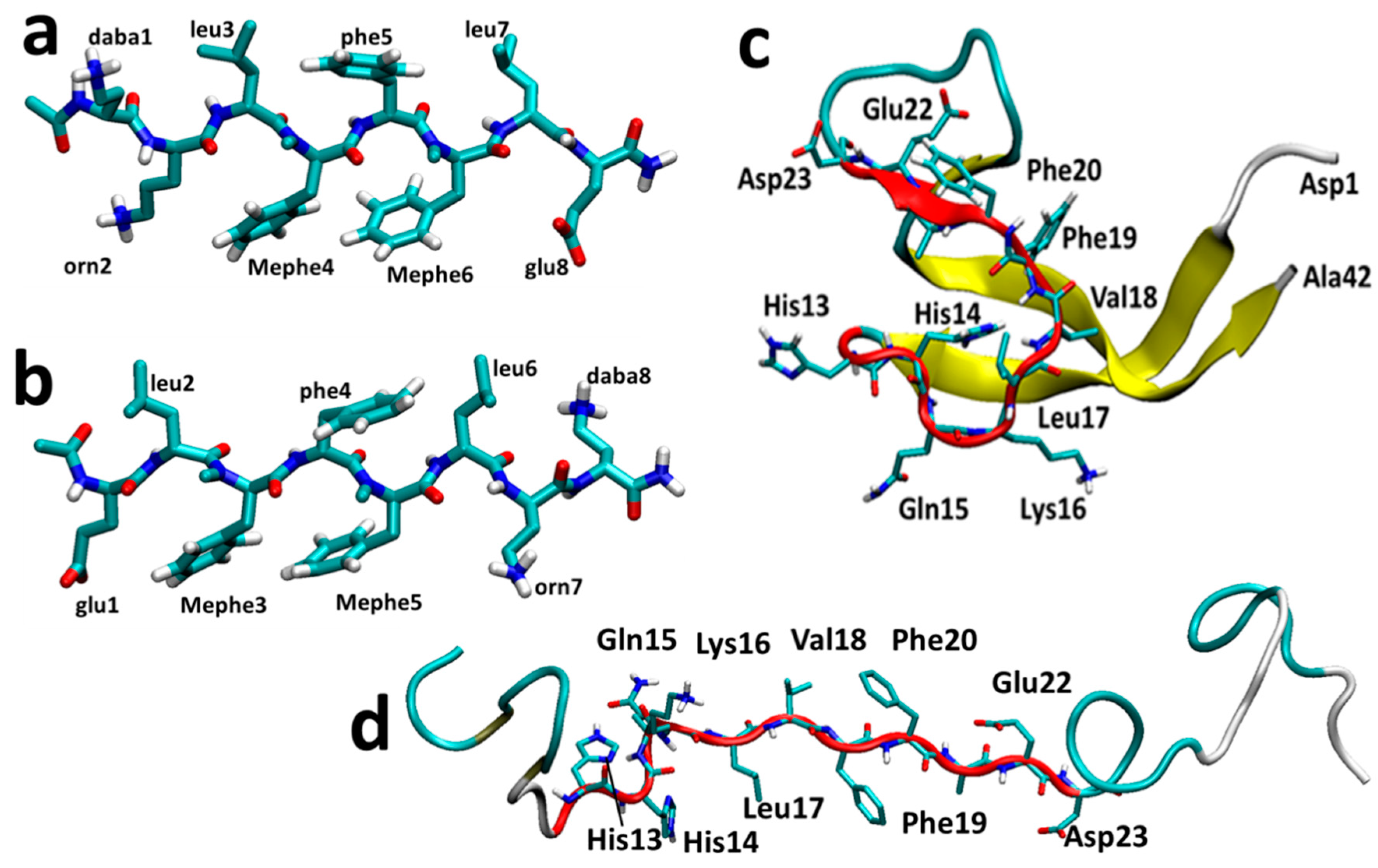
3.1. The Monomers
3.2. The Binary Complexes, SGB1-R, SGD1-R, SGB1-SGB1 and SGD1-SGD1
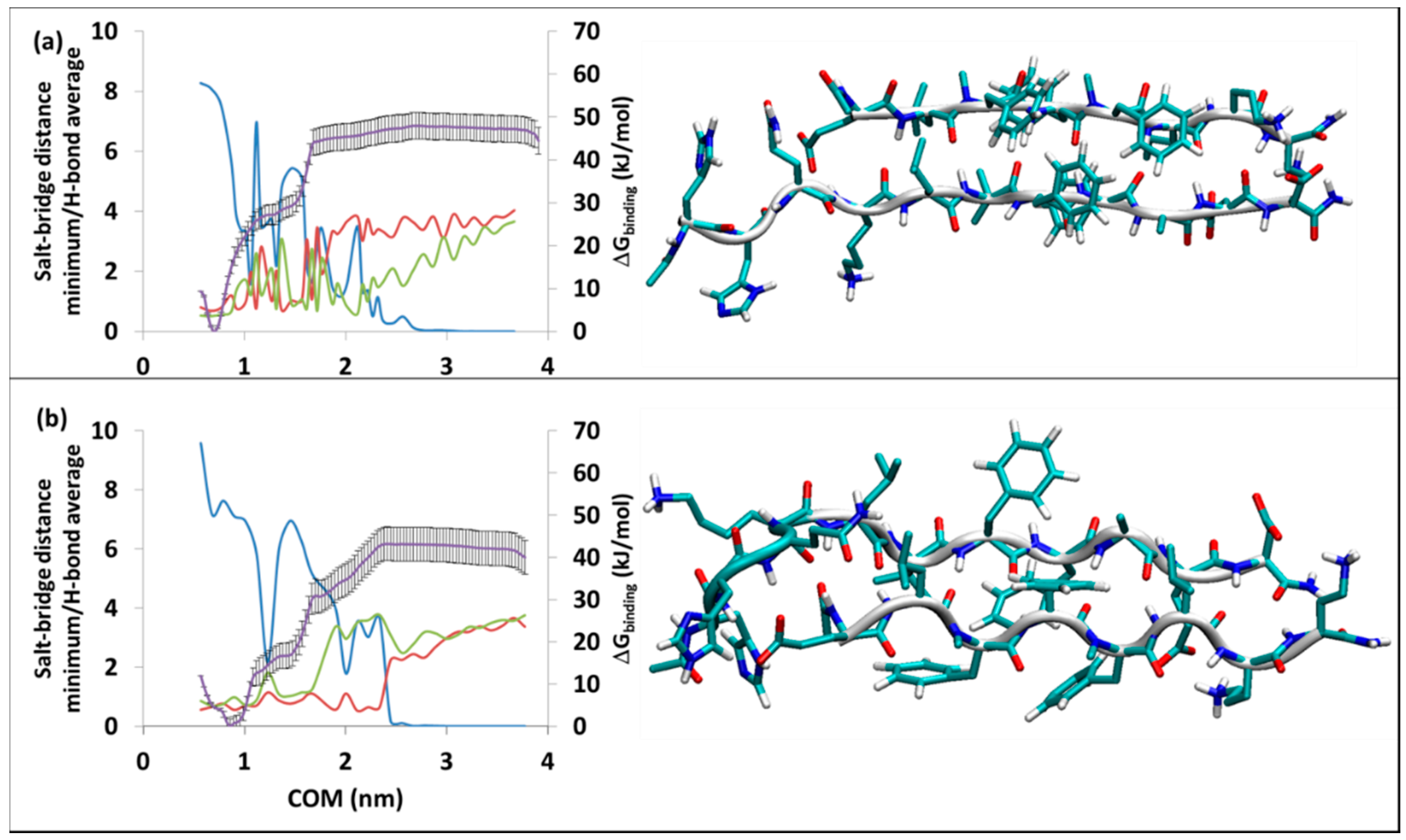
3.2.1. The RT-SGB1 and RB-SGB1
3.2.2. The RT-SGD1 and RB-SGD1
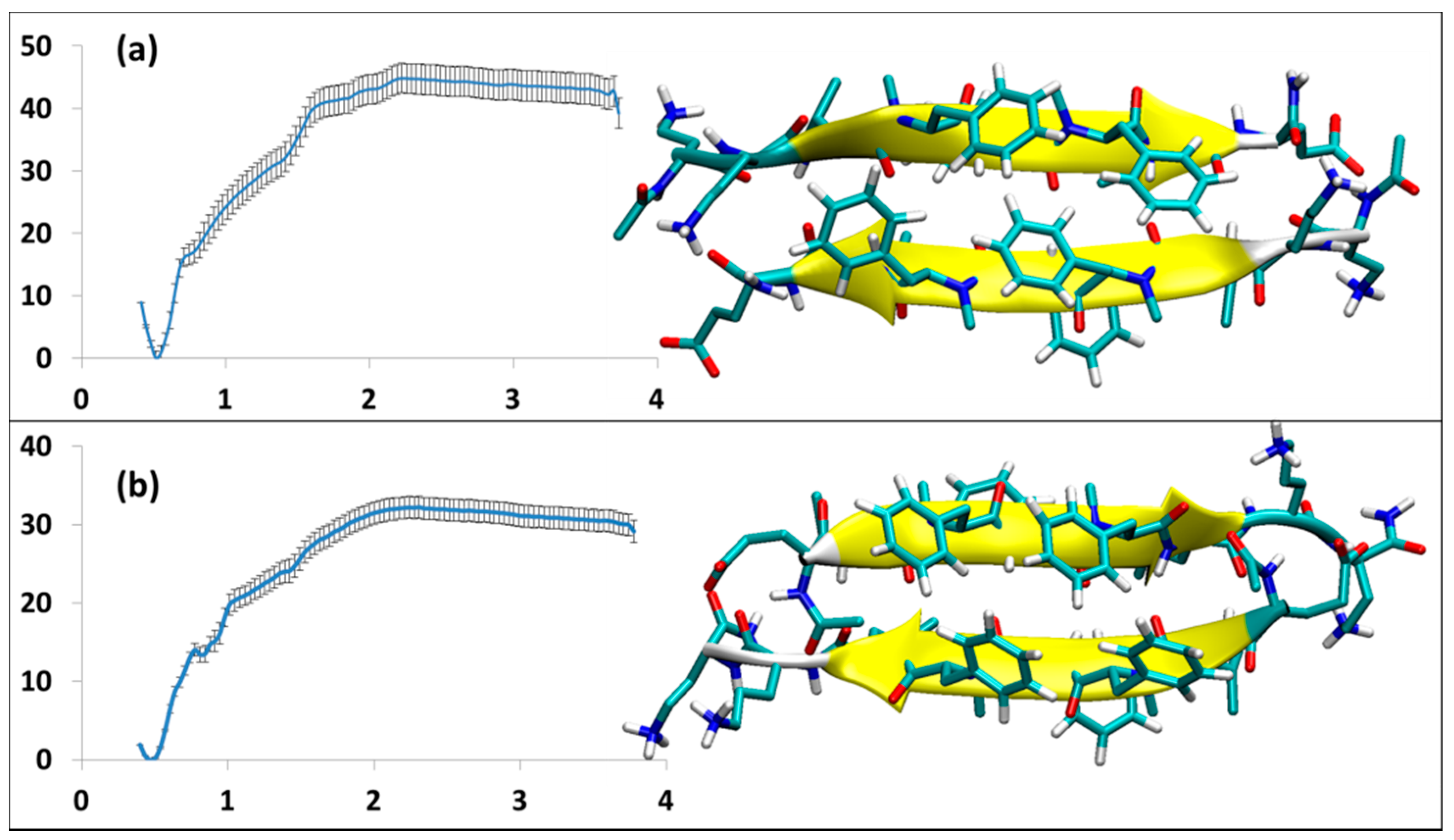
3.2.3. SGB1 and SGD1 Homodimers
3.3. The Effective ∆Geff
4. Results for Aβ42-SGB1 and Aβ42-SGD1
4.1. Aβ42-SGB1
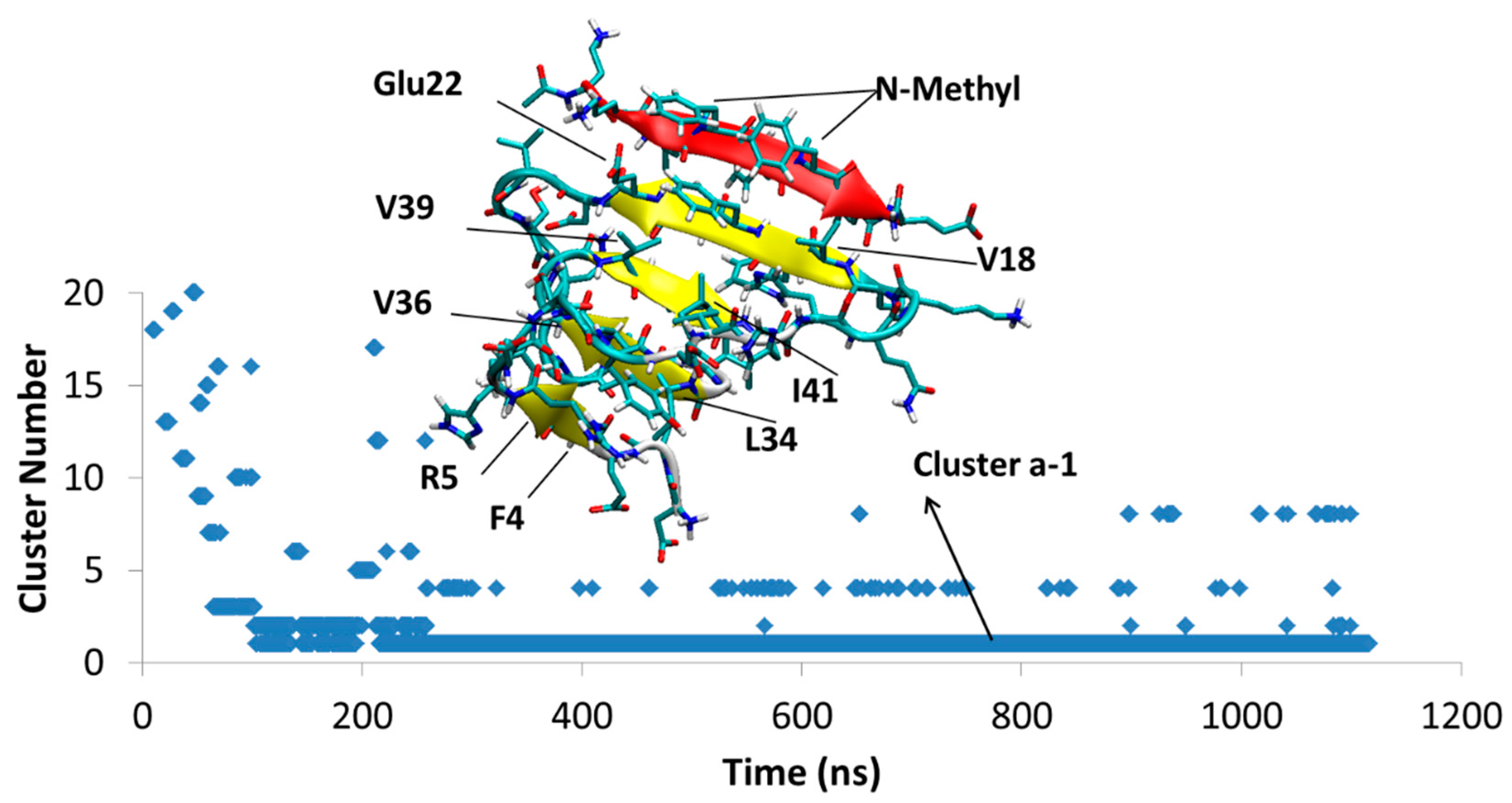
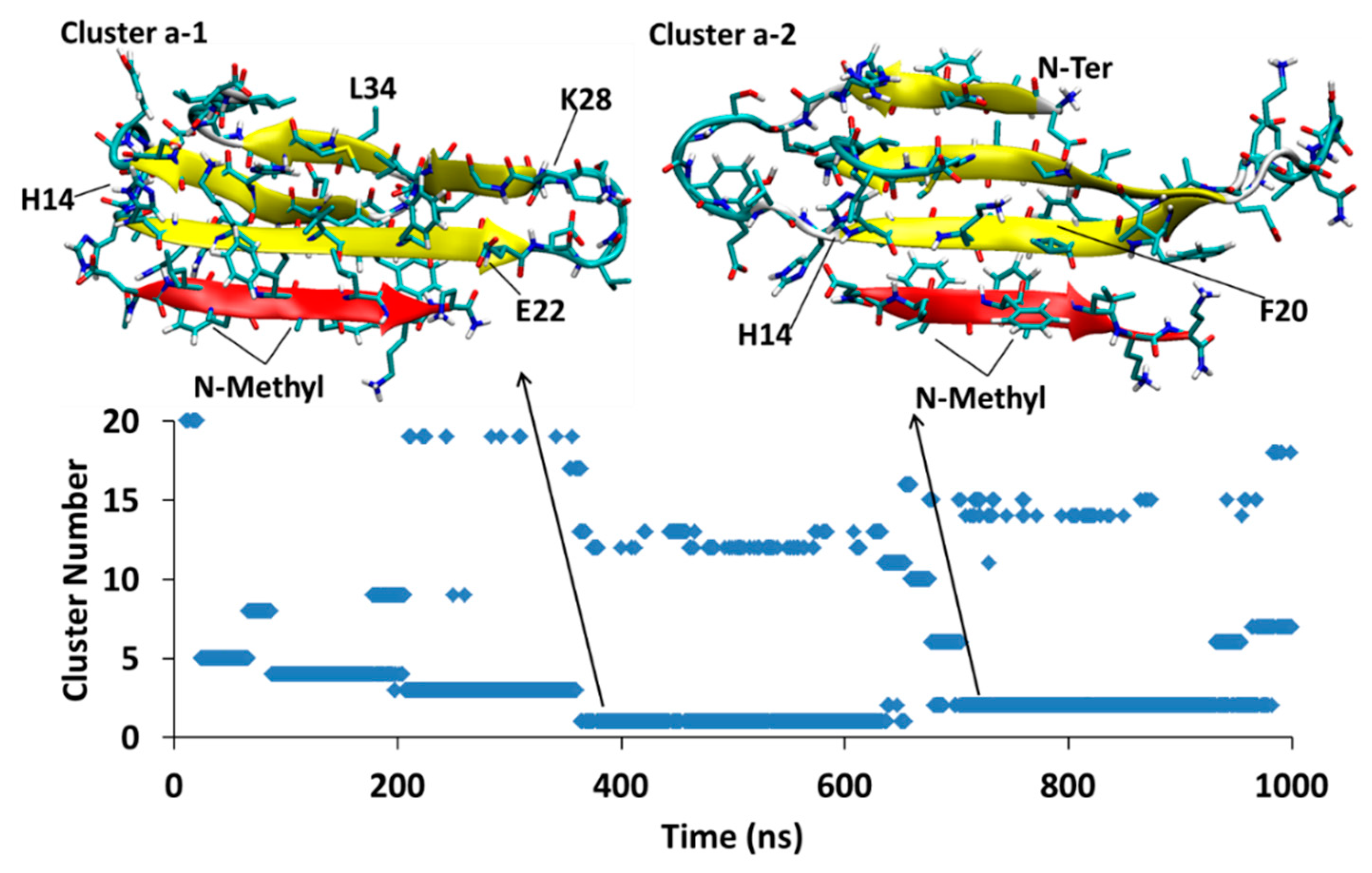
4.1.1. AβT-SGB1-a
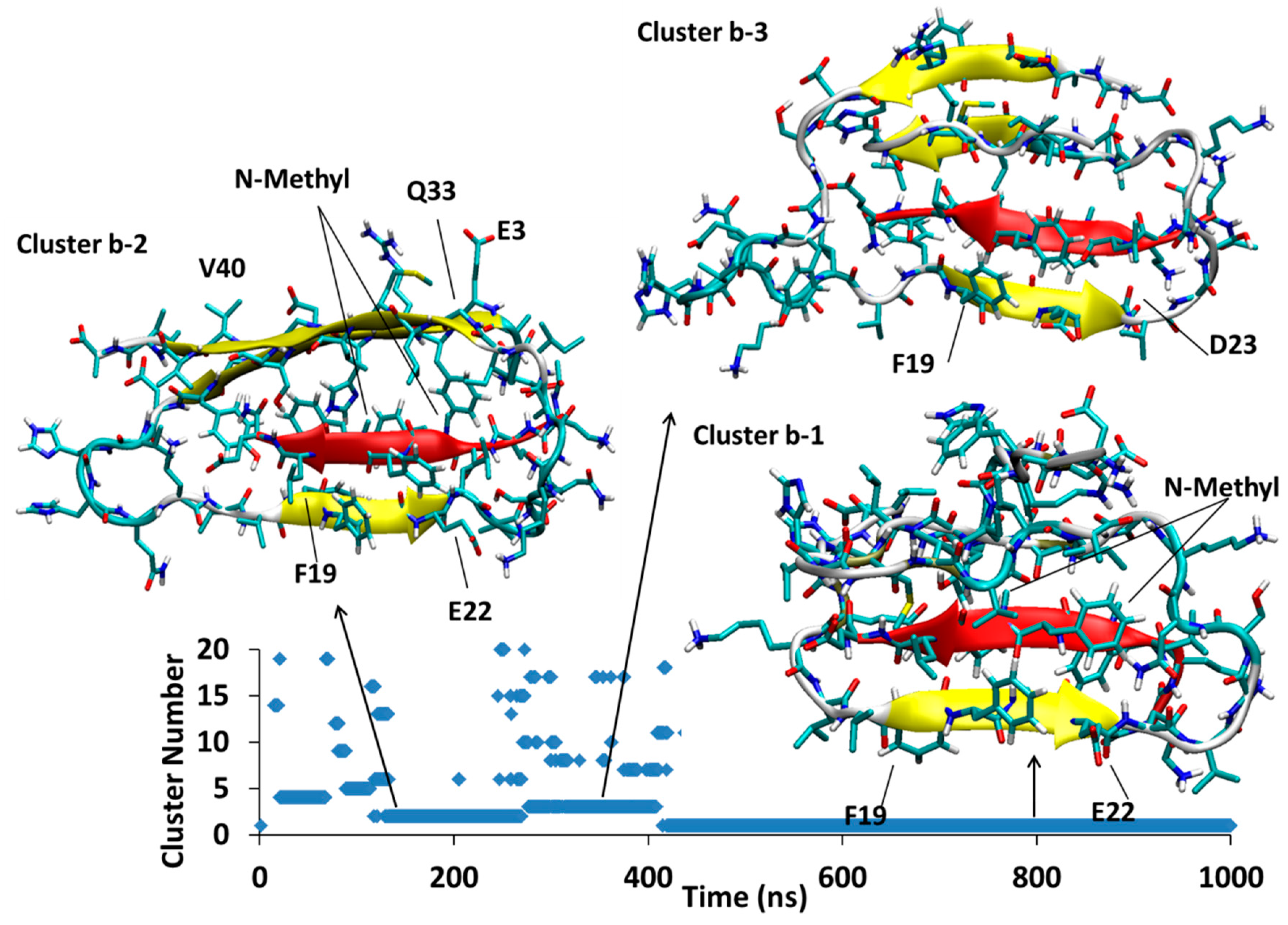
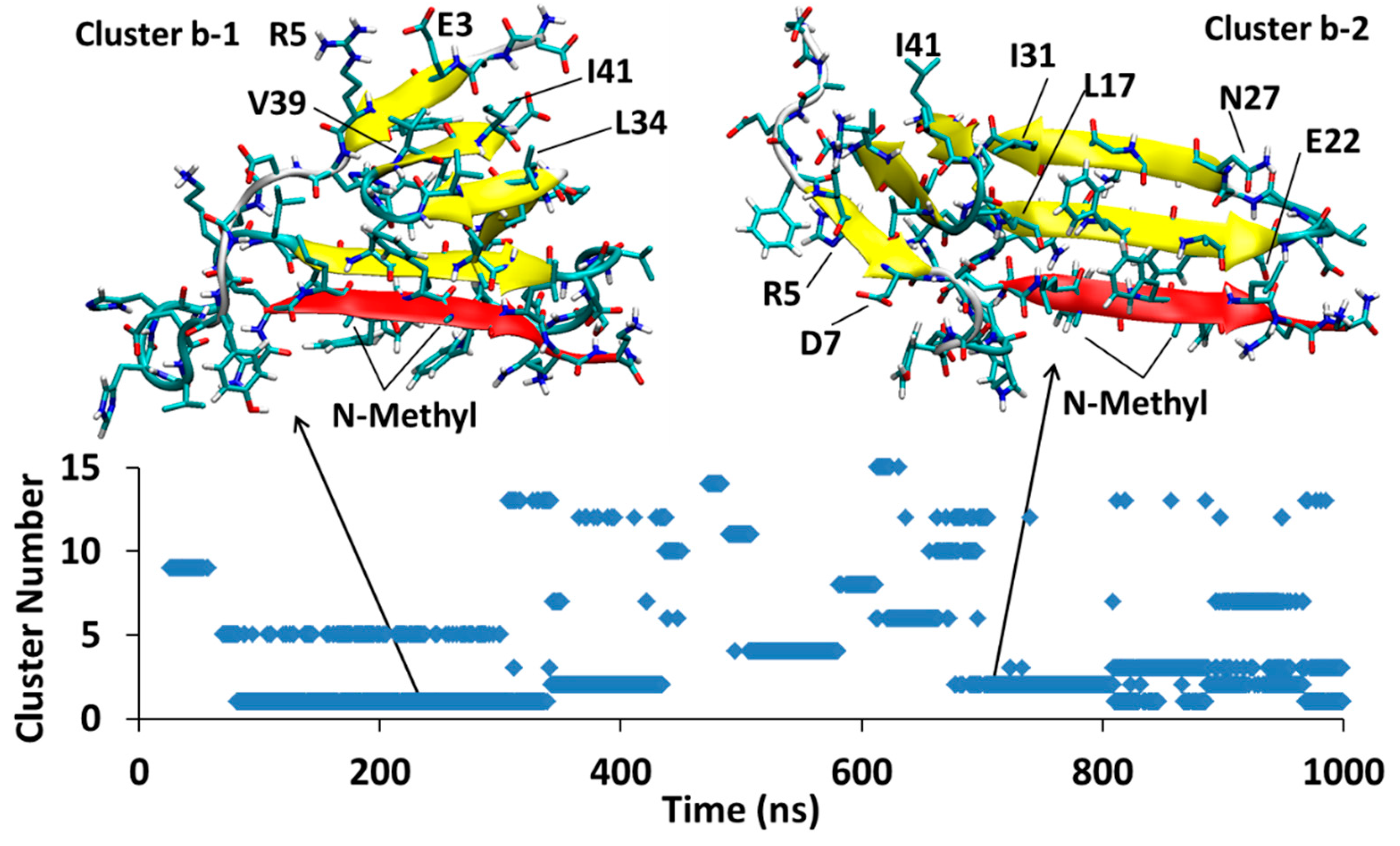
4.1.2. AβB-SGB1-b
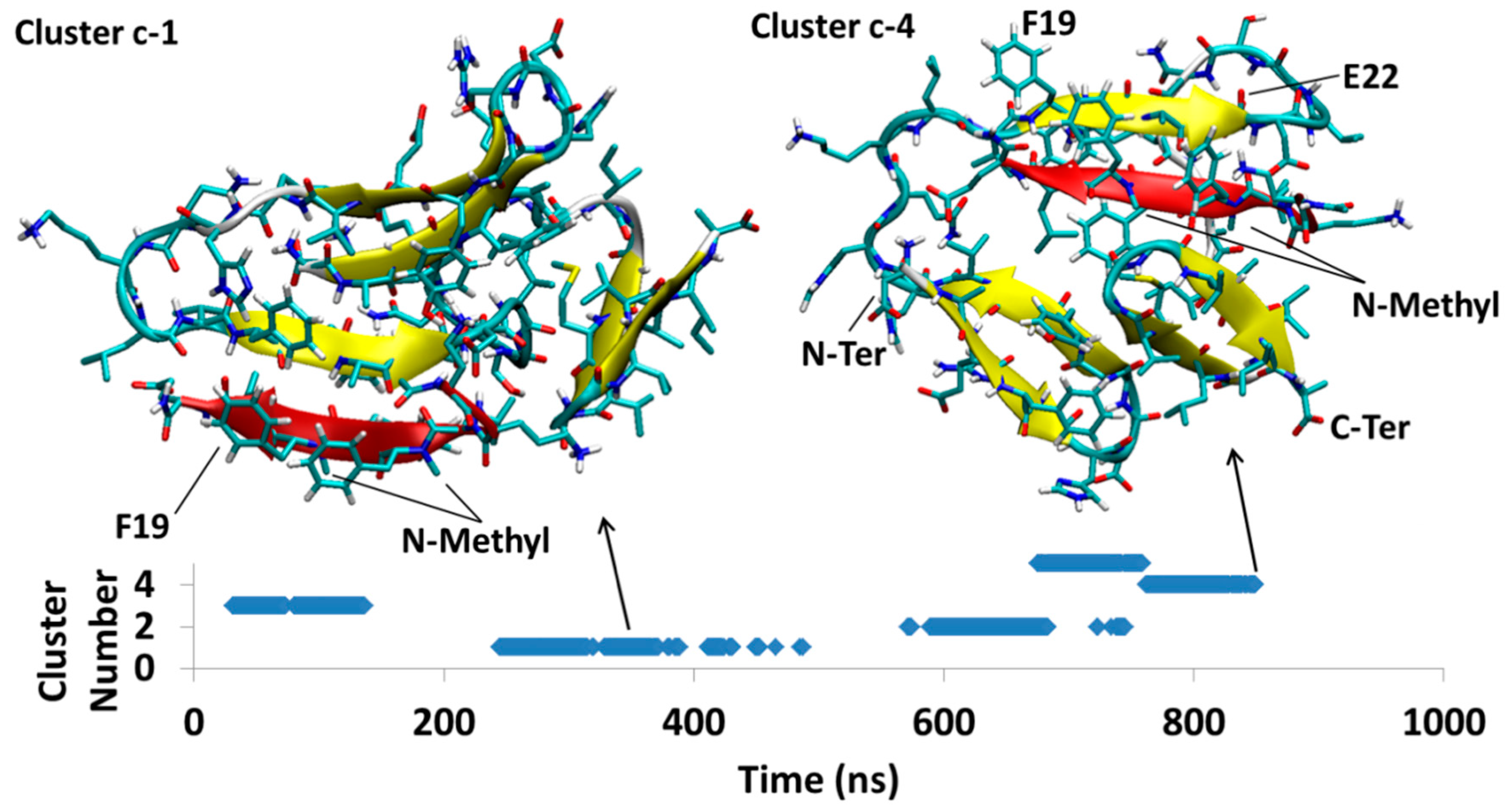
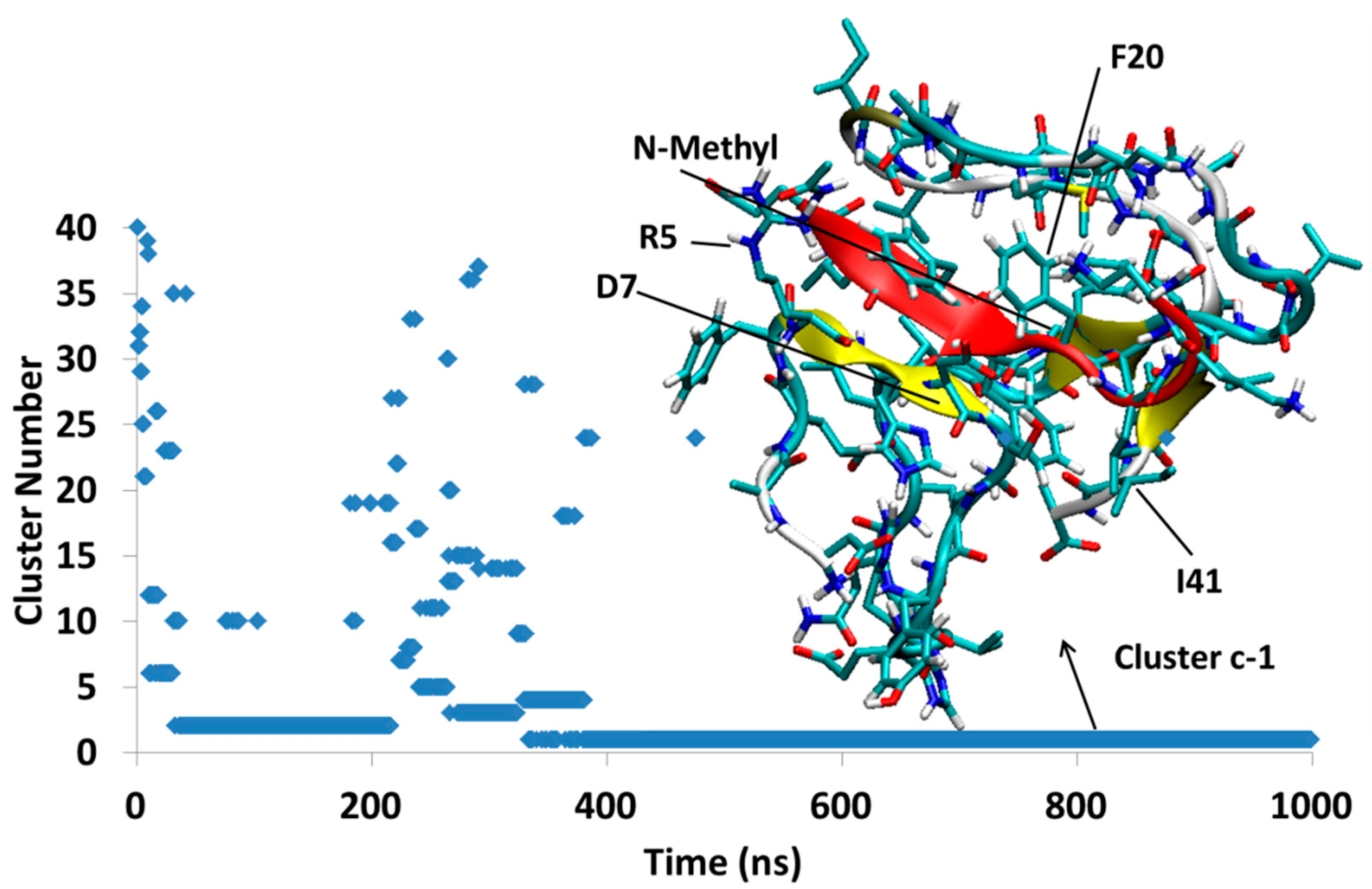
4.1.3. AβB-SGB1-c
4.1.4. Relative Energies of the Aβ42-SGB1 Complexes
4.2. Aβ42-SGD1
4.2.1. AβT-SGD1-a
4.2.2. AβB-SGD1-b
4.2.3. Aβ-SGD1-c
4.2.4. AβB-SGD1-d
4.2.5. Relative Energies of the Aβ42-SGD1 Complexes
5. Discussion
5.1. The PP-R Complexes
5.2. The PP-Aβ Complexes
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Querfurth, H.W.; Laferla, F.M. Alzheimer’s Disease. N. Engl. J. Med. Rev. 2010, 4, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2015 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2015, 11, 332–384. [Google Scholar] [CrossRef]
- Hardy, J. The Amyloid Hypothesis for Alzheimer’s Disease: A Critical Reappraisal. J. Neurochem. 2009, 110, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Rauk, A. The Chemistry of Alzheimer’s Disease. Chem. Soc. Rev. 2009, 38, 2698–2715. [Google Scholar] [CrossRef] [PubMed]
- Rauk, A. Why Is the Amyloid Beta Peptide of Alzheimer’s Disease Neurotoxic? Dalton Trans. 2008, 10, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Anand, U.; Mukherjee, M. Exploring the Self-Assembly of a Short Aromatic Aβ16–24 Peptide. Langmuir 2013, 29, 2713–2721. [Google Scholar] [CrossRef] [PubMed]
- Cecchini, M.; Curcio, R.; Pappalardo, M.; Melki, R.; Caflisch, A. A Molecular Dynamics Approach to the Structural Characterization of Amyloid Aggregation. J. Mol. Biol. 2006, 357, 1306–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mousseau, N.; Derreumaux, P. Exploring the Early Steps of Amyloid Peptide Aggregation by Computers. Acc. Chem. Res. 2005, 38, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Selkoe, D.J. Soluble Protein Oligomers in Neurodegeneration: Lessons from the Alzheimer’s Amyloid Beta-Peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Härd, T.; Lendel, C. Inhibition of Amyloid Formation. J. Mol. Biol. 2012, 421, 441–465. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.L.; Johnson, B.R.G.; Urbanc, B.; Jenkins, A.T.A.; Connell, S.D.A.; Serpell, L.C. Aβ42 Oligomers but Not Fibrils, Simultaneously Bind to and Cause Damage to Ganglioside-Containing Lipid Membranes. Biochem. J. 2011, 439, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Cecchi, C.; Stefani, M. The Amyloid-Cell Membrane System. The Interplay between the Biophysical Features of Oligomers/fibrils and Cell Membrane Defines Amyloid Toxicity. Biophys. Chem. 2013, 182, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Winklhofer, K.F.; Tatzelt, J.; Haass, C. The Two Faces of Protein Misfolding: Gain- and Loss-of-Function in Neurodegenerative Diseases. EMBO J. 2008, 27, 336–349. [Google Scholar] [CrossRef] [PubMed]
- Treusch, S.; Cyr, D.M.; Lindquist, S. Amyloid Deposits: Protection against Toxic Protein Species? Cell Cycle 2009, 8, 1668–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benilova, I.; Karran, E.; De Strooper, B. The Toxic Aβ Oligomer and Alzheimer’s Disease: An Emperor in Need of Clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Cerf, E.; Sarroukh, R.; Tamamizu-Kato, S.; Breydo, L.; Derclaye, S.; Dufrêne, Y.F.; Narayanaswami, V.; Goormaghtigh, E.; Ruysschaert, J.-M.; Raussens, V. Antiparallel Beta-Sheet: A Signature Structure of the Oligomeric Amyloid Beta-Peptide. Biochem. J. 2009, 421, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Condron, M.M.; Teplow, D.B. Structure – Neurotoxicity Relationships of Amyloid Beta Protein Oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 14745. [Google Scholar] [CrossRef] [PubMed]
- Tjernberg, L.O.; Näslund, J.; Lindqvist, F.; Johansson, J.; Karlström, R.; Thyberg, J.; Terenius, L.; Nordstedt, C. Arrest of Beta-Amyloid Fibril Formation by a Pentapeptide Ligand. J. Biol. Chem. 1996, 271, 8545–8548. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, G.G.; Cavalli, A.; Pellarin, R.; Caflisch, A. Prediction of Aggregation Rate and Aggregation-Prone Segments in Polypeptide Sequences. Protein Sci. 2005, 14, 2723–2734. [Google Scholar] [CrossRef] [PubMed]
- Bitan, G.; Vollers, S.S.; Teplow, D.B. Elucidation of Primary Structure Elements Controlling Early Amyloid b-Protein Oligomerization. J. Biol. Chem. 2003, 278, 34882–34889. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, K.L.; Gordon, D.J.; Petkova, A.T.; Tycko, R.; Meredith, S.C. Aβ 40-lactam(D23/K28) Models a Conformation Highly Favorable for Nucleation of Amyloid. Biochemistry 2005, 44, 6003–6014. [Google Scholar] [CrossRef] [PubMed]
- Lazo, N.D.; Grant, M.A.; Condron, M.C.; Rigby, A.C.; Teplow, D.B. On the Nucleation of Amyloid β-Protein Monomer Folding. Protein Sci. 2005, 14, 1581–1596. [Google Scholar] [CrossRef] [PubMed]
- Mothana, B.; Roy, S.; Rauk, A. Molecular Dynamics Study of the Interaction of Aβ13–23 with β-Sheet Inhibitors. Arkivoc 2009, 2009, 116–134. [Google Scholar]
- Raffa, D.F.; Rickard, G. a.; Rauk, A. Ab Initio Modelling of the Structure and Redox Behaviour of Copper(I) Bound to a His-His Model Peptide: Relevance to the b-Amyloid Peptide of Alzheimer’s Disease. J. Biol. Inorg. Chem. 2007, 12, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Raffa, D.F.; Rauk, A. Molecular Dynamics Study of the Beta Amyloid Peptide of Alzheimer’s Disease and Its Divalent Copper Complexes. J. Phys. Chem. B 2007, 111, 3789–3799. [Google Scholar] [CrossRef] [PubMed]
- Curtain, C.C.; Ali, F.; Volitakis, I.; Cherny, R.A.; Norton, R.S.; Beyreuther, K.; Barrow, C.J.; Masters, C.L.; Bush, A.I.; Barnham, K.J. Alzheimer’s Disease Amyloid-β Binds Copper and Zinc to Generate an Allosterically Ordered Membrane-Penetrating Structure Containing Superoxide Dismutase-like Subunits. J. Biol. Chem. 2001, 276, 20466–20473. [Google Scholar] [CrossRef] [PubMed]
- Karr, J.W.; Akintoye, H.; Kaupp, L.J.; Szalai, V.A. N-Terminal Deletions Modify the Cu2+ Binding Site in Amyloid-b. Biochemistry 2005, 44, 5478–5487. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.T.; Howlett, G.; Barrow, C.J. Histidine-13 Is a Crucial Residue in the Zinc Ion-Induced Aggregation of the Aβ Peptide of Alzheimer’s Disease. Biochemistry 1999, 38, 9373–9378. [Google Scholar] [CrossRef] [PubMed]
- 29. Samir Roy. Novel Beta-Amyloid Oligomerization Inhibitors. Ph.D. Thesis, University of Calgary, Calgary, Canada, December 2010.
- Mehrazma, B.; Petoyan, A.; Opare, S.K.A.; Rauk, A. Interaction of the N-AcAβ13–23NH2 Segment of the Beta Amyloid Peptide with Beta-Sheet-Blocking Peptides: Site and Edge Specificity. Can. J. Chem. 2016, 6, 583–592. [Google Scholar] [CrossRef]
- Mehrazma, B.; Robinson, M.; Opare, S.K.A.; Petoyan, A.; Lou, J.; Hane, F.T.; Rauk, A.; Leonenko, Z. Pseudo-Peptide Amyloid-β Blocking Inhibitors: Molecular Dynamics and Single Molecule Force Spectroscopy Study. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 1707–1718. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Kindy, M.S.; Baumann, M.; Frangione, B. Inhibition of Alzheimer’s Amyloidosis by Peptides That Prevent Beta-Sheet Conformation. Biochem. Biophys. Res. Commun. 1996, 226, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Poduslo, J.F.; Curran, G.L.; Kumar, A.; Frangione, B.; Soto, C. β-Sheet Breaker Peptide Inhibitor of Alzheimer’s Amyloidogenesis with Increased Blood—Brain Barrier Permeability and Resistance to Proteolytic Degradation in Plasma. J. Neurobiol. 1999, 39, 371–382. [Google Scholar] [CrossRef]
- Findeis, M.A.; Musso, G.M.; Arico-Muendel, C.C.; Benjamin, H.W.; Hundal, A.M.; Lee, J.J.; Chin, J.; Kelley, M.; Wakefield, J.; Hayward, N.J.; et al. Modified-Peptide Inhibitors of Amyloid β-Peptide Polymerization. Biochemistry 1999, 38, 6791–6800. [Google Scholar] [CrossRef] [PubMed]
- Zawadzke, L.E.; Berg, J.M. A Racemic Protein. J. Am. Chem. Soc. 1992, 114, 4002–4003. [Google Scholar] [CrossRef]
- Dintzis, H.M.; Symer, D.E.; Dintzis, R.Z.; Zawadzke, L.E.; Berg, J.M. A Comparison of the Immunogenicity of a Pair of Enantiomeric Proteins. Proteins Struct. Funct. Bioinform. 1993, 16, 306–308. [Google Scholar] [CrossRef] [PubMed]
- Kokkoni, N.; Stott, K.; Amijee, H.; Mason, J.M.; Doig, A.J. N-Methylated Peptide Inhibitors of β-Amyloid Aggregation and Toxicity. Optimization of the Inhibitor Structure. Biochemistry 2006, 45, 9906–9918. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.J.; Tappe, R.; Meredith, S.C. Design and Characterization of a Membrane Permeable N-Methyl Amino Acid-Containing Peptide That Inhibits Abeta1–40 Fibrillogenesis. J. Pept. Res. 2002, 60, 37–55. [Google Scholar] [CrossRef] [PubMed]
- Grillo-Bosch, D.; Carulla, N.; Cruz, M.; Sánchez, L.; Pujol-Pina, R.; Madurga, S.; Rabanal, F.; Giralt, E. Retro-Enantio N-Methylated Peptides as β-Amyloid Aggregation Inhibitors. Chem. Med. 2009, 4, 1488–1494. [Google Scholar] [CrossRef] [PubMed]
- Chebaro, Y.; Derreumaux, P. Targeting the Early Steps of ApiG-22 Protofibril Disassembly by N-Methylated Inhibitors: A Numerical Study. Proteins Struct. Funct. Bioinform. 2009, 75, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Albericio, F.; Serratosa, J.; Rabanal, F.; Giralt, E. Inhibition of β-Amyloid Toxicity by Short Peptides Containing N-Methyl Amino Acids. J. Pept. Res. 2004, 324–328. [Google Scholar]
- Kumar, J.; Sim, V. d-Amino Acid-Based Peptide Inhibitors as Early or Preventative Therapy in Alzheimer Disease. Prion 2014, 8, 1–6. [Google Scholar] [CrossRef]
- Chalifour, R.J.; McLaughlin, R.W.; Lavoie, L.; Morissette, C.; Tremblay, N.; Boulé, M.; Sarazin, P.; Stéa, D.; Lacombe, D.; Tremblay, P.; et al. Stereoselective Interactions of Peptide Inhibitors with the β-Amyloid Peptide. J. Biol. Chem. 2003, 278, 34874–34881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesehan, K.; Buder, K.; Linke, R.P.; Patt, S.; Stoldt, M.; Unger, E.; Schmitt, B.; Bucci, E.; Willbold, D. Selection of d-Amino-Acid Peptides That Bind to Alzheimer’s Disease Amyloid Peptide abeta1-42 by Mirror Image Phage Display. Chem. Biochem. 2003, 4, 748–753. [Google Scholar]
- Wiesehan, K.; Stöhr, J.; Nagel-Steger, L.; Van Groen, T.; Riesner, D.; Willbold, D. Inhibition of Cytotoxicity and Amyloid Fibril Formation by a d-Amino Acid Peptide That Specifically Binds to Alzheimer’s Disease Amyloid Peptide. Protein Eng. Des. Sel. 2008, 21, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Van Groen, T.; Wiesehan, K.; Funke, S.A.; Kadish, I.; Nagel-Steger, L.; Willbold, D. Reduction of Alzheimer’s Disease Amyloid Plaque Load in Transgenic Mice by D3, a D-Enantiomeric Peptide Identified by Mirror Image Phage Display. ChemMedChem Chem. Enabling Drug Dis. 2008, 3, 1848–1852. [Google Scholar] [CrossRef] [PubMed]
- Aileen Funke, S.; Van Groen, T.; Kadish, I.; Bartnik, D.; Nagel-Steger, L.; Brener, O.; Sehl, T.; Batra-Safferling, R.; Moriscot, C.; Schoehn, G.; et al. Oral Treatment with the D-Enantiomeric Peptide D3 Improves the Pathology and Behavior of Alzheimer’s Disease Transgenic Mice. ACS Chem. Neurosci. 2010, 1, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Parthsarathy, V.; McClean, P.L.; Hölscher, C.; Taylor, M.; Tinker, C.; Jones, G.; Kolosov, O.; Salvati, E.; Gregori, M.; Masserini, M.; et al. A Novel Retro-Inverso Peptide Inhibitor Reduces Amyloid Deposition, Oxidation and Inflammation and Stimulates Neurogenesis in the APPswe/PS1ΔE9 Mouse Model of Alzheimer’s Disease. PLoS ONE 2013, 8, e54769. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Gr. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; Van Gunsteren, W.F. A Biomolecular Force Field Based on the Free Enthalpy of Hydration and Solvation: The GROMOS Force-Field Parameter Sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Hoover, W.G. Canonical Dynamics: Equilibrium Phase-Space Distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Nosé, S. A Molecular Dynamics Method for Simulations in the Canonical Ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Nosé, S.; Klein, M.L. Constant Pressure Molecular Dynamics for Molecular Systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Opare, S.K.A.; Petoyan, A.; Mehrazma, B.; Rauk, A. Molecular Dynamics Study of the Monomers and Dimers of N-AcAβ13–23 NH2: On the Effect of pH on the Aggregation of the Amyloid Beta Peptide of Alzheimer ’s Disease. Can. J. Chem. 2015, 94, 273–281. [Google Scholar] [CrossRef]
- Ghoorah, A.W.; Devignes, M.D.; Smaïl-Tabbone, M.; Ritchie, D.W. Protein Docking Using Case-Based Reasoning. Proteins Struct. Funct. Bioinform. 2013, 81, 2150–2158. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, D.W.; Venkatraman, V. Ultra-Fast FFT Protein Docking on Graphics Processors. Bioinformatics 2010, 26, 2398–2405. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, D.W.; Kozakov, D.; Vajda, S. Accelerating and Focusing Protein-Protein Docking Correlations Using Multi-Dimensional Rotational FFT Generating Functions. Bioinformatics 2008, 24, 1865–1873. [Google Scholar] [CrossRef] [PubMed]
- Macindoe, G.; Mavridis, L.; Venkatraman, V.; Devignes, M.D.; Ritchie, D.W. HexServer: An FFT-Based Protein Docking Server Powered by Graphics Processors. Nucleic Acids Res. 2010, 38, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; Hermans, J. Interaction Models for Water in Relation to Protein Hydration. Intermol. Forces 1981, 331–342. [Google Scholar]
- Steffen, C.; Thomas, K.; Huniar, U.; Hellweg, A.; Rubner, O.; Schroer, A. TmoleX—A Graphical User Interface for TURBOMOLE. J. Comput. Chem. 2010, 31, 2967–2970. [Google Scholar] [CrossRef] [PubMed]
- Hub, J.S.; De Groot, B.L.; Van Der Spoel, D. g-Whams-a Free Weighted Histogram Analysis Implementation Including Robust Error and Autocorrelation Estimates. J. Chem. Theory Comput. 2010, 6, 3713–3720. [Google Scholar] [CrossRef]
- Aqvist, J.; Medina, C.; Samuelsson, J.E. A New Method for Predicting Binding Affinity in Computer-Aided. Drug Des. Protein Eng. 1994, 7, 385–391. [Google Scholar] [CrossRef]
- Hansson, T.; Marelius, J.; Aqvist, J. Ligand Binding Affinity Prediction by Linear Interaction Energy Methods. J. Comput. Aided Mol. Des. 1998, 12, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Almlöf, M.; Carlsson, J.; Åqvist, J. Improving the Accuracy of the Linear Interaction Energy Method for Solvation Free Energies. J. Chem. Theory Comput. 2007, 3, 2162–2175. [Google Scholar] [CrossRef] [PubMed]
- Miranda, W.E.; Noskov, S.Y.; Valiente, P.A. Improving the LIE Method for Binding Free Energy Calculations of Protein-Ligand Complexes. J. Chem. Inf. Model. 2015, 55, 1867–1877. [Google Scholar] [CrossRef] [PubMed]
- Hess, B. Determining the Shear Viscosity of Model Liquids from Molecular Dynamics Simulations. J. Chem. Phys. 2002, 116, 209–217. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
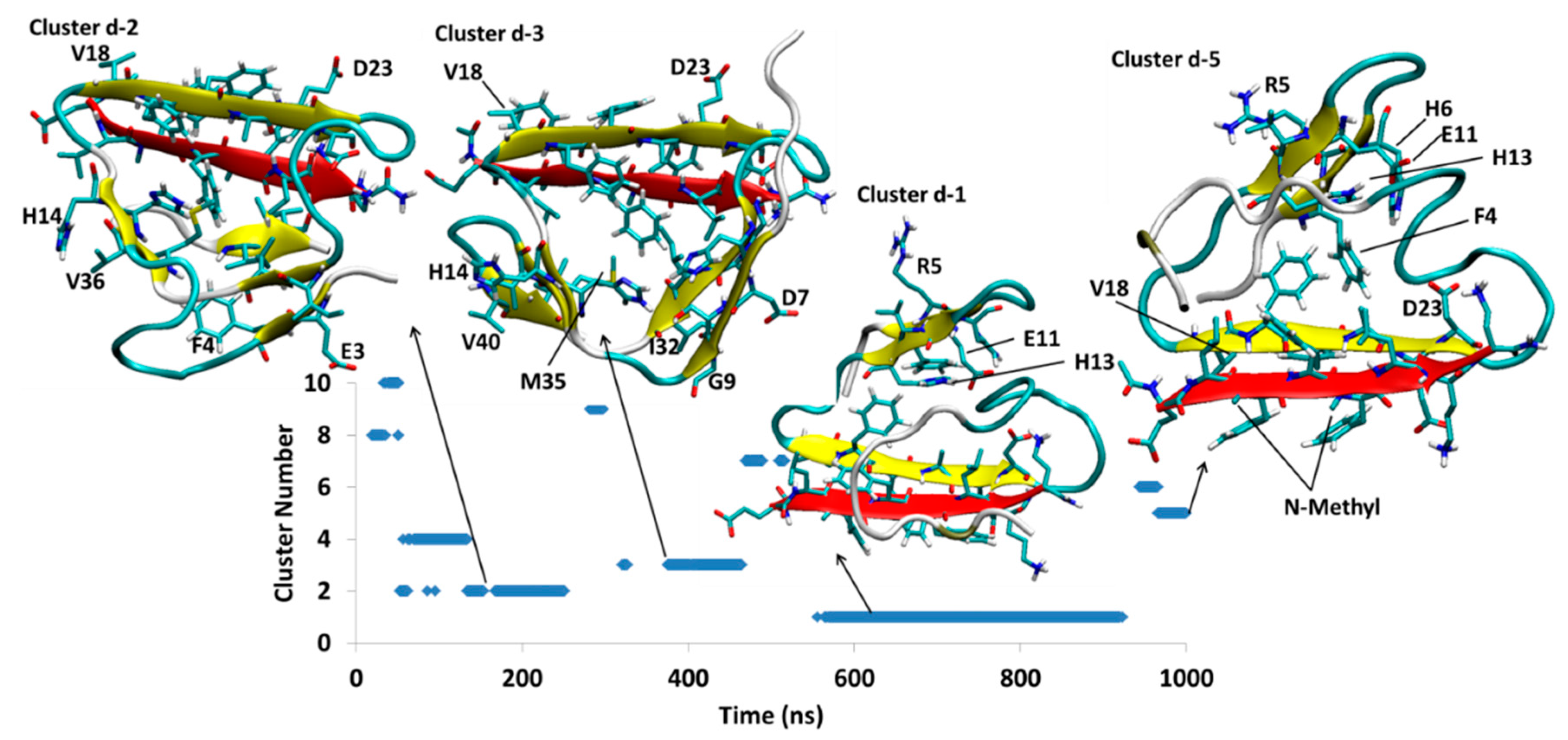{kind=link}
| Complex | Registry a | pi | H-Bond b | ∆Gbinding (kJ/mol) | Effective ∆Geff c (kJ/mol) |
|---|---|---|---|---|---|
| d-amino acid PPs (this work) | |||||
| RT-SGB1 | 23 | 0.99 | 8.8 | −57 ± 3 | −16 |
| RB-SGB1 | 24 | 0.89 | 7.9 | −62 ± 3 | −26 |
| RT-SGD1 | NA f | 0.73 | −50 ± 3 | −15 | |
| RB-SGD1 | NA f | 0.93 | 9.6 | −43 ± 4 | −1 |
| SGB1-SGB1 | 9 | 0.99 | −45 ± 3 | ||
| SGD1-SGD1 | 8 | 0.80 | 7.1 | −32 ± 2 | |
| l-amino acid PPs: | |||||
| RB-RB d | 38 | 0.29 | 10.3 | −53 ± 3 | - |
| RT-SGA3 d | 24 | 0.35 | 8.4 | −56 ± 3 | −3 |
| RB-SGA3 d | 21 | 0.31 | 7.8 | −47 ± 2 | +5 |
| RT-SGC1 e | NA f | 0.80 | 8.1 | −53 ± 3 | −27 |
| RB-SGC1 e | NA f | 0.92 | 8.2 | −50 ± 2 | −21 |
| SGA3-SGA3 d | 10 | 0.72 | - | −46 ± 4 | |
| SGC1-SGC1 e | 8 | 0.97 | 6.7 | −26 ± 3 |
| Cluster # | Pi b | ΔGgas-PBSA c | ΔGLIE-D d | ΔGLIE-DR e |
|---|---|---|---|---|
| Ba1 | 0.8 | 78 ± 29 | −33 ± 4 | −26 ± 41 |
| Bb2 | 0.13 | −14 ± 33 | −58 ± 7 | −102 ± 43 |
| Bb3 | 0.11 | −16 ± 141 | −67 ± 4 | −106 ± 41 |
| Bb1 | 0.58 | −37 ± 32 | −64 ± 4 | −101 ± 41 |
| Bc1 | 0.12 | 100 ± 29 | −33 ± 3 | −37 ± 41 |
| Bc4 | 0.08 | 50 ± 44 | −60 ± 5 | −91 ± 41 |
| Cluster # | Pi b | ΔGgas-PBSA c | ΔGLIE-D d | ΔGLIE-DR e |
|---|---|---|---|---|
| Da1 | 0.24 | 56 ± 30 | −37 ± 3 | −35 ± 40 |
| Da2 | 0.23 | 45 ± 36 | −35 ± 3 | −40 ± 42 |
| Db1 | 0.25 | 16 ± 43 | −48 ± 4 | −62 ± 41 |
| Db2 | 0.23 | 11 ± 62 | −47 ± 5 | −59 ± 41 |
| Dc1 | 0.62 | 374 ± 29 | −67 ± 3 | −111 ± 40 |
| Dd2 | 0.09 | −58 ± 31 | −56 ± 3 | −69 ± 41 |
| Dd3 | 0.08 | −41 ± 32 | −48 ± 4 | −62 ± 44 |
| Dd1 | 0.33 | 58 ± 32 | −54 ± 3 | −75 ± 41 |
| Dd5 | 0.03 | −21 ± 31 | −41 ± 3 | −42 ± 41 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehrazma, B.; Opare, S.; Petoyan, A.; Rauk, A. d-Amino Acid Pseudopeptides as Potential Amyloid-Beta Aggregation Inhibitors. Molecules 2018, 23, 2387. https://doi.org/10.3390/molecules23092387
Mehrazma B, Opare S, Petoyan A, Rauk A. d-Amino Acid Pseudopeptides as Potential Amyloid-Beta Aggregation Inhibitors. Molecules. 2018; 23(9):2387. https://doi.org/10.3390/molecules23092387
Chicago/Turabian StyleMehrazma, Banafsheh, Stanley Opare, Anahit Petoyan, and Arvi Rauk. 2018. "d-Amino Acid Pseudopeptides as Potential Amyloid-Beta Aggregation Inhibitors" Molecules 23, no. 9: 2387. https://doi.org/10.3390/molecules23092387
APA StyleMehrazma, B., Opare, S., Petoyan, A., & Rauk, A. (2018). d-Amino Acid Pseudopeptides as Potential Amyloid-Beta Aggregation Inhibitors. Molecules, 23(9), 2387. https://doi.org/10.3390/molecules23092387