
Recent Advances in the Addition of Amide/Sulfonamide Bonds to Alkynes


1
Antibiotics Research and Re-evaluation Key Laboratory of Sichuan Province, Sichuan Industrial Institute of Antibiotics, Chengdu University, 168 Hua Guan Road, Chengdu 610052, China
2
State Key Laboratory of Drug Research and CAS Key Laboratory of Receptor Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China
3
University of Chinese Academy of Sciences, No.19A Yuquan Road, Beijing 100049, China
*
Author to whom correspondence should be addressed.
Molecules 2019, 24(1), 164; https://doi.org/10.3390/molecules24010164
Submission received: 11 December 2018
/
Revised: 27 December 2018
/
Accepted: 27 December 2018
/
Published: 4 January 2019
(This article belongs to the Special Issue Amide Bond Activation)
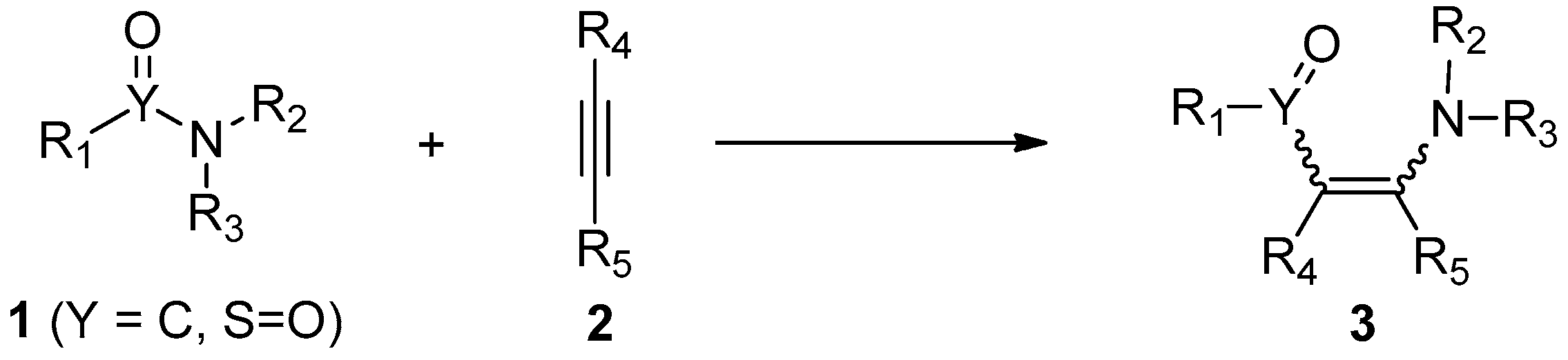{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
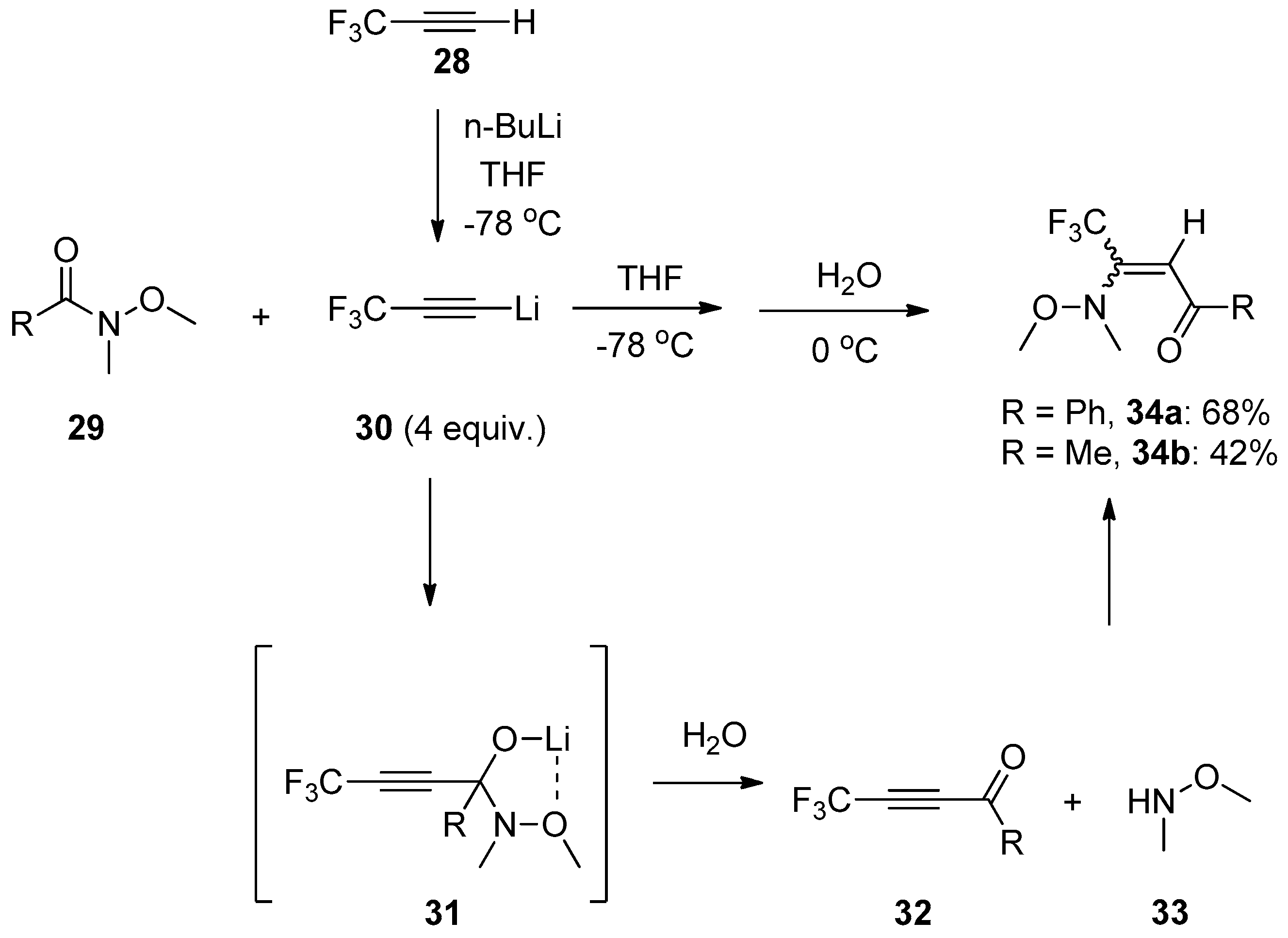{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
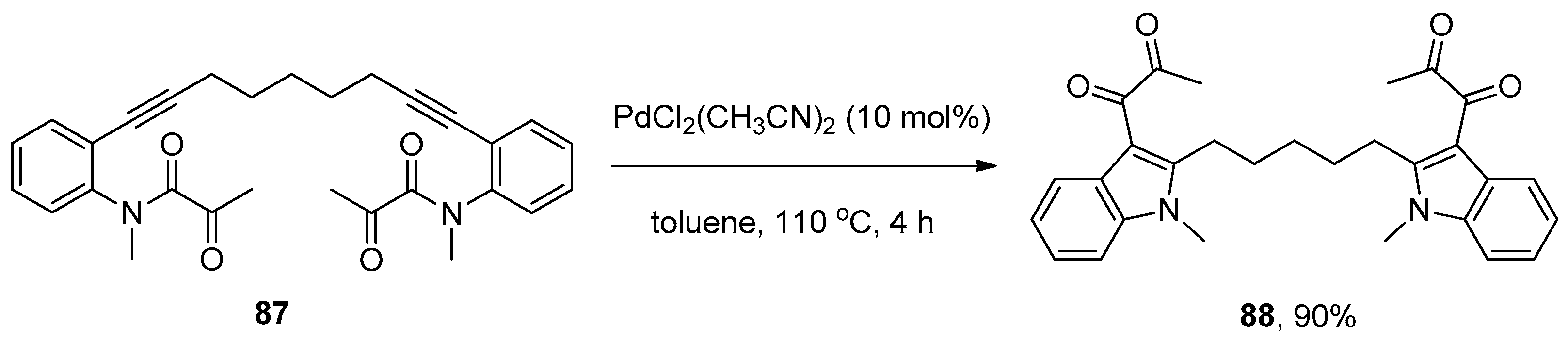{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The addition of amide/sulfonamide bonds to alkynes is not only one of the most important strategies for the direct functionalization of carbon–carbon triple bonds, but also a powerful tool for the downstream transformations of amides/sulfonamides. The present review provides a comprehensive summary of amide/sulfonamide bond addition to alkynes, including direct and metal-free aminoacylation, based-promoted aminoacylation, transition-metal-catalyzed aminoacylation, organocatalytic aminoacylation and transition-metal-catalyzed aminosulfonylation of alkynes up to December 2018. The reaction conditions, regio- and stereoselectivities, and mechanisms are discussed and summarized in detail.
1. Introduction
The addition of atom–atom bonds to alkynes has become an important strategy for the functionalization of carbon–carbon triple bonds [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16]. These intermolecular and intramolecular addition reactions provide a facile and efficient access to highly functionalized alkenes and cyclic compounds, respectively, in a high atom- and step-economic manner. Considering the large occurrence of amide/sulfonamide motifs in natural products and pharmaceutical agents, the addition of amide/sulfonamide bonds to alkynes, namely aminoacylation/aminosulfonylation of alkynes, is particularly important. Because they allow the direct downstream transformations of amides/sulfonamides by the insertion of carbon–carbon triple bonds into the amide/sulfonamide bonds, they thus produce more complex and skeletally different addition molecules (Scheme 1). In addition, the aminoacylation/aminosulfonylation of alkynes also constitutes a tool for the structural modification of compounds carrying amide/sulfonamide bonds, especially for peptides, which are an important class of drugs used in the clinic [17,18,19]. Besides, amide/sulfonamide bond addition to alkynes, which constructs one C–C/S and one C–N bond in a single step featuring high atom- and step-economy, is in accordance with the concept of “green and sustainable chemistry”.
It should be noted that the addition of amide/sulfonamide bonds to alkynes has not been reviewed before. Moreover, amide/sulfonamide bond addition to alkynes has achieved many important developments in recent decades, especially in transition-metal-catalyzed and organocatalytic processes. Therefore, a review focused on the aminoacylation/aminosulfonylation of alkynes would enrich the knowledge of synthetic chemists who are interested in amide/sulfonamide bond activation. The aim of the present review is to provide a systematical and comprehensive summary on the addition of amide/sulfonamide bonds to alkynes, including direct and catalyst-free aminoacylation, based-promoted aminoacylation, transition-metal-catalyzed aminoacylation, organocatalytic aminoacylation, and transition-metal-catalyzed aminosulfonylation of alkynes up to December 2018. We hope this review will serve as a handy reference for chemists interested in the addition of amide/sulfonamide bonds to alkynes, and will encourage further developments in this field in overcoming the remaining challenges.
2. Addition of Amide Bonds to Alkynes
2.1. Direct Addition of Amide Bonds to Alkynes without Catalysts and Additives
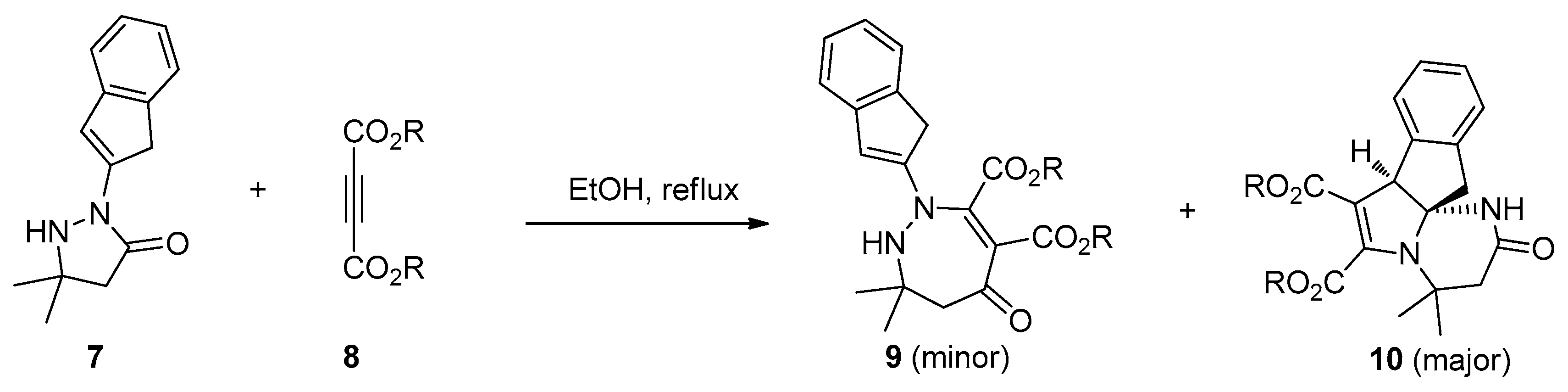
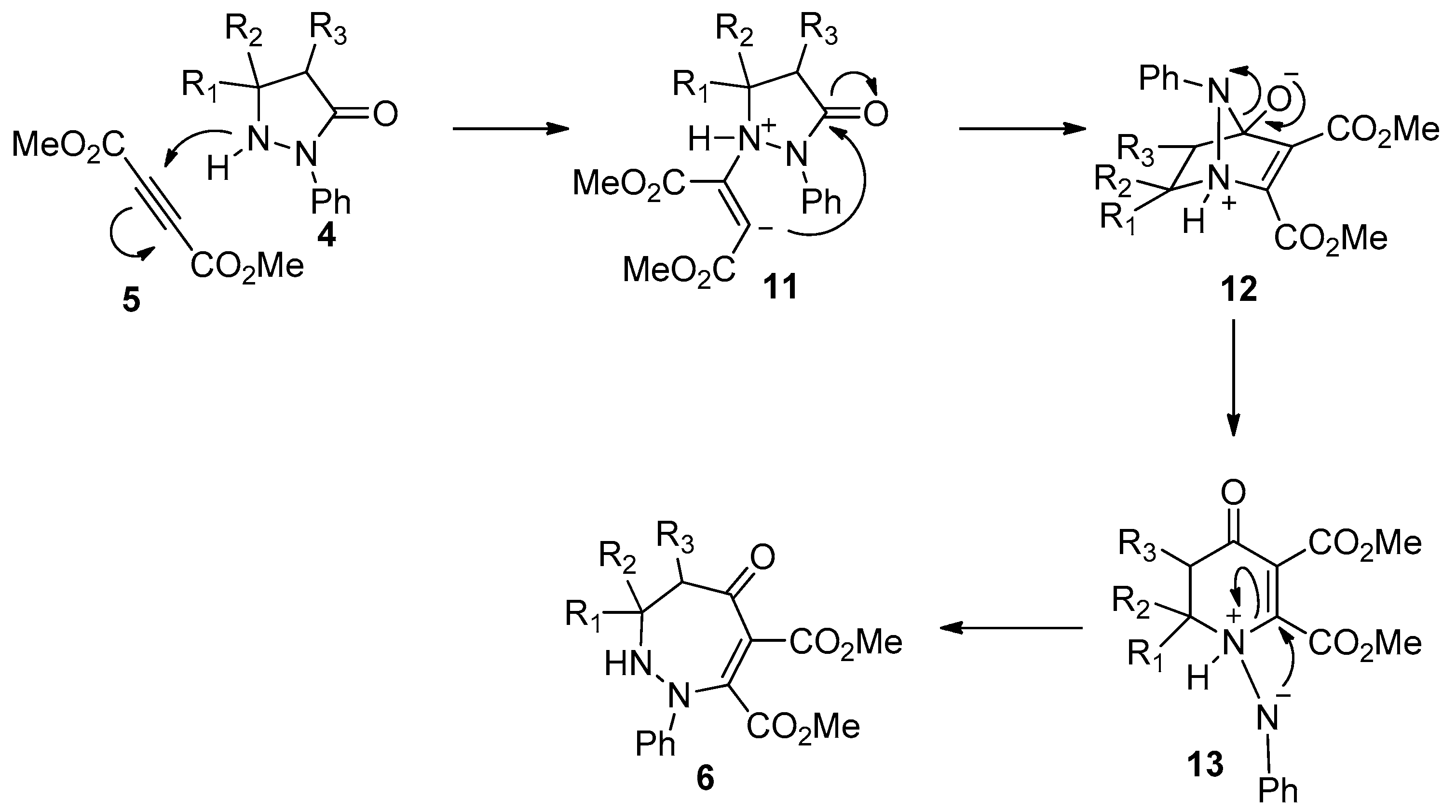
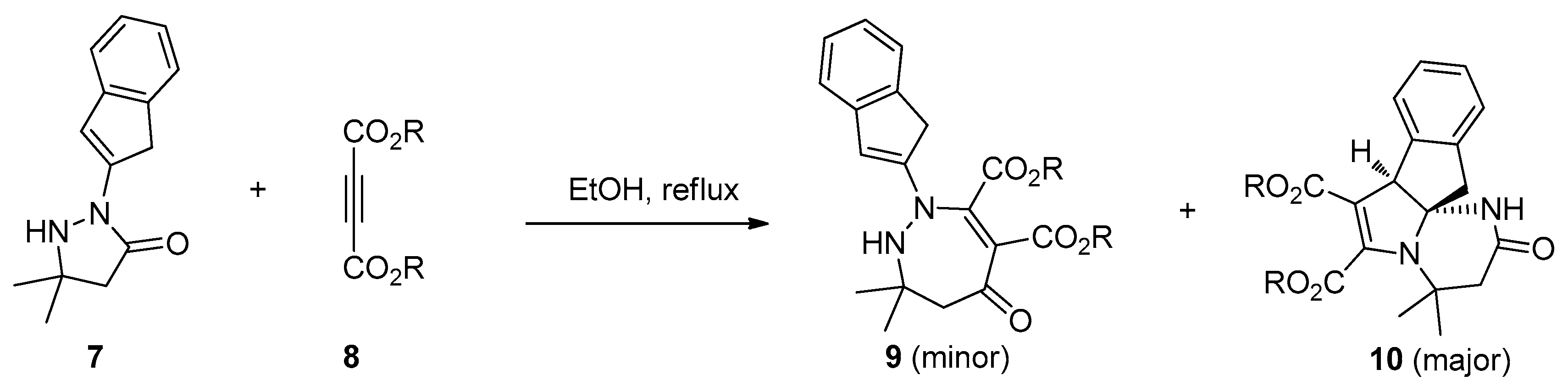
The first example of amide bond addition to alkynes was a catalyst- and additive-free process as reported by Eğe’s group in 1976 [20]. They found the treatment of active 2-phenylpyrazolidin-3-ones 4 with dimethyl acetylenedicarboxylate 5 in CH3CN under reflux led to the formation of the interesting ring expansion products 1,2-diazepin-5-ones 6, albeit with unsatisfactory selectivities and yields (Scheme 2). The main byproducts of this reaction were the cis- and trans- Michael-type addition products. Besides, this transformation was strongly influenced by the solvent used. Polar but nonprotic solvents such as acetone and acetonitrile gave the best results while few products were obtained in protic solvents such as ethanol. Similar results were also observed in Svete and Stanovnik’s research on the addition reactions between 5,5-dimethyl-2-(1H-indenyl-2)-3-pyrazolidinones 7 and acetylenedicarboxylates 8 (Scheme 3) [21]. A plausible reaction mechanism was outlined in Scheme 4. The Michael addition of N1 of the pyrazolidinones to acetylenedicarboxylate generates the carbanionic intermediate 11, which attacks the carbonyl group across the ring to give the bicyclic amino-acetal intermediate 12. The following ring opening of 12 affords the zwitterionic intermediate 13, which undergoes ring expansion to produce the addition products 6.
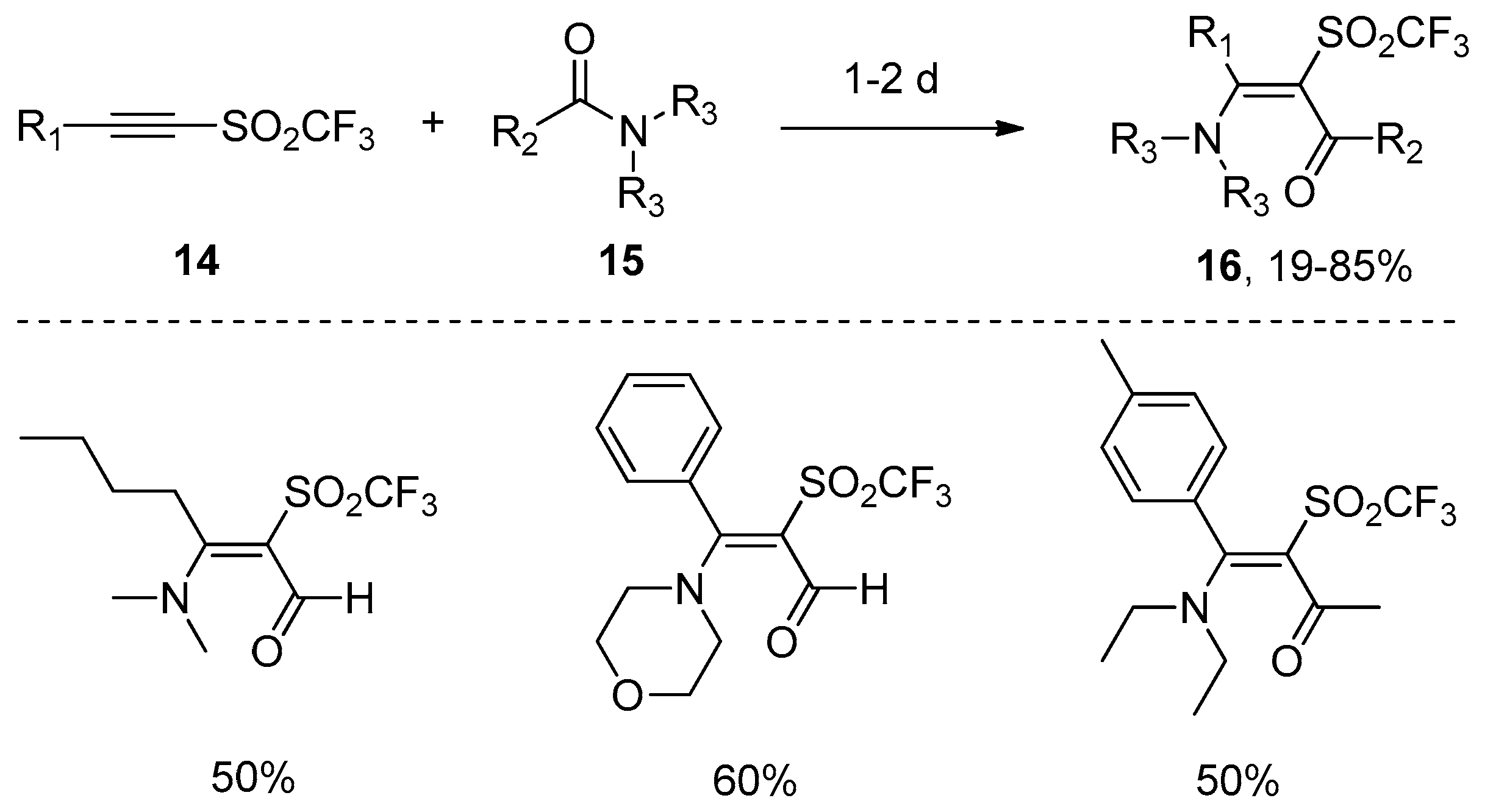
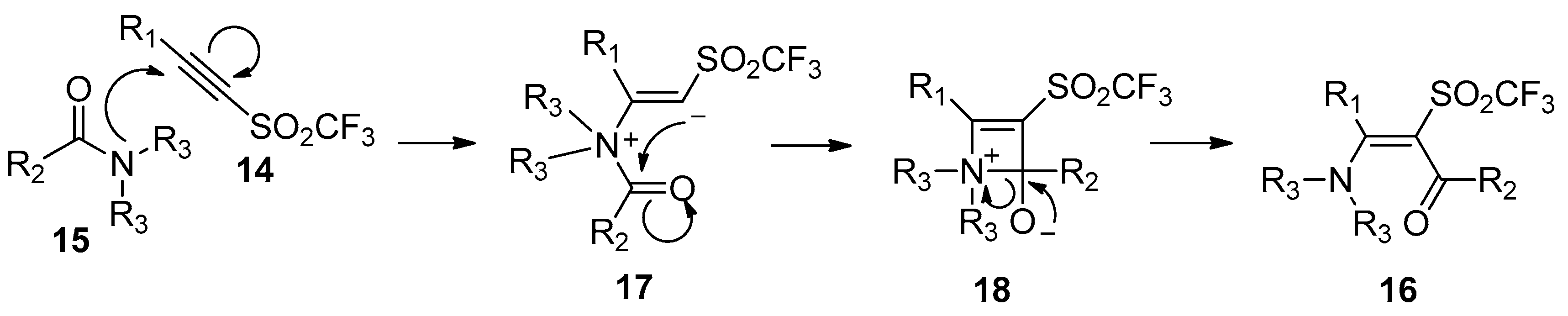
In contrast, Hanack’s group developed a more general and practical addition of amide bonds to carbon–carbon triple bonds without any catalysts and additives in 1989 [22]. Amides 15 were directly added to alkynyl trifluoromethyl sulfones 14 to afford the cis-adducts 16 with excellent regioselectivity and good yields, despite the fact that a long reaction time was required (Scheme 5). This protocol showed advantages such as simple operation, broad substrate scope, and the avoidance of metals and additives. A plausible mechanism was proposed in Scheme 6. The Michael reaction of nitrogen atom of the amides to alkynyl trifluoromethyl sulfones yields a zwitterion 17, which undergoes cyclization to form intermediate 18. The subsequent rupture of the carbon–nitrogen bond of 18 gives the products 16. The regio- and stereoselectivity observed in this reaction could be well explained by this mechanism.
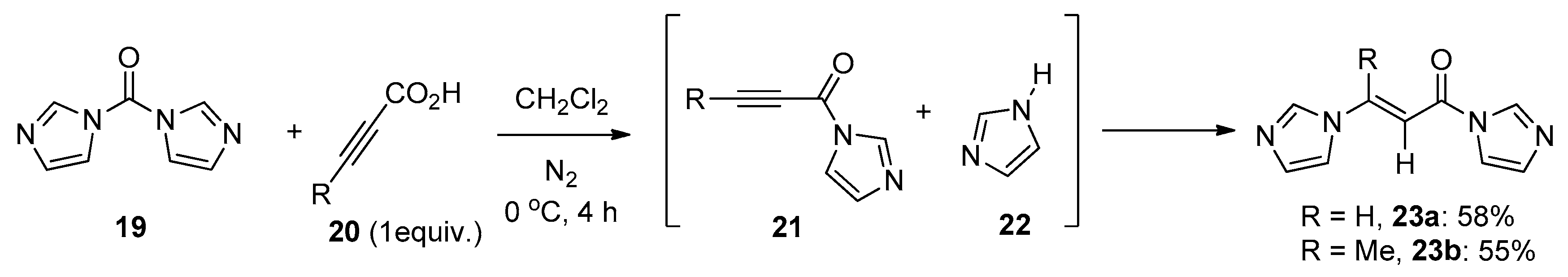
What seems particularly interesting is the addition of 1,1′-carbonyldiimidazole (CDI) 19 to alkynoic acids 20 with the release of CO2, as reported by Knölker and co-workers in 1993 (Scheme 7) [23]. This reaction proceeded well under mild conditions to provide the E-adducts 23 in moderate yields. The reaction of CDI 19 with alkynoic acids 20 generated intermediate 21 and imidazole 22 with the release of CO2, and the subsequent addition of the imidazole 22 to the electron deficient alkyne 21 stereoselectively produced the products 23.
2.2. Base-Promoted Addition of Amide Bonds to Alkynes
In 1987, Suzuki and Tsuchihashi disclosed a sequential process for the preparation of enaminones 27 through the insertion of lithium (triphenylsilyl)acetylide into amides (Scheme 8) [24]. Acyclic amides reacted smoothly to give the E-enaminones in high yields, while lower yields of the desired ring expansion products were obtained when cyclic amides were used as the substrates. It is worth noting that the triphenyl group on silicon was essential for the transformation as other silylacetylides failed to give the enaminone products. The possible reaction pathway may involve the initial formation of the silylalkynone, the subsequent Michael addition of in situ-formed lithium amide and the final protiodesilylation.
Subsequently, Jeong et al. successfully realized the addition of Weinreb amides 29 to the carbon–carbon triple bond of trifluoropropynyl lithium 30 in a one-pot two-step pathway [25,26,27], providing a Z/E mixture of β-trifluoromethyl enaminones 34 in moderate yields (Scheme 9). It should be noticed that an excessive amount of trifluoropropynyl lithium was required to consume Weinreb amides completely. The reaction temperature had a decisive impact on the outcome of this transformation since quenching the reaction with H2O at room temperature failed to give the enaminones but gave recovery of Weinreb amides. Besides, the use of N,N-dimethylbenzamide instead of N-methoxy-N-methylbenzamide under optimal conditions did not provide the desired product at all, only the recovery of starting material. This indicated that the N-methoxy group in Weinreb amides played an indispensable role in this reaction. The proposed mechanism involved the key intermediate 31, which was formed from the addition of trifluoropropynyl lithium with Weinreb amides. Then 31 was quenched by H2O to give the ynone intermediate 32, which rapidly reacted with N-methoxy-N-methylamine 33 generated from the reaction to give the products 34. The N-methoxy group in Weinreb amides was essential because the oxygen could coordinate with the lithium cation to stabilize the key intermediate 31. Particularly, the fact that trapping 31 with trimethylsilyl chloride afforded the corresponding siloxane derivative in a high yield further demonstrated the mechanism.
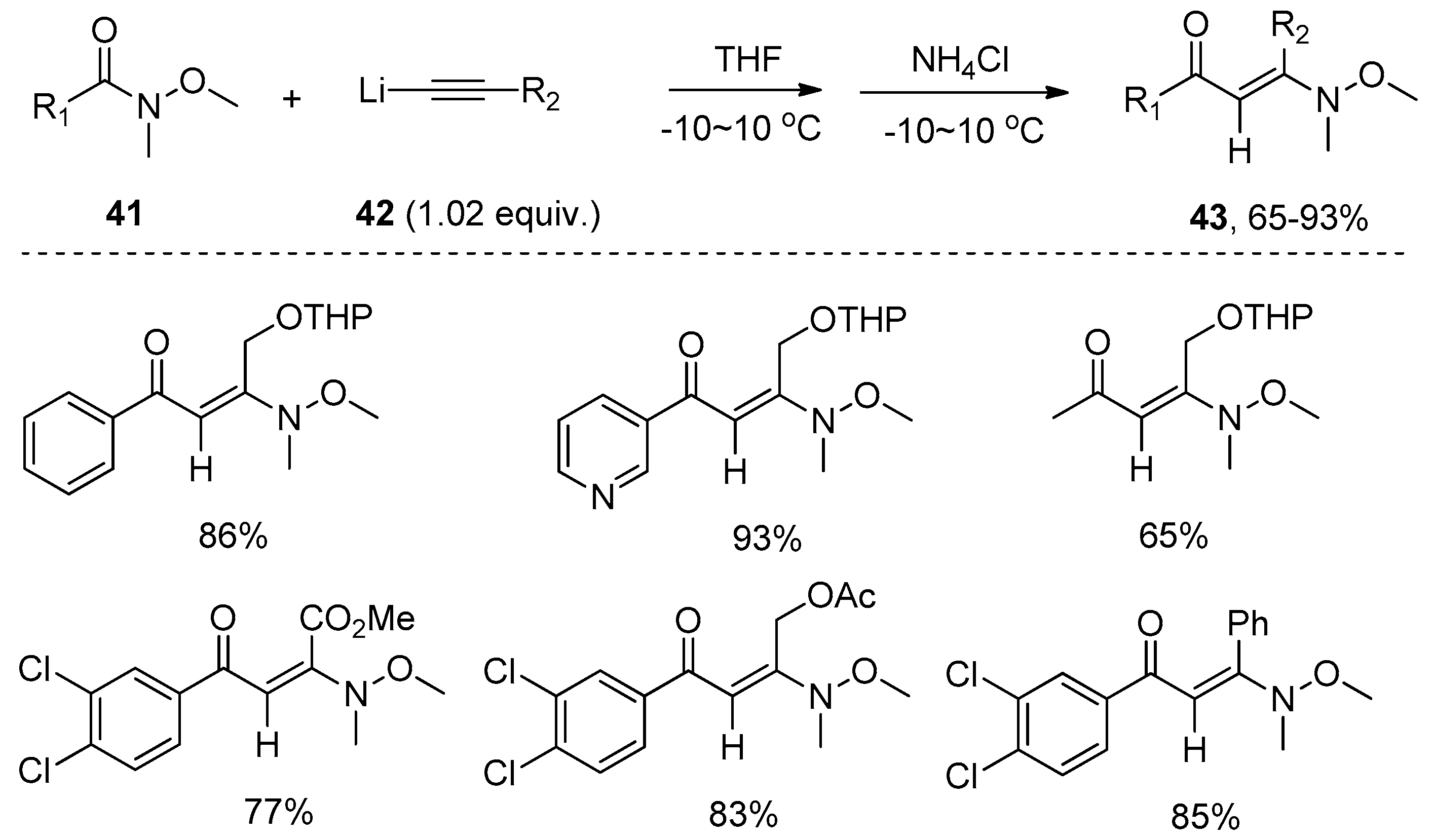
Soon afterwards, the group of Nielsen reported the insertion of sodium acetylide of ethyl propynoate 35 into Weinreb amides 29 to produce the 1,2-addition products 37 as the major products (Scheme 10a) [28]. The selectivity of the 1,2-addition products 37 over 1,1-addition products 38 depended on the R group of the Weinreb amides. Substrates with bigger R substituents showed higher selectivity than those with smaller ones. For example, substituents such as phenyl showed excellent selectivity, providing the 1,2-addition adduct as the single product in high yield. However, substrates carrying bulky substituents such as tert-butyl or 2,4-dimethoxyphenyl did not undergo this transformation. Notably, the tertiary enaminones 37 preferentially adopted E-geometry in all cases, suggesting the 1,2-addition reactions proceeded in a highly trans-selective manner. In addition, the β-enaminoketoesters 37 were employed by the authors to react with hydrazines 39 under microwave irradiation to construct pyrazoles 40 through a regioselective cyclocondensation (Scheme 10b). Similarly, Choudhury’s group reported a one-pot sequential process consisting of nucleophilic substitution of the lithiated acetylides with Weinreb amides, and a following Michael reaction of the extruded N-methoxy-N-methylamine to a carbon–carbon triple bond after quenching with saturated NH4Cl, producing the E-β-enamino ketones 43 as the single geometrical isomer in high yields (Scheme 11) [29].
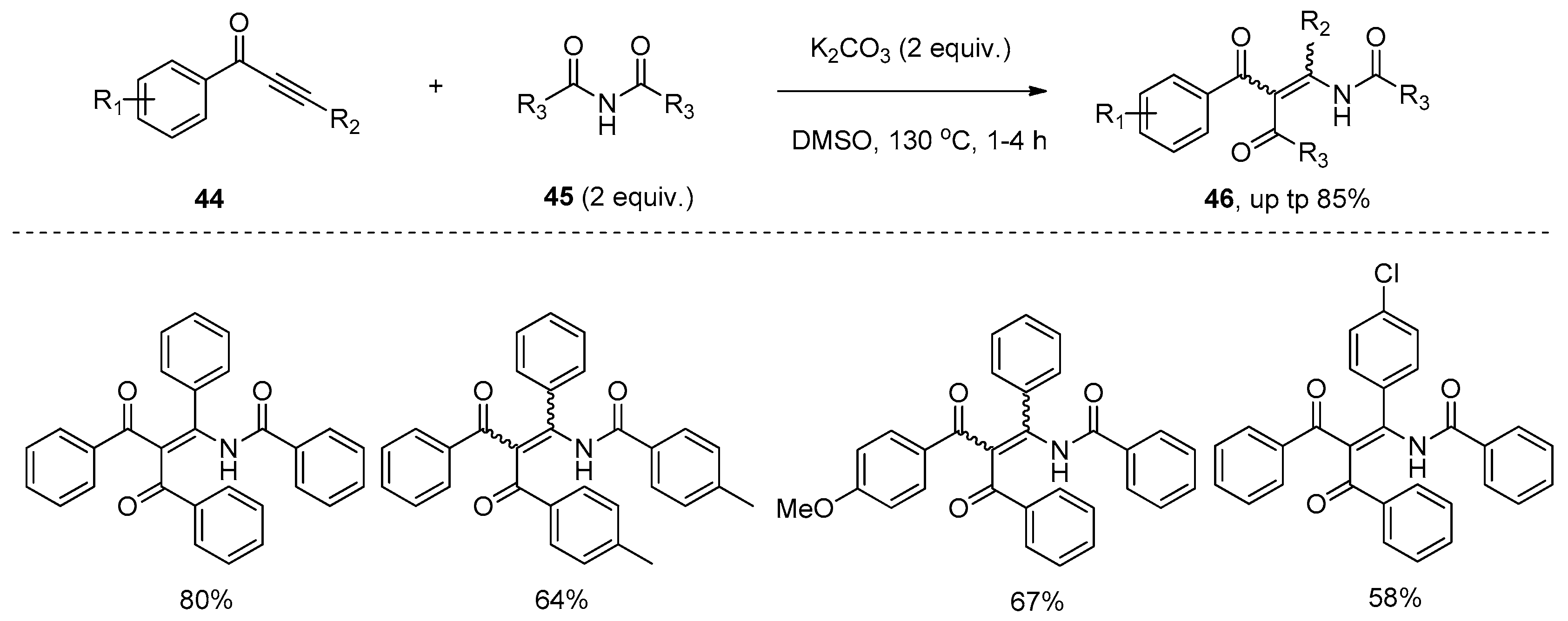
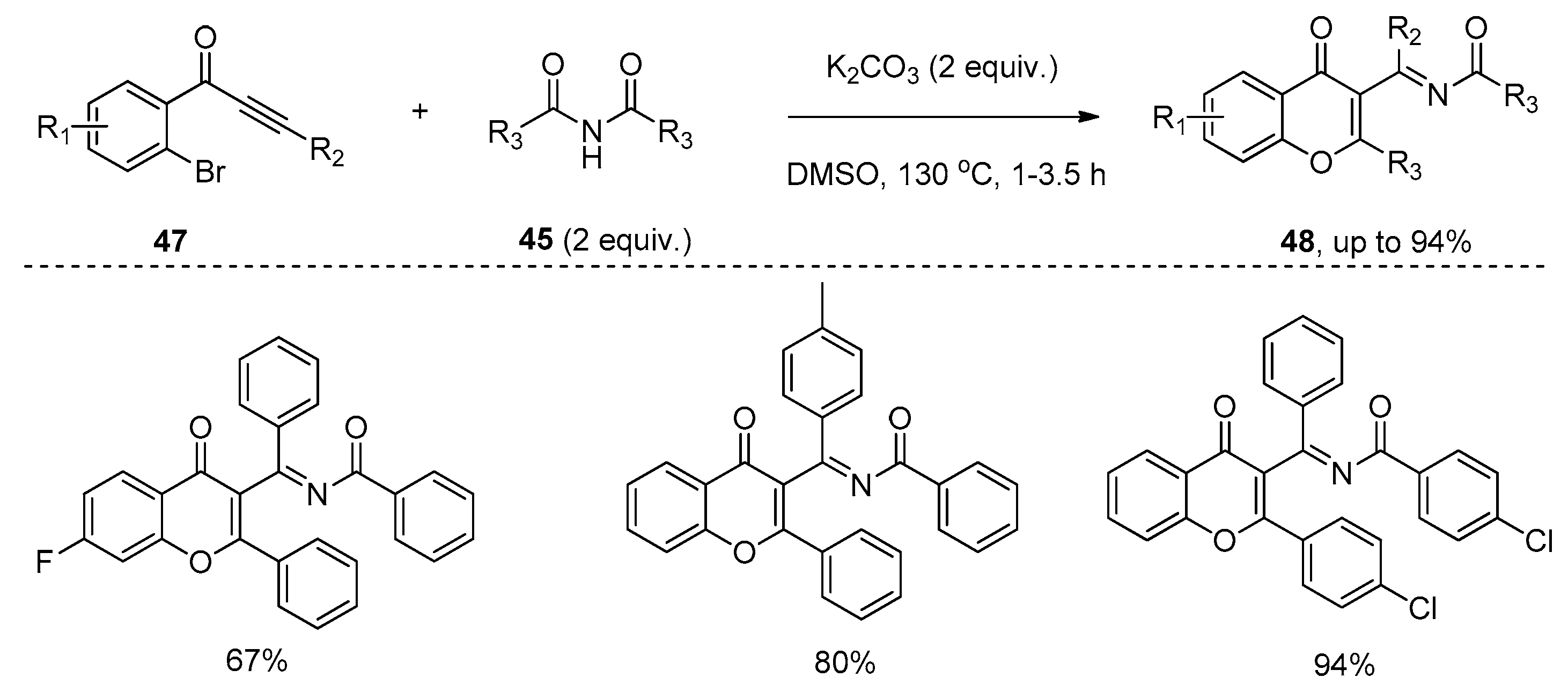
Very recently, Li and co-workers reported the addition of the amide bond of imides 45 to the carbon–carbon triple bond of alkynones 44 under basic conditions (Scheme 12) [30]. This addition reaction proceeded smoothly with the addition of a base such as K2CO3 in DMSO at high temperature, affording the corresponding tetra-substituted enamides 46 in good yields. Although this transformation suffered from unsatisfactory stereoselectivities, excellent regioselectivities were observed. The acyl group and amide group were dominantly located at the α-position and β-position of the carbonyl, respectively. Interestingly, in the reactions of alkynones 47 bearing an ortho-bromo-substituted aryl ring, highly functional chromones 48 were selectively formed in good to high yields via the O-cyclization pathway (Scheme 13). Control experiments showed that the base played an important role. It could deprotonate the imides 45 to form a nitrogen anion, which undergoes a Michael-type addition to the alkynones 49 to produce the anion intermediate 50. Then intermediate 50 undergoes an intramolecular nucleophilic addition/ring-opening sequence to provide intermediate 52. Hydrolysis of intermediate 52 generates enamides 46 (X = H), or imine–enamine tautomerization of intermediate 52 followed by nucleophilic aromatic substitution (SNAr) to give the chromones 48 (X = Br) (Scheme 14).
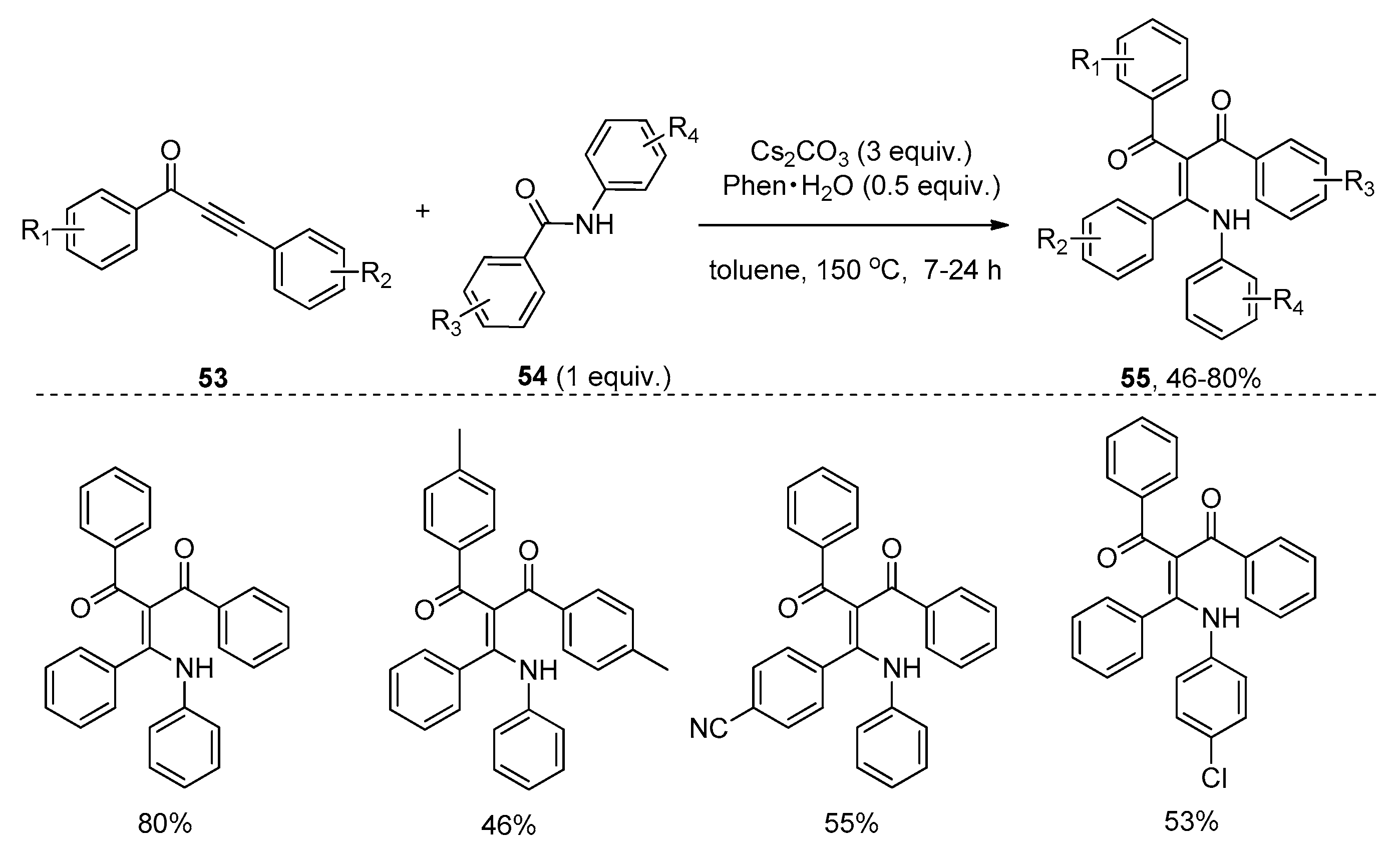
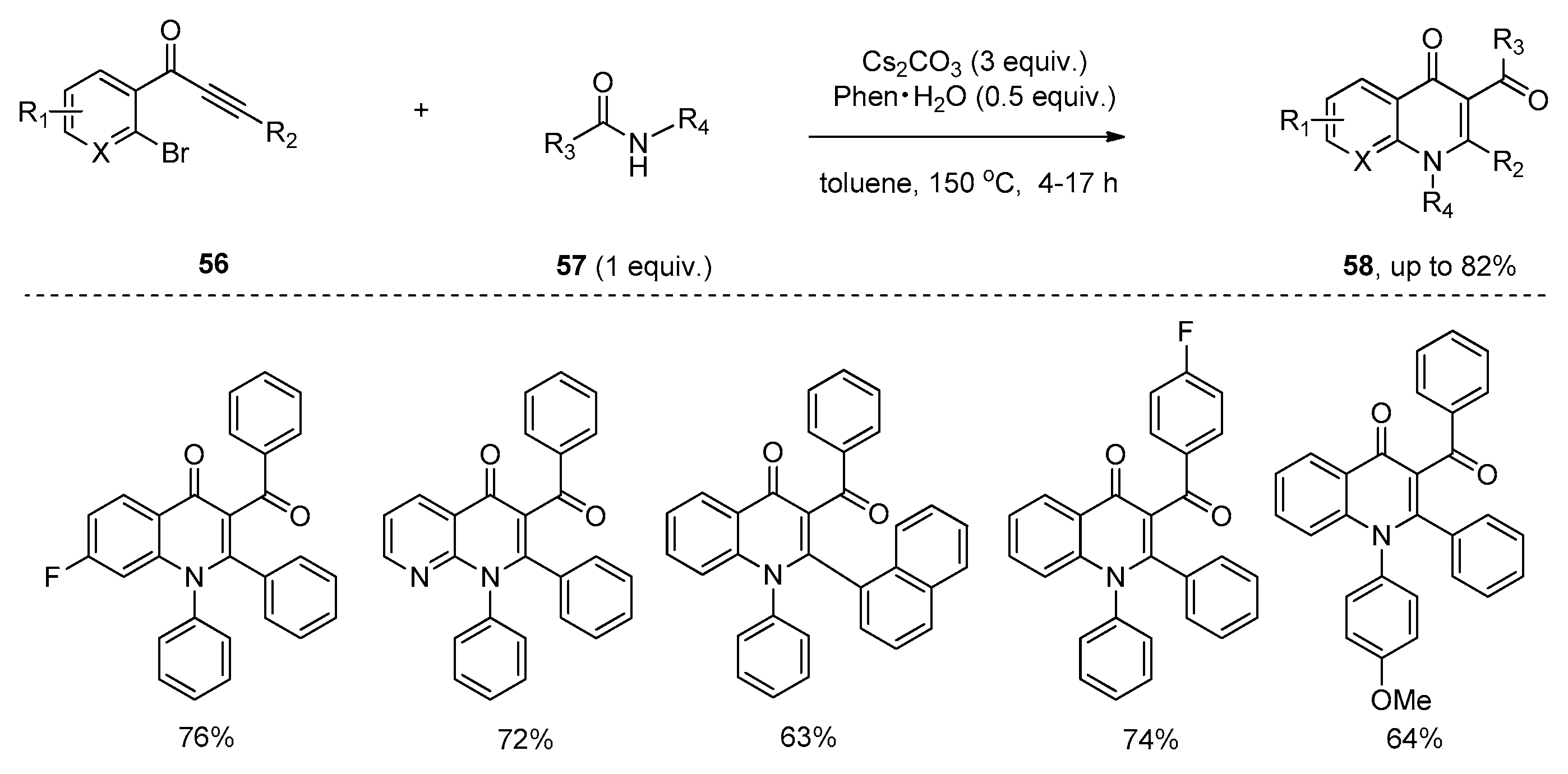
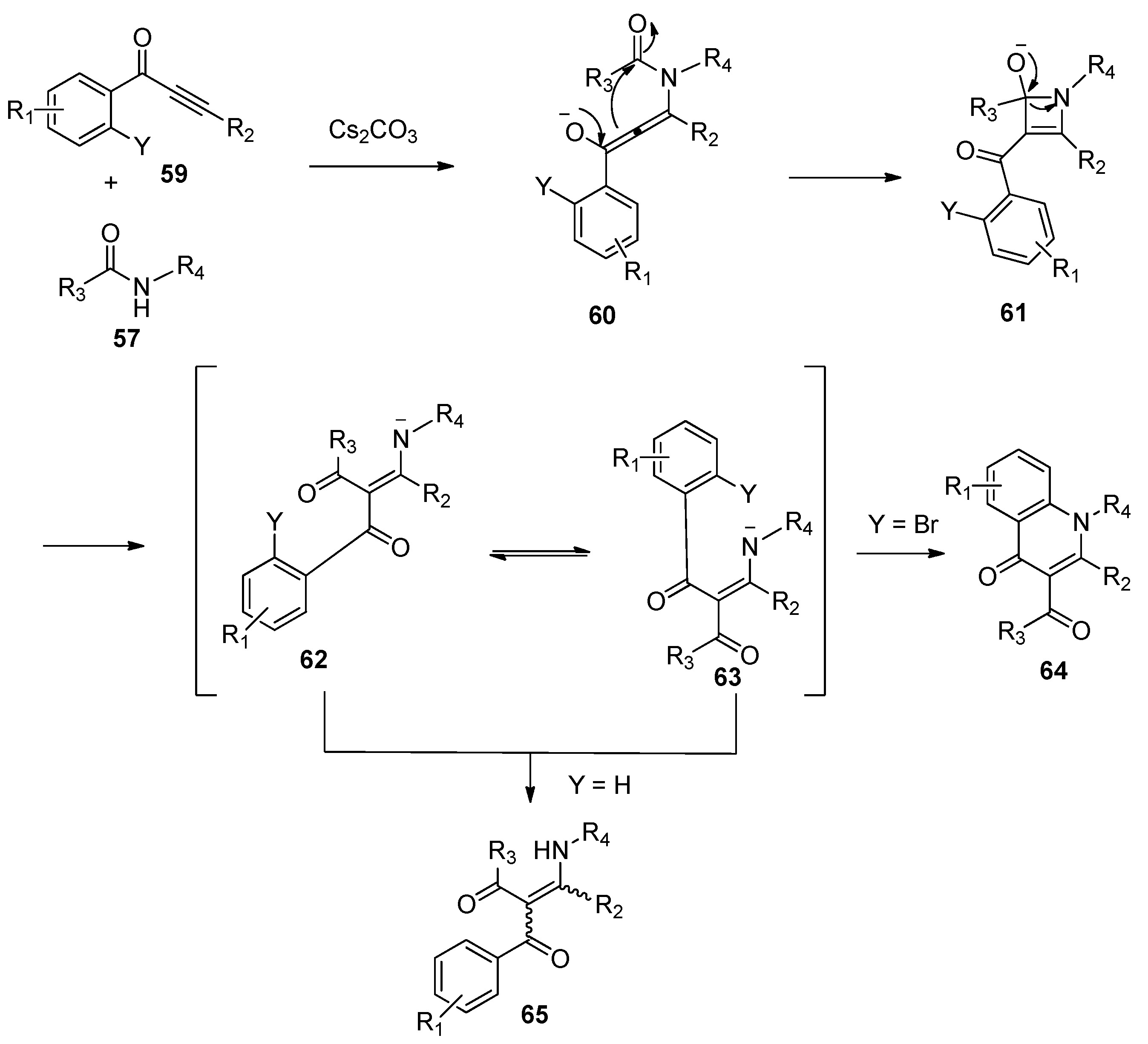
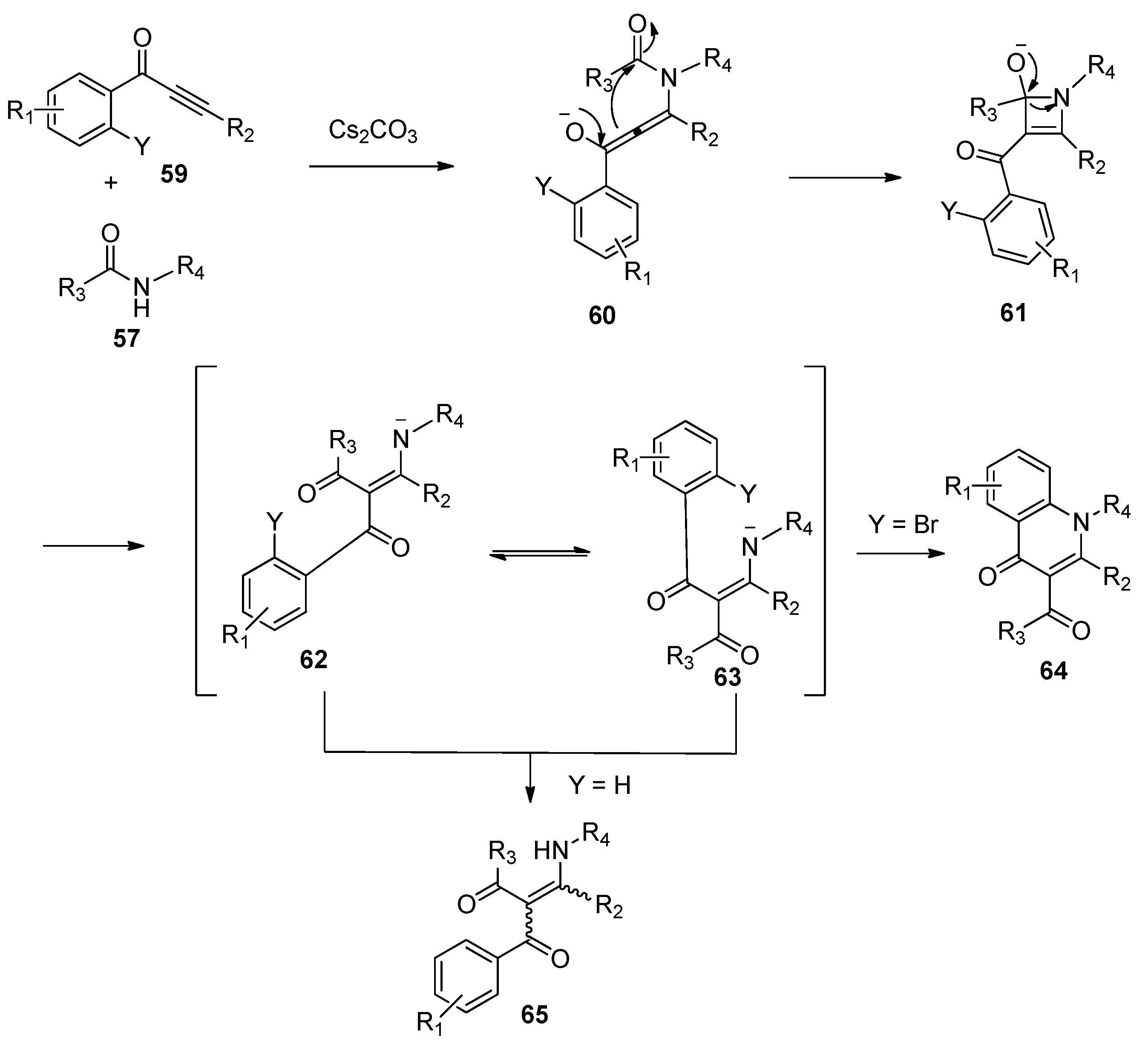
Soon afterwards, Li and co-workers presented the insertion of alkynones 53 into the amide bond of amide 54 promoted by Cs2CO3 (Scheme 15), providing the functionalized enaminones 55 with high stereoselectivity and excellent regioselectivity [31]. It should be noted that the combination of Cs2CO3 and 1,10-phenanthroline hydrate (Phen∙H2O) is essential to obtain good yields in a short reaction time. The authors hypothesized that 1,10-phenanthroline hydrate may act as a metal ion chelator, which increased the basicity of Cs2CO3 to accelerate the reaction. Similarly, 3-carbonyl-4-quinolinones 58 were selectively formed via a subsequent N-cyclization pathway in the cases of alkynones 56 bearing an ortho-bromo-substituted aryl ring (Scheme 16). The proposed reaction mechanism is outlined in Scheme 17. The Michael-type addition of amides 57 to alkynones 59 under basic conditions yields an allenol intermediate 60. The subsequent intramolecular nucleophilic addition gives a highly reactive cyclobutenol intermediate 61, which undergoes ring opening to produce a formal alkyne insertion intermediate 62. Then intermediate 62 undergoes imine-enamine tautomerization to provide intermediate 63, which undergoes a nucleophilic aromatic substitution (SNAr) to afford the quinolinone products 64 (Y = Br). In contrast, the protonation of intermediate 62 or 63 leads to the formation of the enaminone products 65 (Y = H).
2.3. Transition-Metal-Catalyzed Addition of Amide Bonds to Alkynes
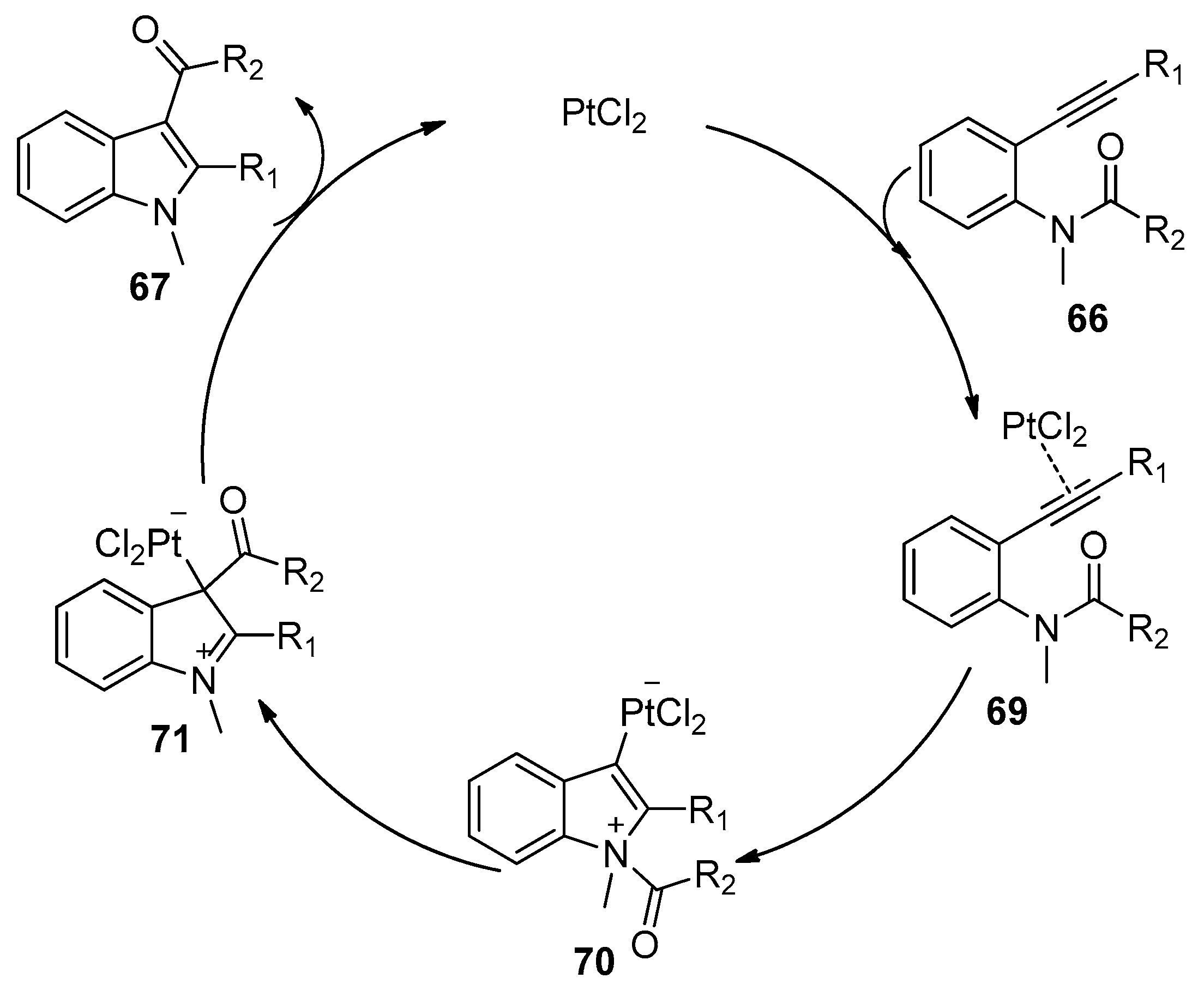
In 2004, Yamamoto’s group reported the platinum catalyzed synthesis of highly functional indoles through an intramolecular amide C–N bond addition to alkynes with the [1,3]-migration of acyl groups (Scheme 18) [32]. PtCl2 showed the highest catalytic activity compared with other platinum catalysts such as PtCl2(CH3CN)2, PtBr2, and Pt(PPh3)4. Various ortho-alkynylanilides 66 bearing diverse alkyl or aryl groups at R1 could be converted into the corresponding indole products 67 with good to high yields with PtCl2. Notably, a variety of acyl groups could undergo intramolecular [1,3]-migration to give 3-acyl-indoles 67. The main drawback of this method is that the desired products 67 are together with deacylated byproducts 68 in most cases. Based on the results of deuterium-labeling experiments and crossover experiments, the authors proposed a catalytic cycle of this intramolecular aminoacylation of alkynes. As shown in Scheme 19, coordination of alkyne moiety to PtCl2 yields the π-complex 69, followed by nucleophilic attack of nitrogen to the alkyne, affording the zwitterionic intermediate 70. An intramolecular [1,3]-migration of the acyl group then gives intermediate 71, which affords the product and regenerates the catalyst. The 3-deacylated byproducts 68 may attribute to the deacylation which takes place through the protonolysis of the C–Pt bond of intermediate 70.
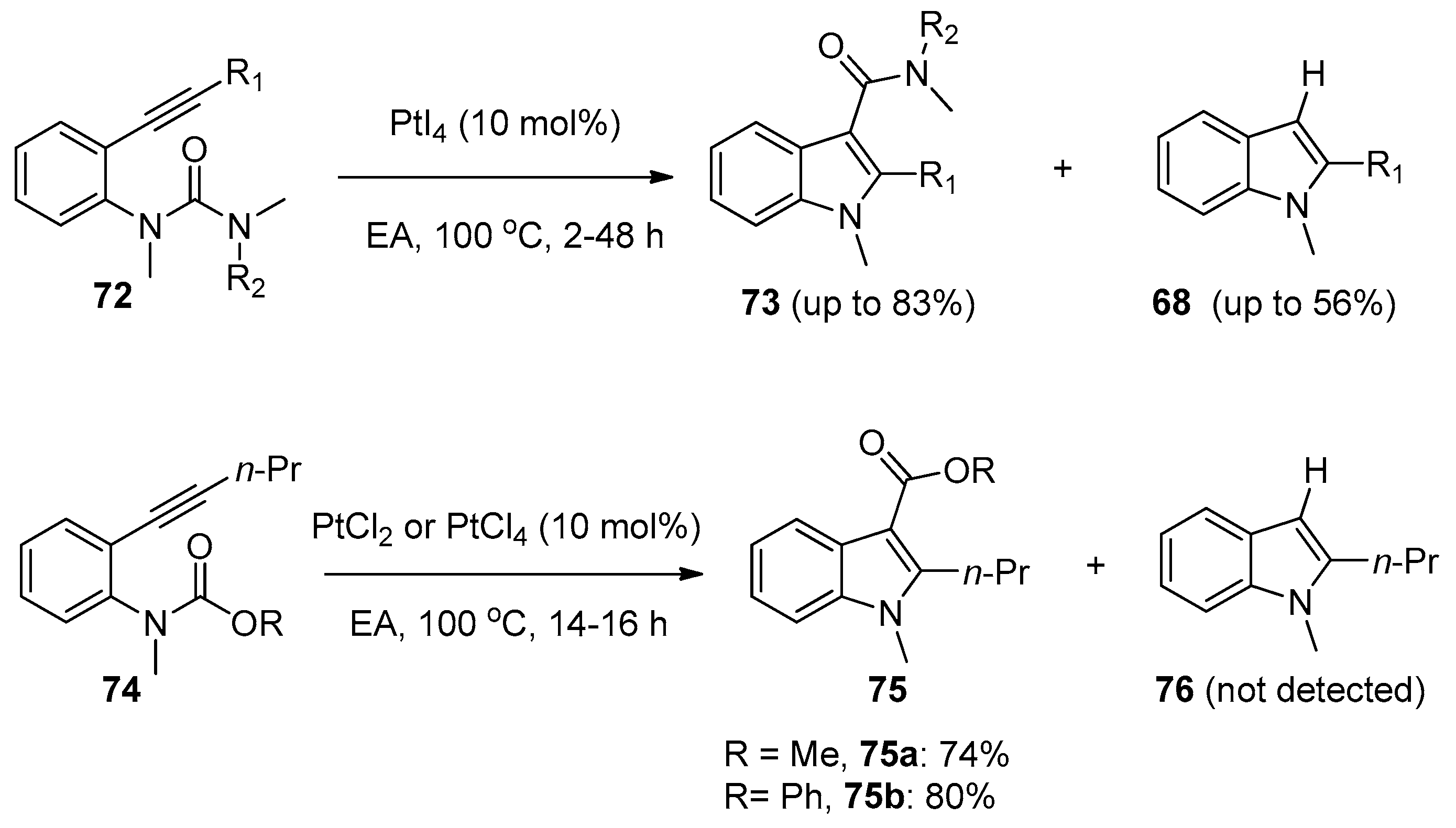
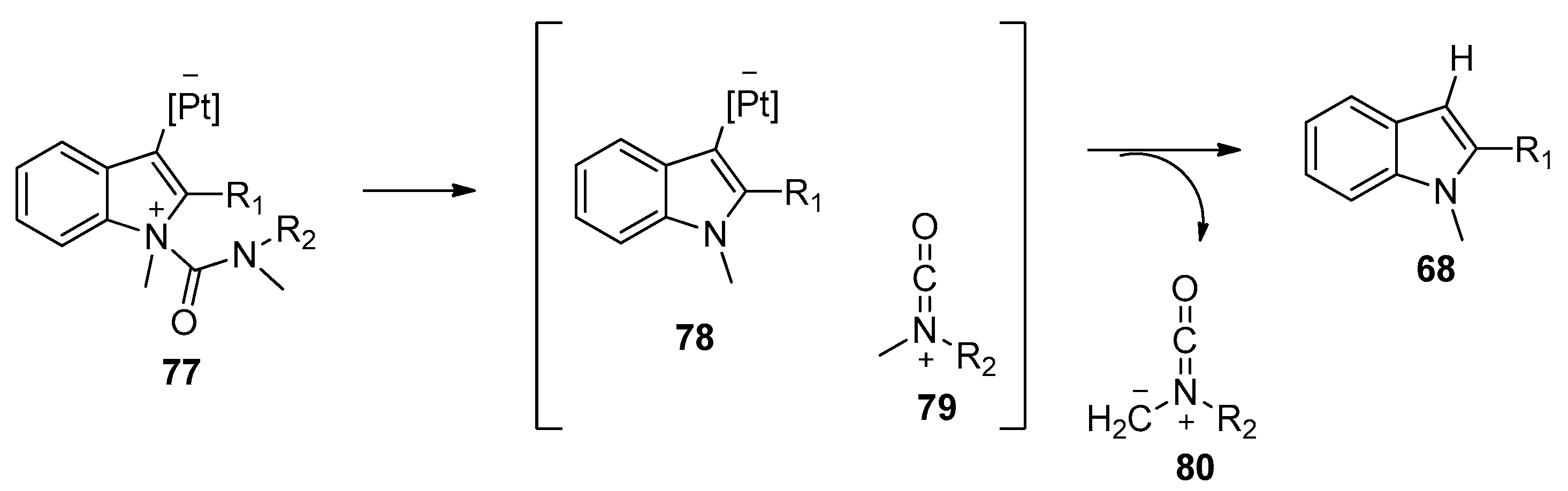
Encouraged by the excellent catalytic performance of platinum catalysts towards the intramolecular aminoacylation of alkynes, Nakamura’s group further studied similar reactions using ortho-alkynylphenylureas or ortho-alkynylphenyl carbamates as substrates (Scheme 20) [33]. The reactions of ortho-alkynylphenylureas 72 having a carbamoyl group attached to the nitrogen atom proceeded successfully under the catalysis of PtI4, providing the desired indole-3-carbamides 73 in moderate to high yields along with the 3-protonated byproducts 68. Interestingly, ortho-alkynylphenyl carbamates 74 could be converted into the corresponding indole-3-carboxylates 75 in good yields without the generation of 3-protonated byproducts 76. The authors proposed a similar mechanism to that of Yamamoto’s group [32]. They assumed the generation of the 3-protonated byproducts 68 in the reactions of ortho-alkynylphenylureas 72 may be attributed to protodemetalation of intermediate 78 by a proton from the methyl moiety of intermediate 79, which was extruded in the reaction (Scheme 21). Notably, this work proved that amide and ester groups could be used as the migrating groups, thus providing an efficient method to synthesize indole-3-carbamides/carboxylates which could not be prepared via Friedel–Crafts electrophilic substitution into the C3-position of the indole ring.
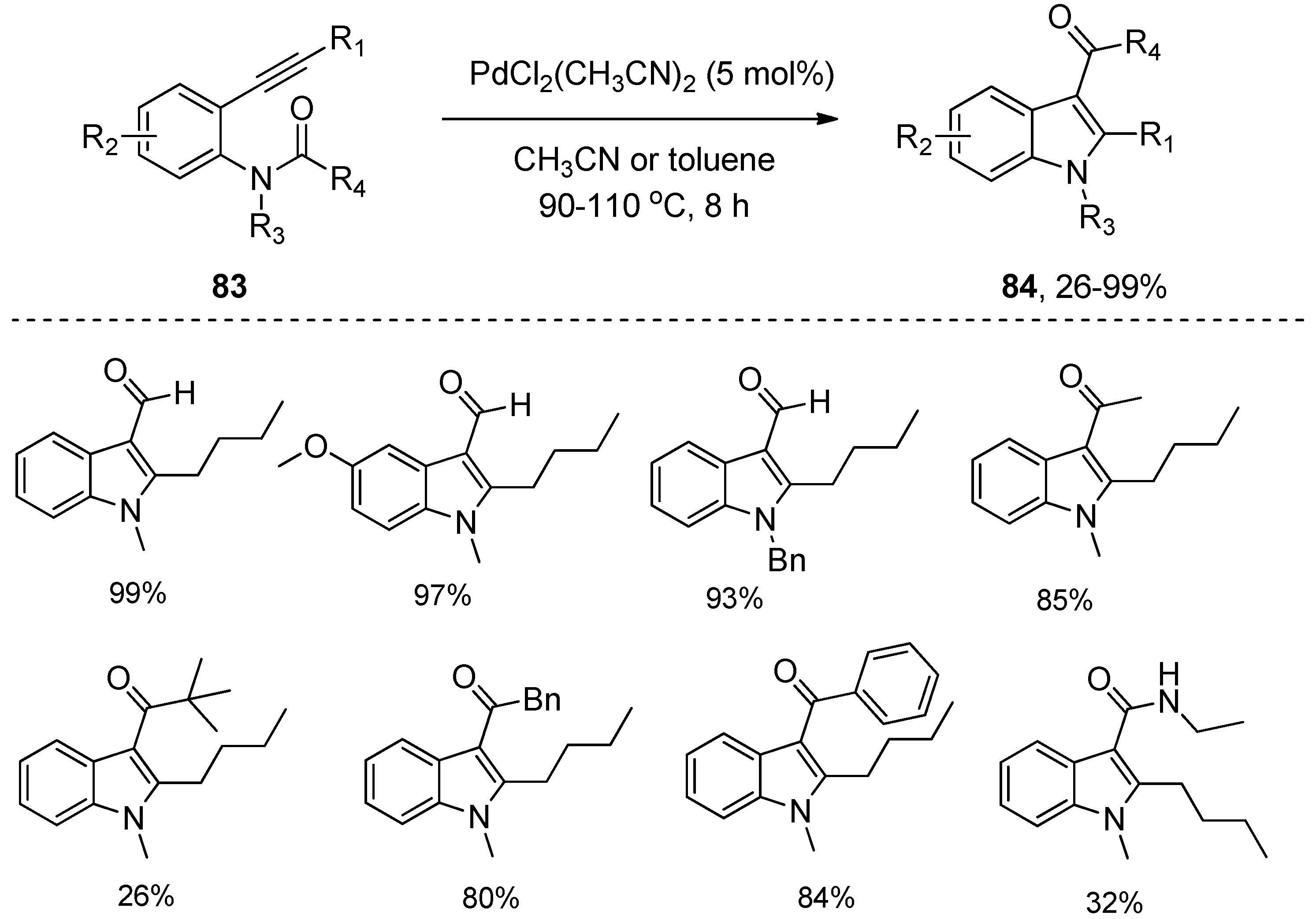
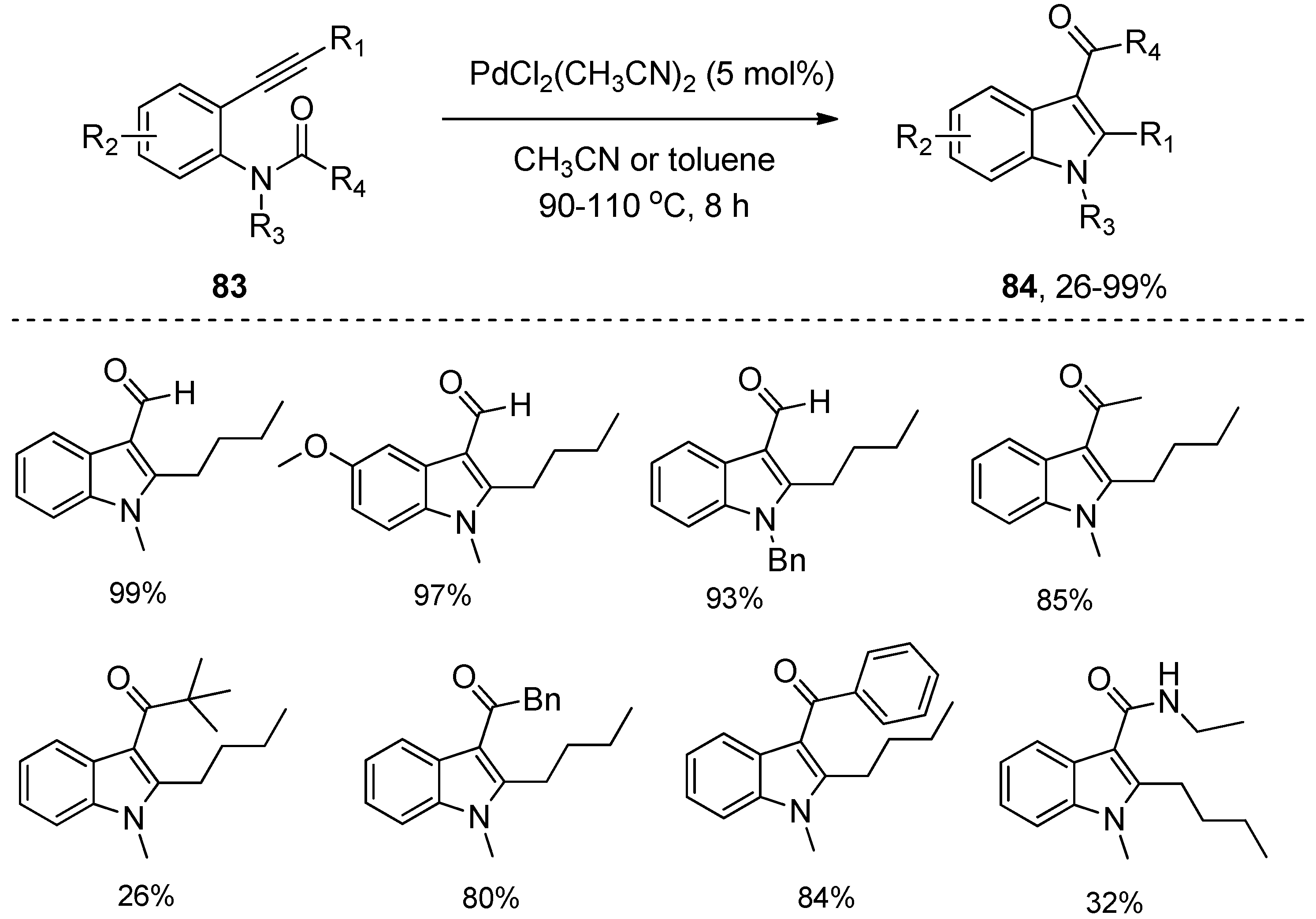
In 2007, Nakamura’s group revealed that PdBr2 could also catalyze the intramolecular amide C–N bond addition to alkynes (Scheme 22), affording the indole adduct 82 from ortho-alkynylanilide 81 in 52% yield [34]. Encouraged by the catalytic performance of PdBr2 towards the intramolecular aminoacylation of alkynes, Liu’s group further screened a series of palladium complexes. They found that PdCl2(CH3CN)2 showed excellent catalytic activity (Scheme 23) [35]. Substrates 83 with alkyl/aryl groups at R1 furnished the corresponding products 84 in good to excellent yields. The protocol was also compatible with substrates 83 bearing electron-donating substituents, halides, and electron-withdrawing substituents at R2, which produced the corresponding products 84 in high yields. In addition, the reactions of substrates 83 with different alkyl substituents at R3 also took place smoothly, providing the desired products 84 in high yields. More importantly, various acyl and amide groups could migrate smoothly and be conveniently introduced at the C3-position of indoles.
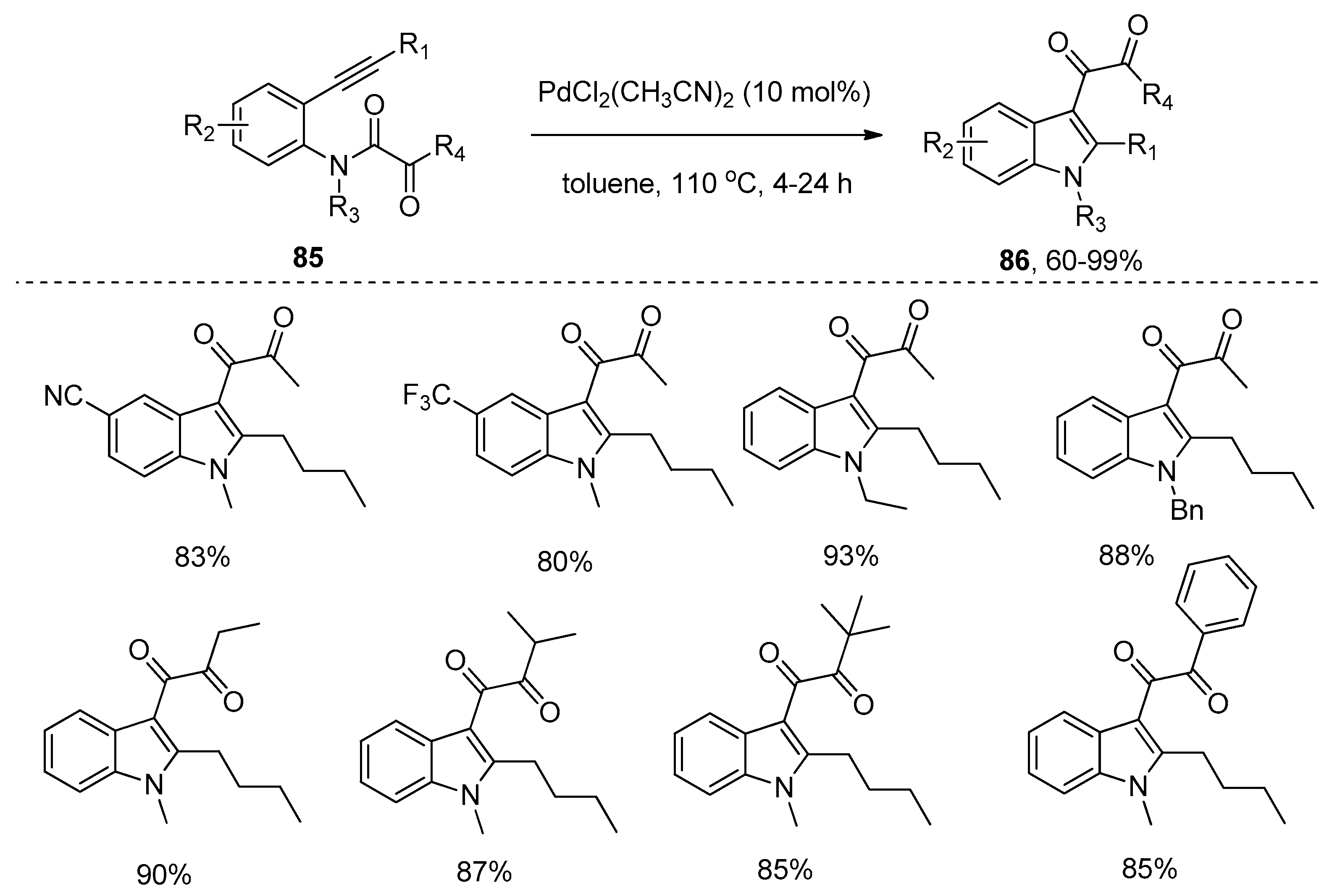
Subsequently, Liu and coworkers further extended their palladium catalytic system to the synthesis of 3-diketoindoles 86 from ortho-alkynyl-N-α-ketoacylanilines 85 via the intramolecular amide bond addition to alkynes (Scheme 24) [36]. Notably, this addition reaction proceeded smoothly to give the high functional 3-diketoindoles 86 with the [1,3]-migration of α-ketoacyl groups, which were used as migrating groups for the first time. Compared with previously reported protocols such as Friedel–Crafts acylation [37], Glyoxylation/Stephens–Castro coupling sequence [38], and the oxidative cross-coupling of indoles [39,40,41], which achieved the synthesis of 3-diketoindoles through the modification of the indole ring, but suffered from poor selectivity, operational complexity, the requirement of strict exclusion of moisture, limited substrate scope and low atom economy, this new method successfully prepared 3-diketoindoles via the construction of an indole ring with valuable features such as operational simplicity, high atom economy, broad substrate scope and high yields. Interestingly, a 3-diketoindole dimer 88 was synthesized in a high yield when substrate 87 was subjected to the optimal reaction conditions (Scheme 25). Finally, the authors proposed a reaction mechanism which is similar to that proposed by Yamamoto’s group [32].
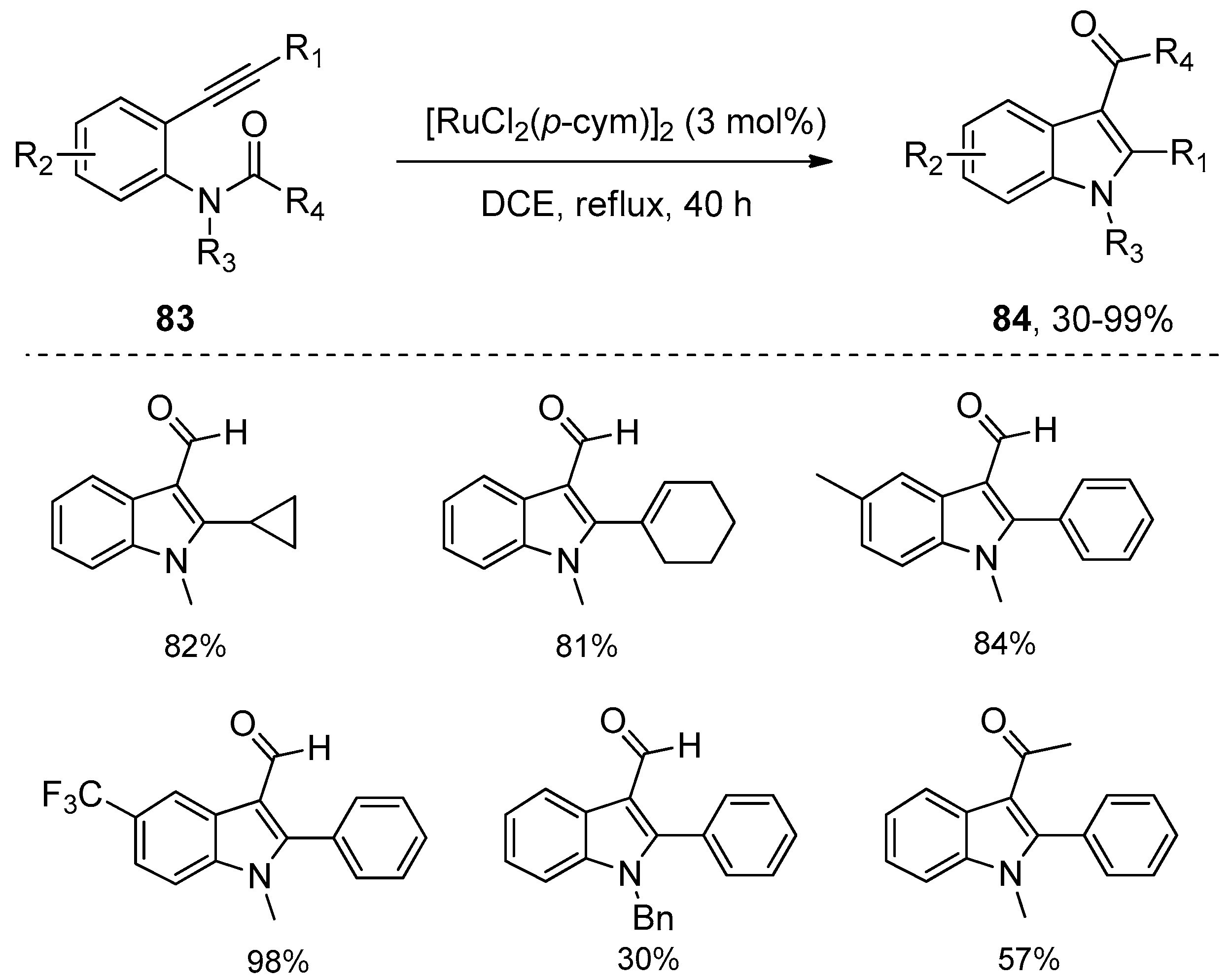
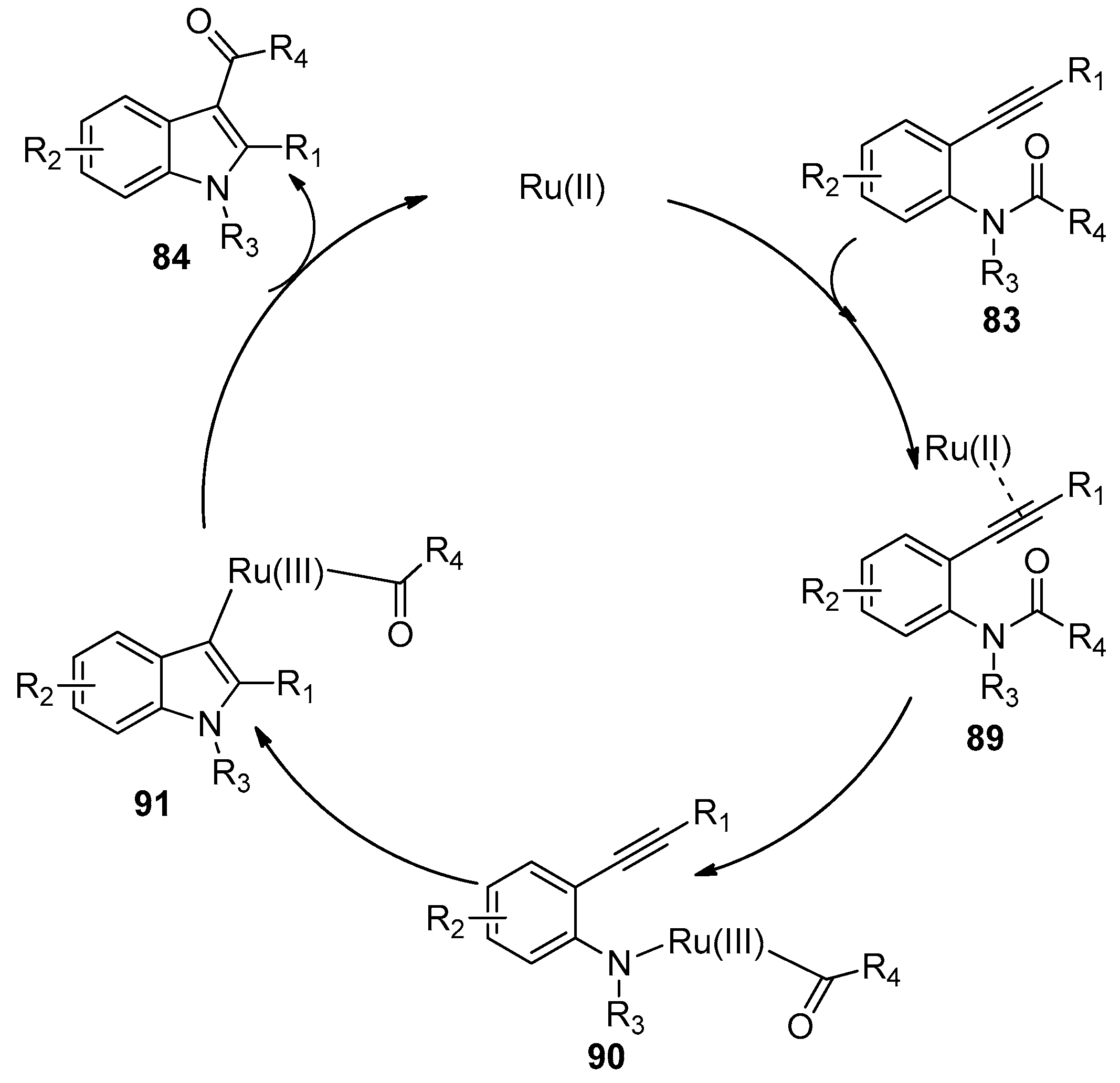
Ruthenium complexes were also found to be efficient catalysts for the intramolecular amide bond addition to alkynes, as was reported by Li’s group in 2012 (Scheme 26) [42]. Their study showed that [RuCl2(p-cym)]2 displayed the highest catalytic activity, with which ortho-alkynylanilides 83 could undergo intramolecular annulation through amide bond addition to the alkyne moiety to synthesize highly functional indoles 84. A variety of substrates 83 carrying diverse functional groups such as olefin, ester, aldehyde were well tolerated and could be converted into the corresponding indole products 84 in moderate to high yields. Despite the fact that a longer reaction time was required compared with platinum or palladium catalytic systems, it is worth noting that no 3-deacylated indoles were observed in all examples. However, the main shortcoming of this method is that unsatisfactory yields were obtained when bigger acyl groups such as acetyl were employed as the migrating groups. Based on the mechanistic study results with deuterium-labeling experiments, the authors hypothesized that the reaction mechanism may involve the complexation of substrates with ruthenium catalyst, the subsequent oxidative addition of ruthenium catalyst across the amide bond, the following addition of the N–Ru bond to carbon–carbon triple bonds, and the final reductive elimination to produce the products and regenerate the catalyst (Scheme 27).
2.4. Addition of Amide Bonds to Alkynes through Organocatalysis
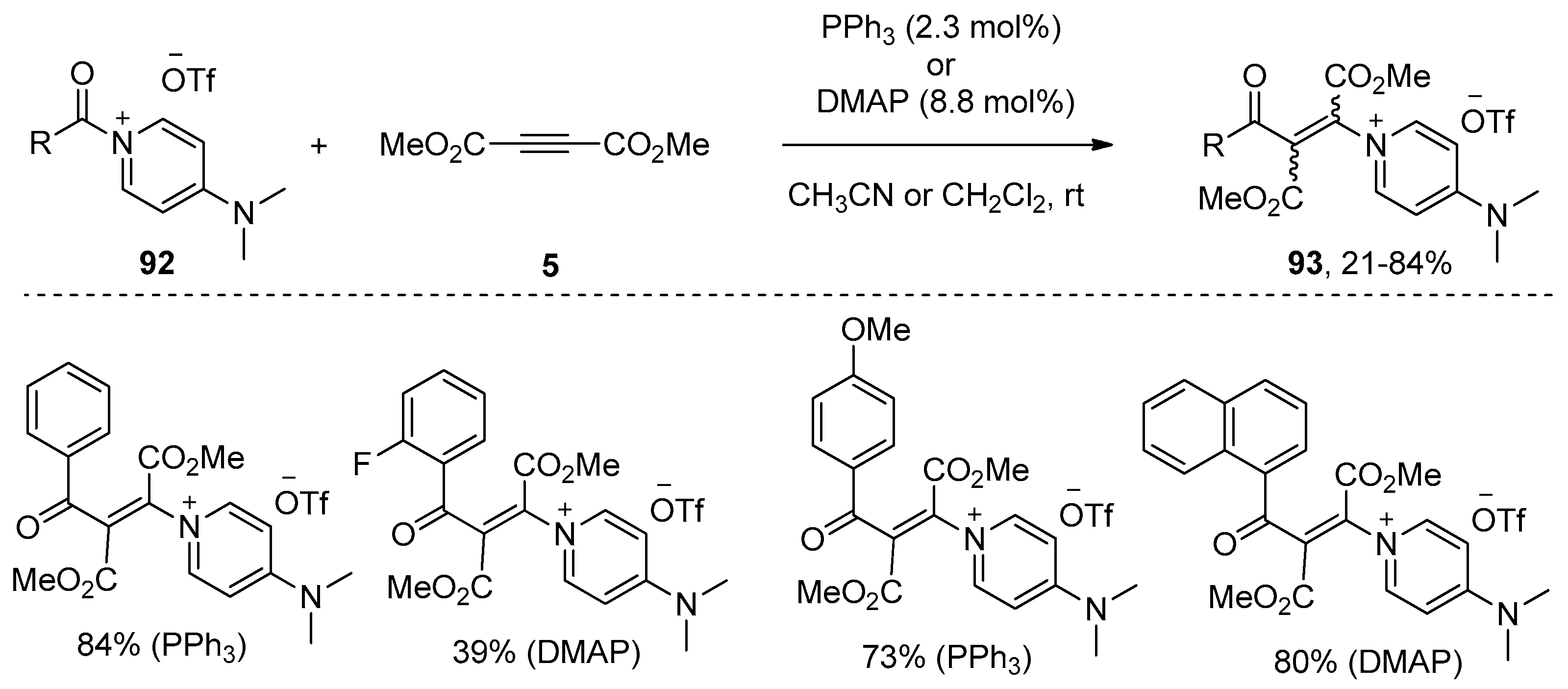
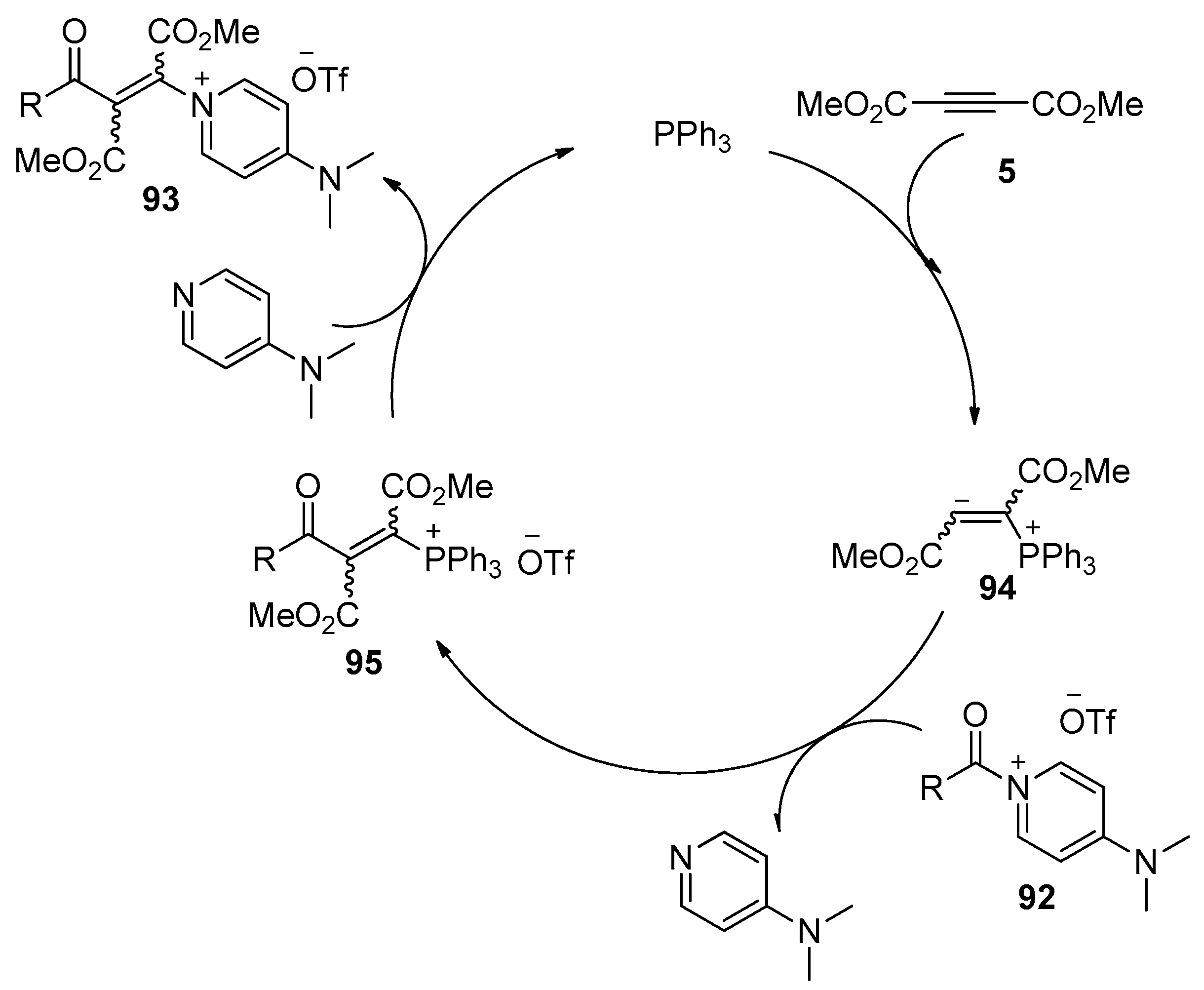
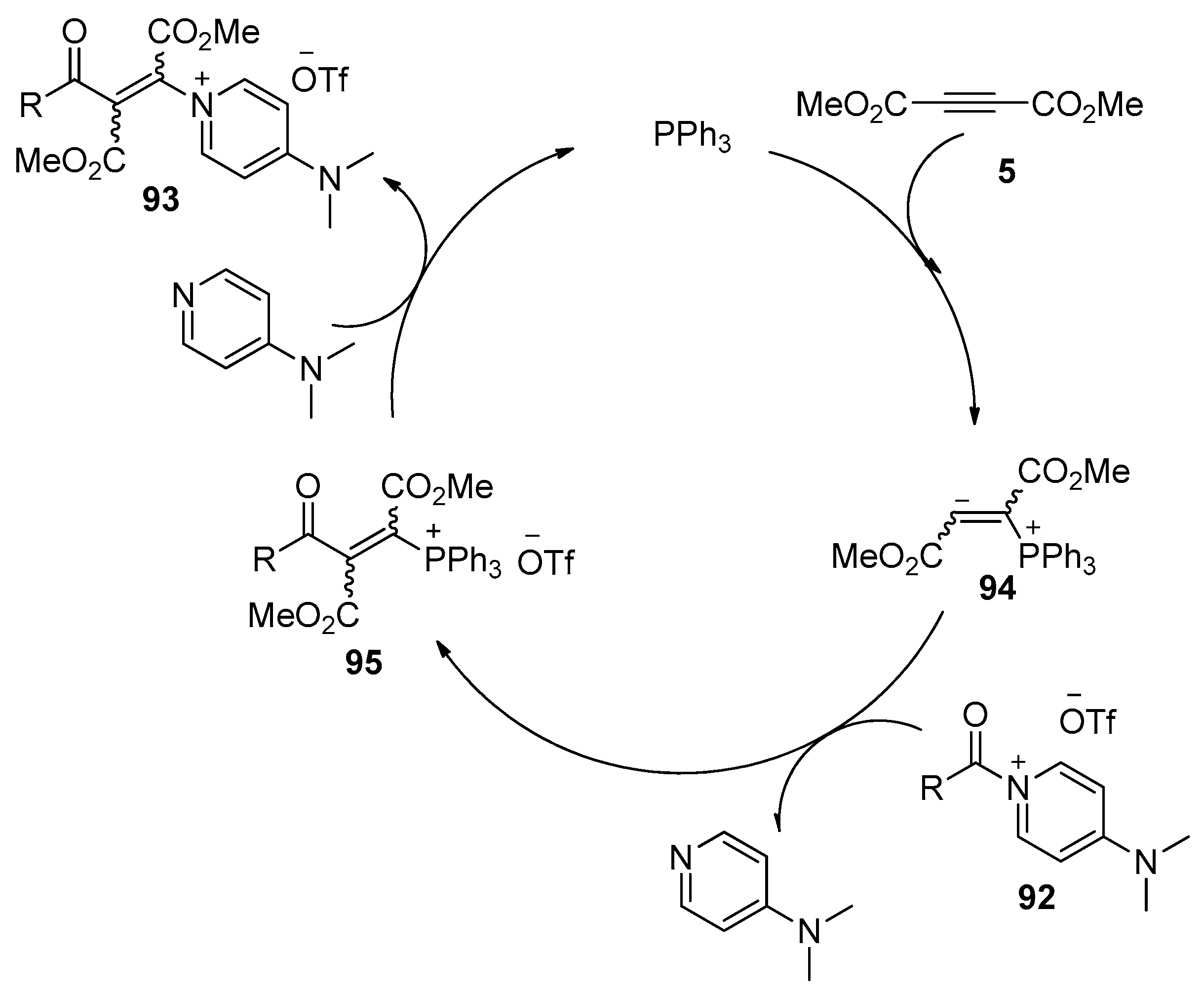
In addition to the metal-catalyzed processes, methods utilizing organocatalysis, which feature advantages such as low cost, environmental economy, and the avoidance of metal contamination, have also been developed in recent years. What seems particularly interesting is the insertion of an electron-deficient alkyne 5 into the amide bond of an acyl-onio salt 92 (Scheme 28), as reported by Weiss and Huber [43]. This reaction could be achieved in the presence of a catalytic amount of small organic molecules such as PPh3 or DMAP, providing the desired β-oniovinylation products 93 in good yields. The stereochemistry of this process depends on the reaction conditions, preferentially E- or Z-stereochemistry was observed, and the Z-isomer is the thermodynamically more stable isomer. More importantly, the onio substituent in the products 93 could be selectively replaced by a number of nucleophiles, such as anilines, phenols, and thiophenols, to prepare Michael systems with donor functions in the β-position, which could be further converted into quinolones, thiochromones, and pyrazoles by intramolecular cyclization. The authors proposed a catalytic cycle for this organocatalytic process. Taking the transformation catalyzed by PPh3 as the example (Scheme 29), the conjugate addition of PPh3 to alkyne 5 produces the zwitterionic intermediate 94, which attacks the electrophilic carbonyl center of 92 to provide intermediate 95 with liberation of 4-dimethylaminopyridine (DMAP). Then intermediate 95 reacts with the liberated DMAP to give the products 93 and regenerates the catalyst.
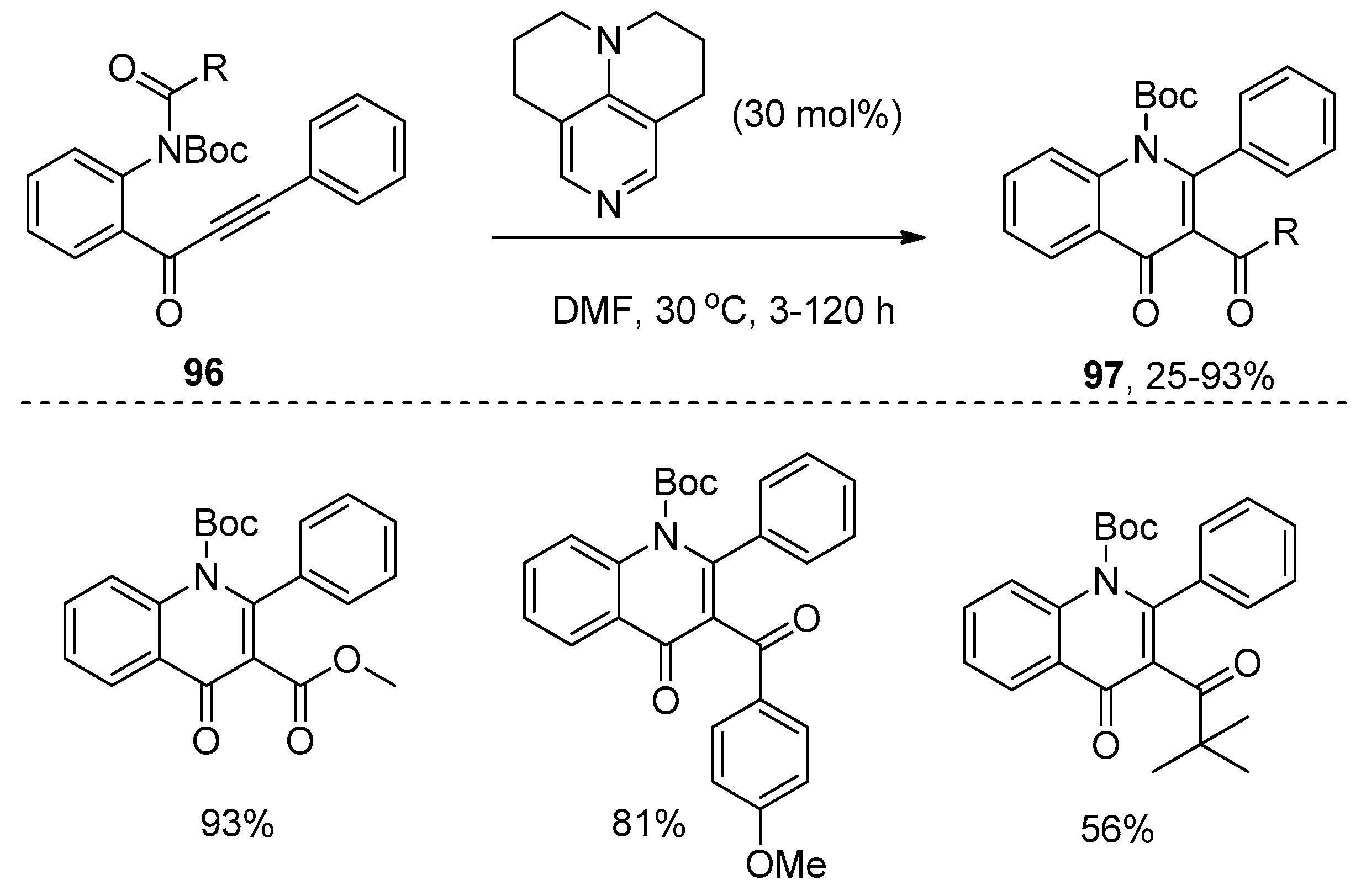
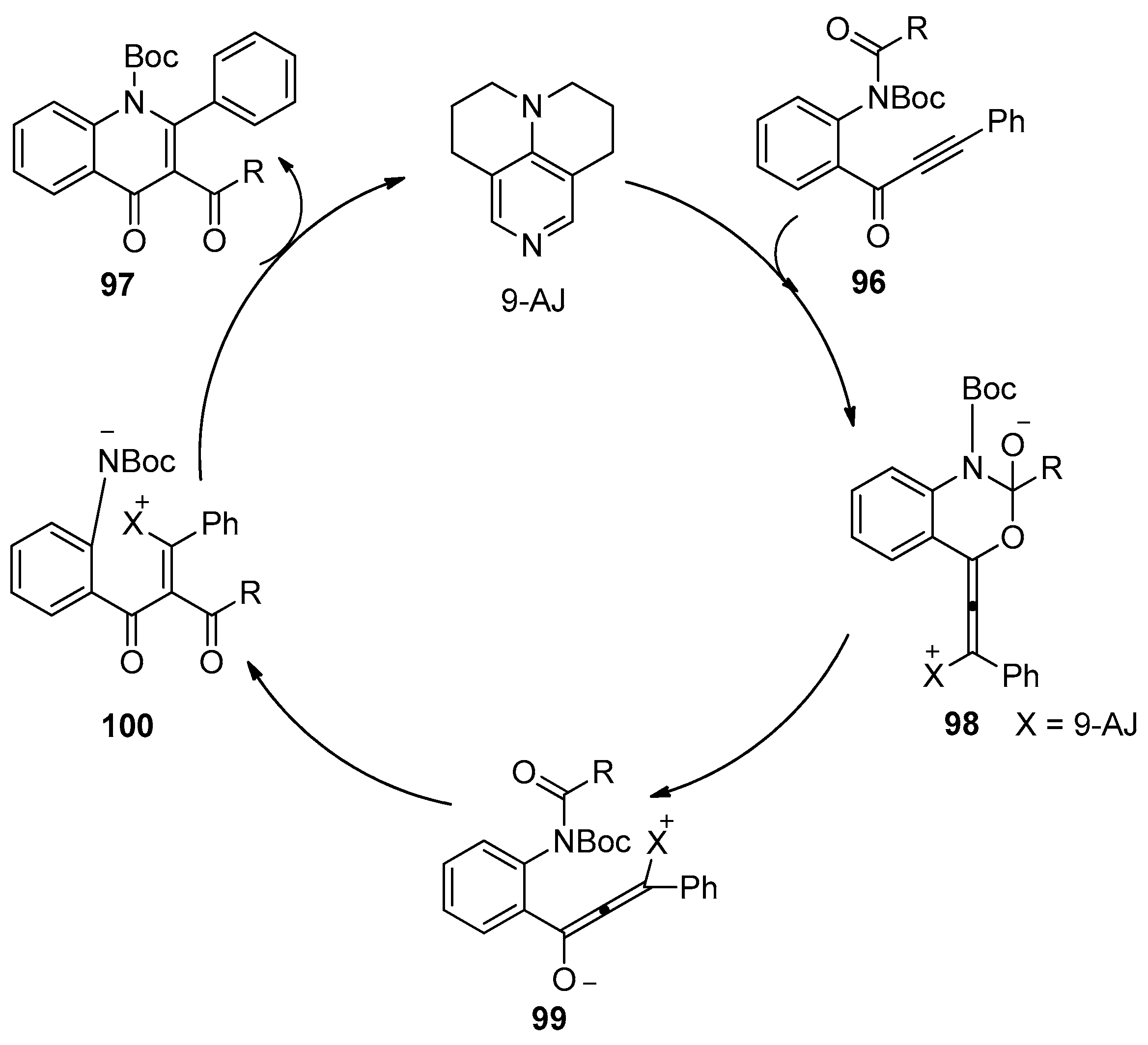
Recently, Doi’s group reported an example of amide addition to alkynes through tertiary amine organocatalysis (Scheme 30) [44]. They found that o-alkynoylaniline derivatives 96 could undergo intramolecular aminoacylation of the carbon–carbon triple bonds successfully under the catalysis of 9-azajulolidine (9-AJ) to afford the 3-acyl-4-quinolinones 97 in moderate to good yields with excellent regioselectivity. Notably, a variety of acyl groups including ester groups could act as migrating groups to be transferred to the C3-positon of the quinolinones. Particularly, the synthesis of pyrrolyl 4-quinolinone alkaloid, quinolactacide, and its analogues were successfully achieved by the authors employing this organocatalytic process. Finally, a plausible reaction mechanism was outlined in Scheme 31. 1,4-addition of 9-AJ to substrates 96 takes place at first, and the subsequent nucleophilic attack of the resulting anion to the acyl group provides intermediate 98, which could be converted into the intermediate 99. Then the acyl group in allenolate 99 could be transferred to the C3-position, thus leading to the formation of enone 100, which undergoes 6-endo cyclization to provide the products 97 with the regeneration of 9-AJ.
3. Transition-Metal-Catalyzed Addition of Sulfonamide Bonds to Alkynes
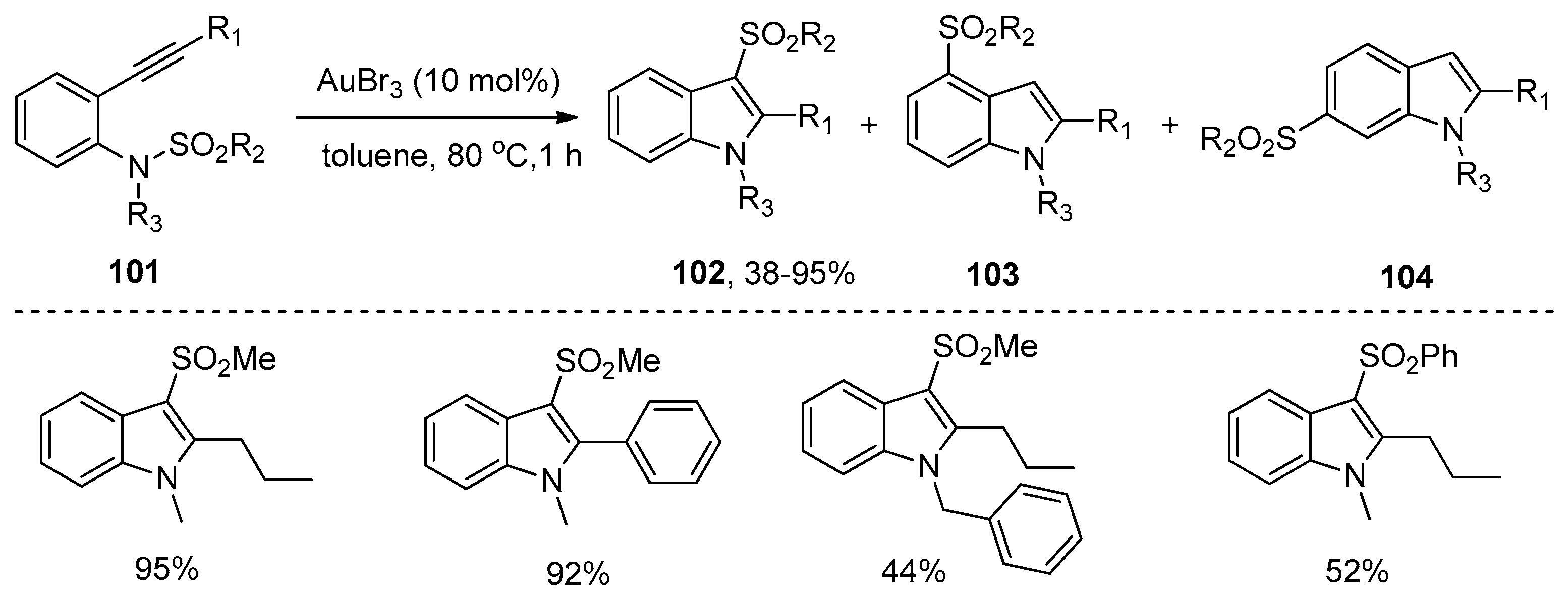
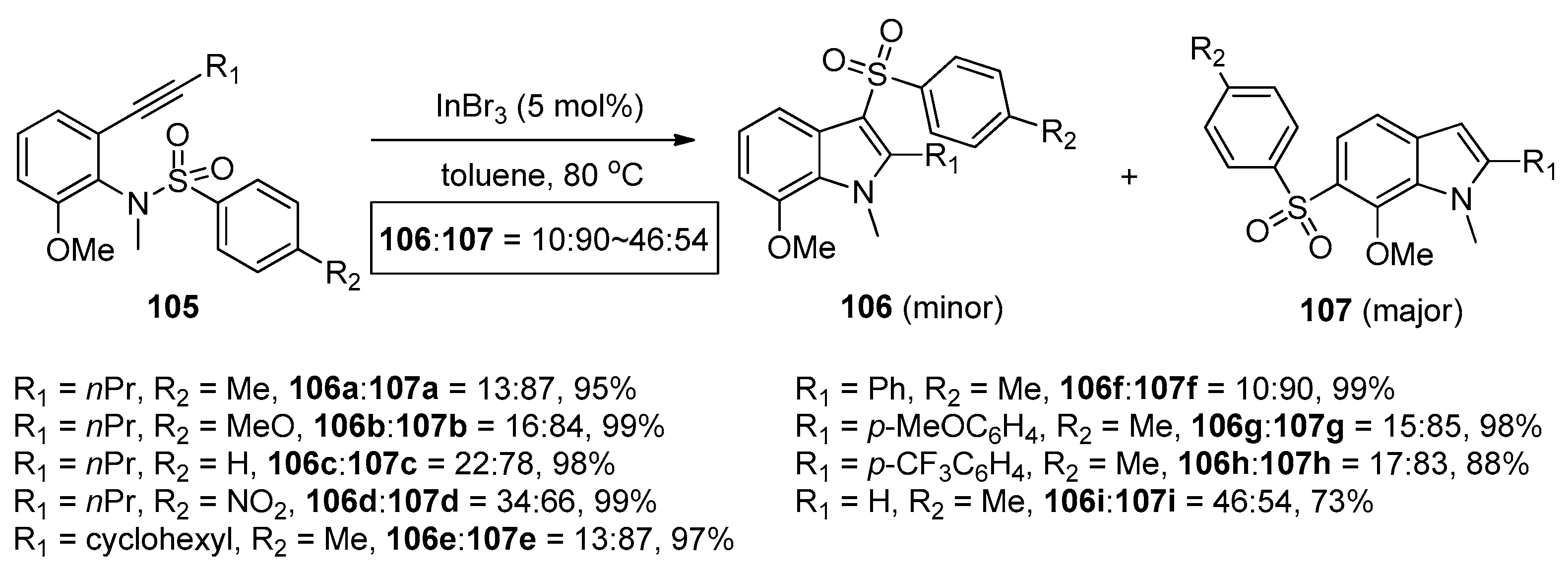
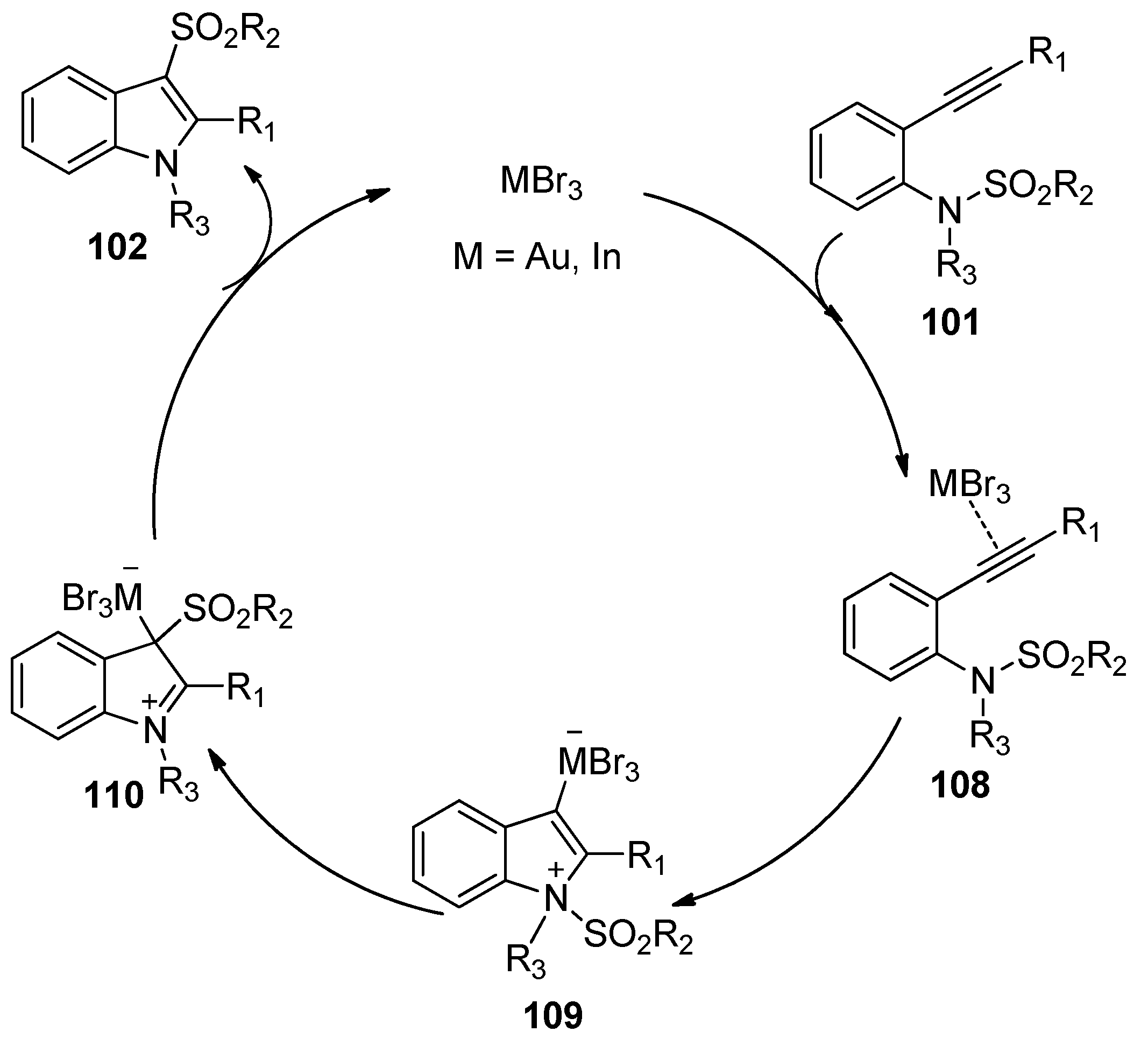
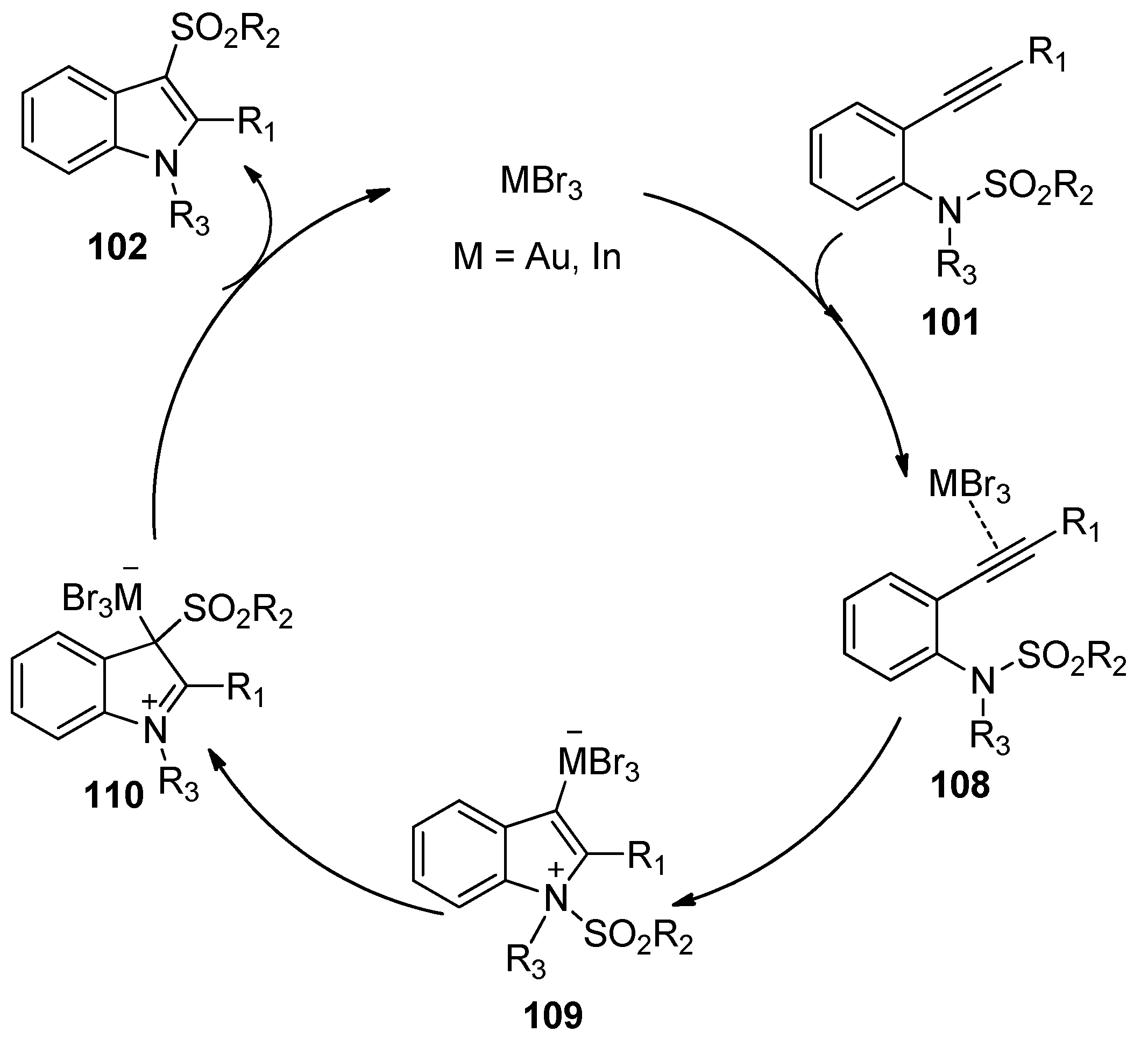
The group of Nakamura reported the first example of the addition of sulfonamides to alkynes in 2007. As shown in Scheme 32, ortho-alkynyl-N-sulfonylanilines 101 could undergo the intramolecular aminosulfonylation of carbon–carbon triple bonds successfully under the catalysis of AuBr3 to give the 3-sulfonylindoles 102 in good to high yields [45,46]. Although small amounts of 4- and 6-sulfonylindoles were obtained as the byproducts in some examples, this process provides a facile and efficient method for the synthesis of 3-sulfonylindoles, which cannot be synthesized directly from the corresponding unsubstituted indoles by electrophilic substitution because the electrophilicity of the sulfonyl groups is much lower than that of the acyl groups and halogens [47,48]. Interestingly, when 2-alkynyl-6-methoxy-N-sulfonylanilines 105 were employed as the substrates, this transformation could also occur under the catalysis of InBr3. However, the intramolecular aminosulfonylation products 106 were obtained as minor products, whereas 6-sulfonylindoles 107 were observed as the major products (Scheme 33). The reaction mechanism of this process may involve the initial coordination of the catalyst to the alkyne moiety, subsequent nucleophilic attack of the nitrogen atom to the carbon–carbon triple bond, the following migration of the sulfonyl group to the C3-position of the indole skeleton, and the final generation of the products with elimination of the catalyst (Scheme 34).
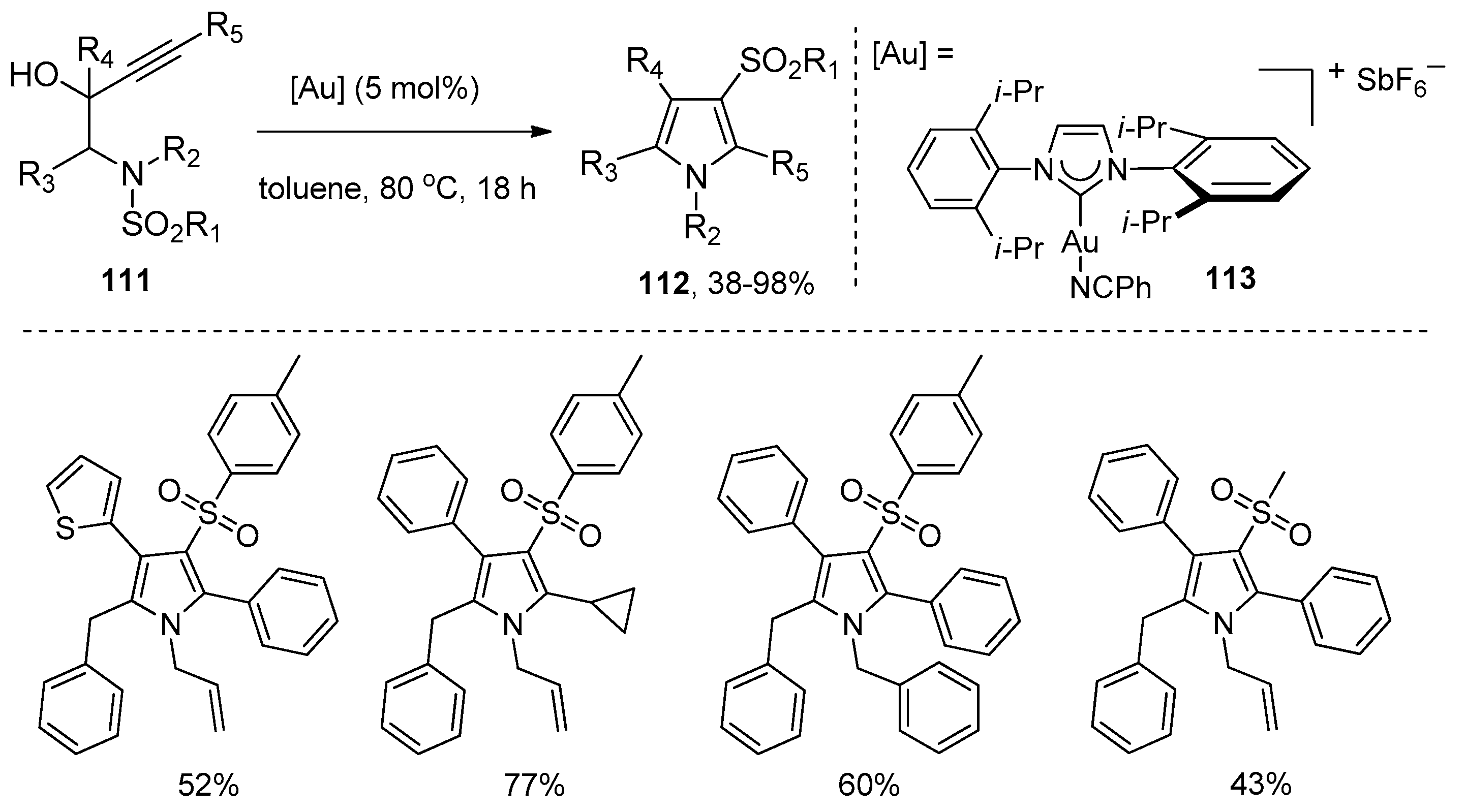
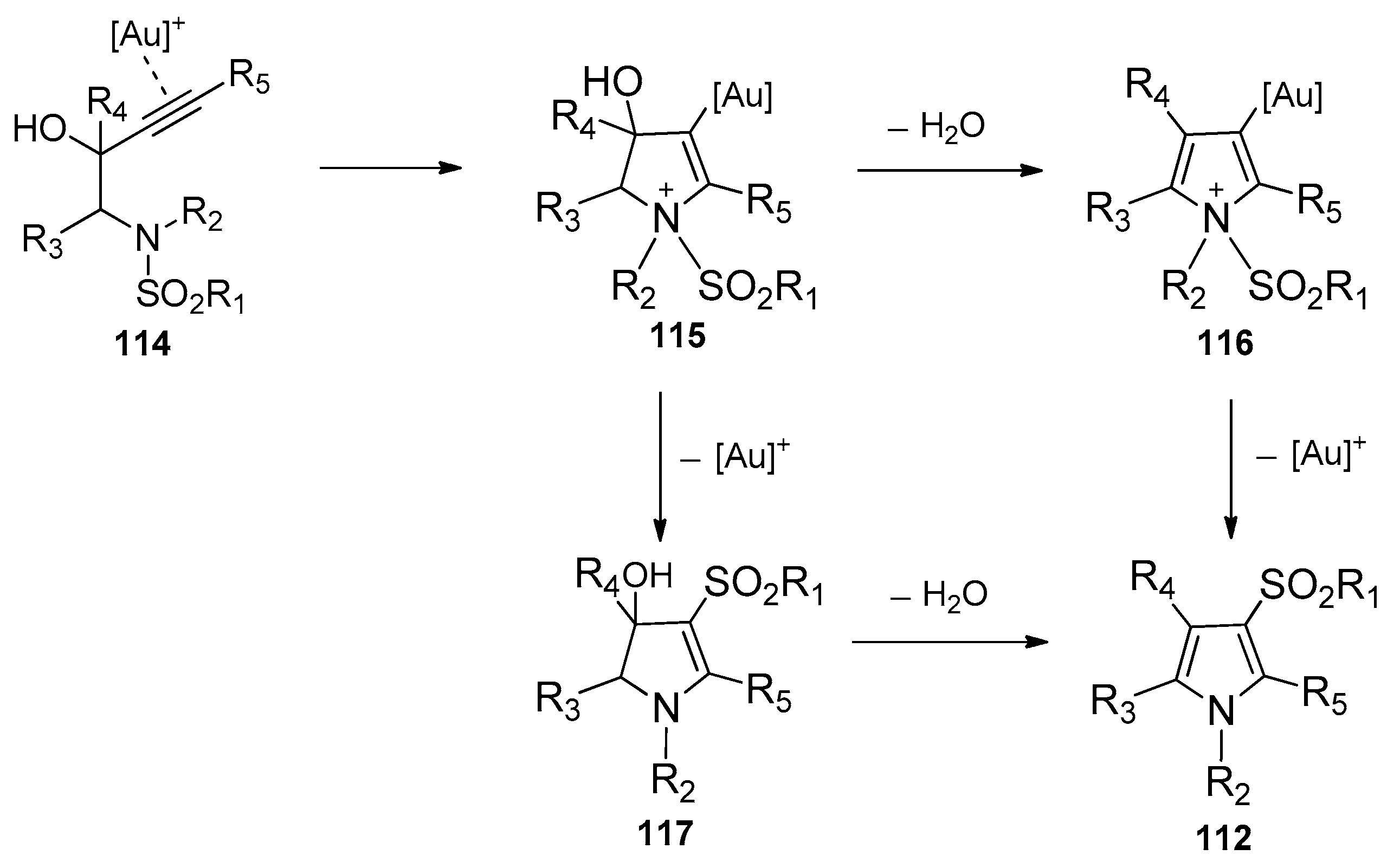
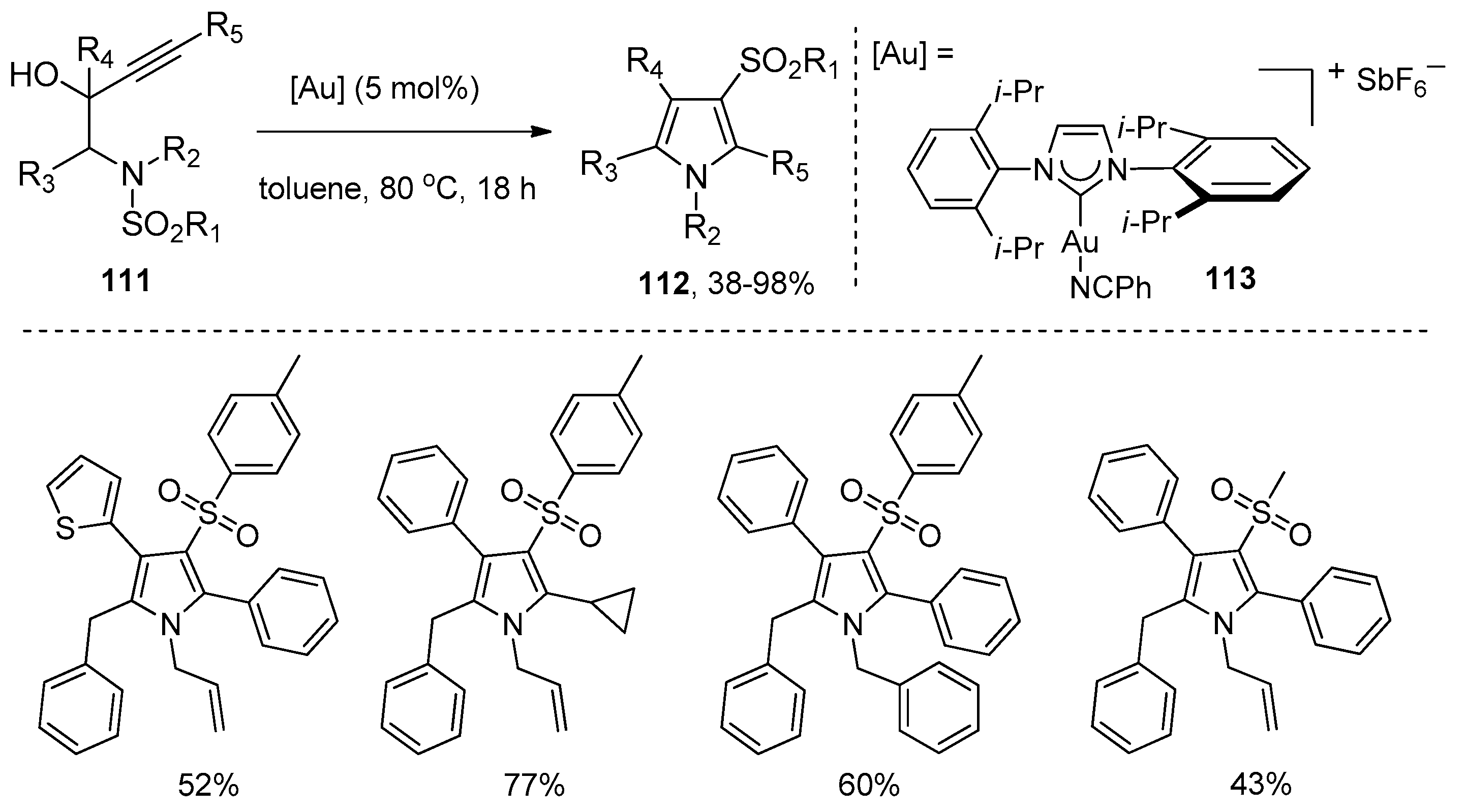
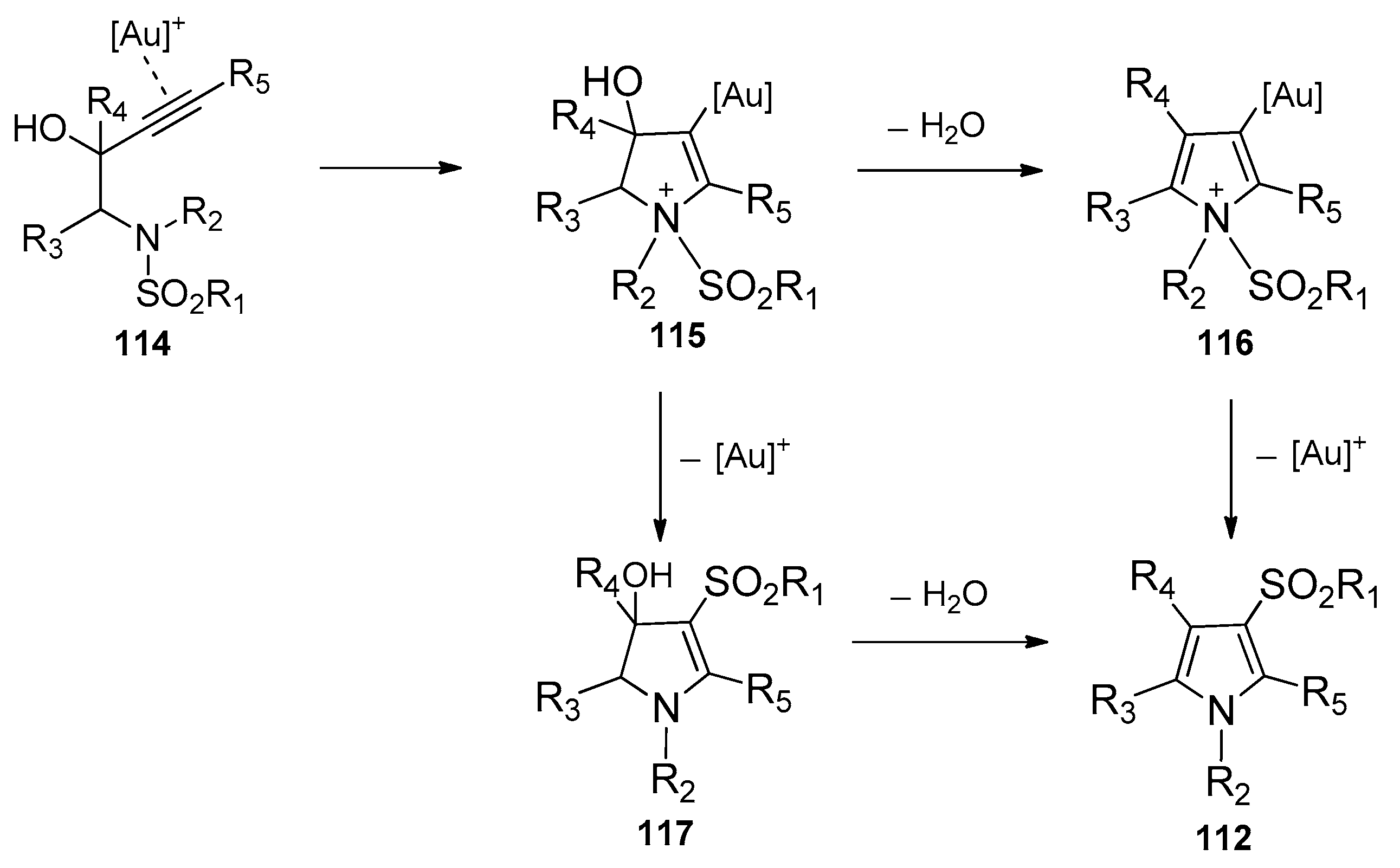
Subsequently, Chan’s group presented a gold-catalyzed domino aminocyclization/1,3-sulfonyl migration of N-substituted N-sulfonyl-aminobut-3-yn-2-ols 111 to synthesize highly functional pyrroles 112 (Scheme 35) [49]. A screening of gold catalysts disclosed that the NHC (N-heterocyclic carbene)-gold(I) complex 113 was found to be the most effective catalyst for this intramolecular aminosulfonylation of alkynes. It could catalyze the conversion of various substrates 111 carrying electron-withdrawing, electron-donating, and sterically demanding groups to the corresponding pyrroles 112 in moderate to high yields. It is worth noting that this method provides an efficient and convenient tool to prepare penta-substituted highly functional pyrroles. The mechanism of this transformation is outlined in Scheme 36. The coordination of gold cation to the alkyne moiety of the substrates gives intermediate 114, which undergoes a nucleophilic attack of the nitrogen atom to the alkynes to produce intermediate 115. At this juncture, dehydration of intermediate 115 yields cationic pyrrole–gold adduct 116, subsequent 1,3-sulfonyl migration of intermediate 116 then results in deauration with the regeneration of the gold catalyst and delivery of the products 112. Alternatively, intermediate 115 could undergo the deaurative 1,3-sulfonyl migration first to afford intermediate 117, which undergoes dehydrative aromatization to produce the products 112.
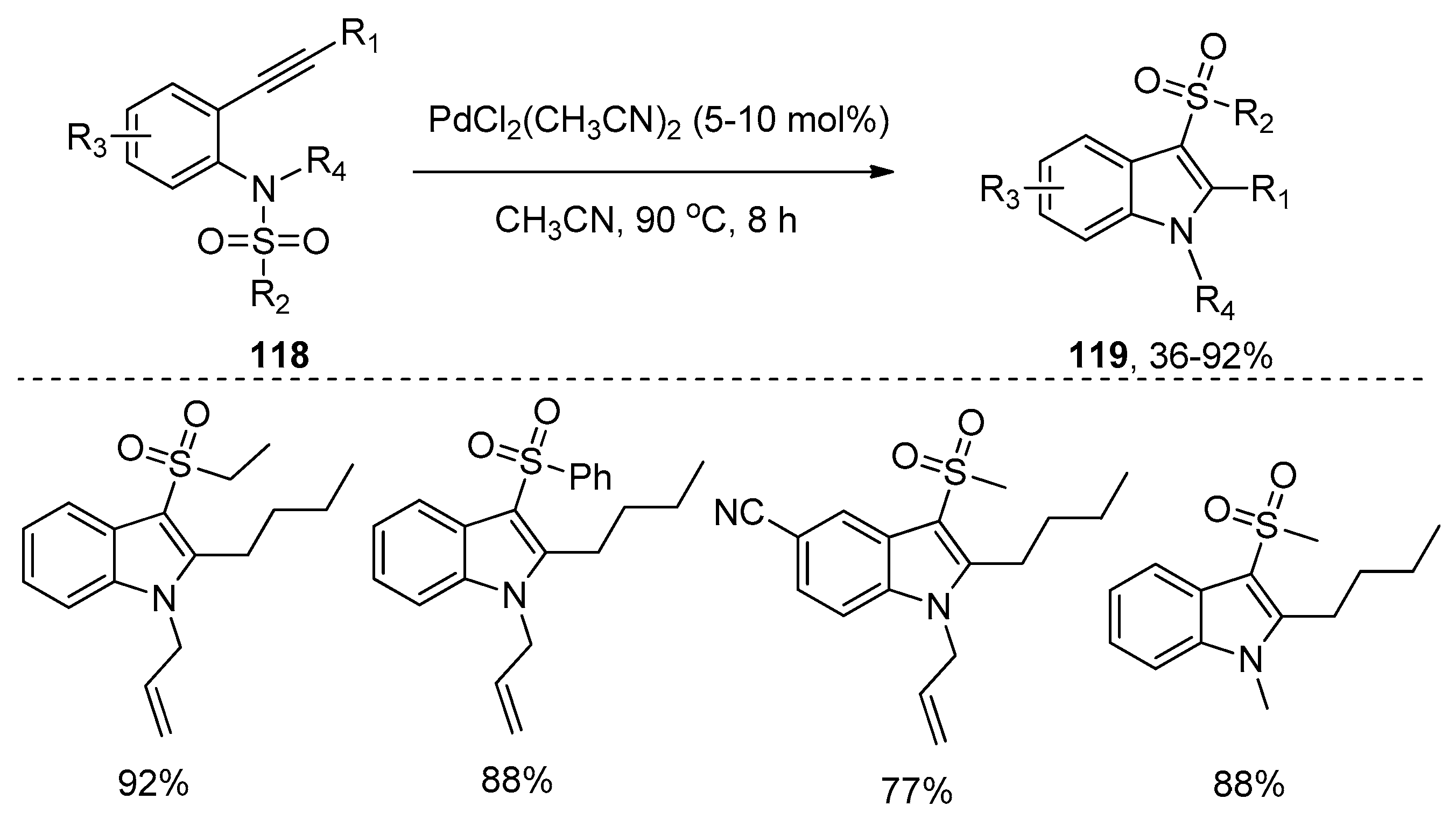
Soon afterwards, Liu’s group reported a more general and efficient intramolecular aminosulfonylation of alkynes to synthesize 3-sulfonylindoles 119 through palladium catalysis (Scheme 37) [35,50]. The reactions took place smoothly with PdCl2(CH3CN)2 in CH3CN at 90 °C to afford 3-sulfonylindoles 119 as the single products without the generation of 4- and 6-sulfonylindole regioisomers, which were obtained as the byproducts in the gold- and indium-catalyzed process. In addition, this protocol features broad substrate scope, good functional group tolerance, and moderate to high yields, thus providing a practical access to functional 3-sulfonylindoles. A plausible mechanism, which is similar to gold- or indium-catalyzed aminosulfonylation of alkynes [45,46], was also proposed by the authors.
4. Conclusions and Perspectives
In this review, we presented a summary of the addition of amide/sulfonamide bonds to alkynes, which has emerged as a highly important tool to functionalize carbon–carbon triple bonds. The aminoacylation/aminosulfonylation of alkynes, which is characterized by high atom- and step-economy in an environmentally-friendly manner, has also become a remarkable method for the downstream transformations of amide/sulfonamides compounds. Notably, the intramolecular aminoacylation/aminosulfonylation of alkynes has provided a facile and efficient protocol for the synthesis of valuable heterocycles such as chromones, quinolinones, indoles, and pyrroles. Despite the remarkable achievements made, there are at least three areas where some critical advances are necessary to make the aminoacylation/aminosulfonylation of alkynes more general and powerful: (a) the intermolecular addition of unactivated amide/sulfonamide bonds to alkynes is still in high demand; (b) as the reported metal-catalyzed process employed expensive metals, the development of cheap metal catalysis as well as organocatalysis, will be a good direction to take; (c) the exploration of tandem reactions involving aminoacylation/aminosulfonylation of alkynes will continue to drive this field considering their efficiency and step-economy in constructing complex heterocyclic compounds.
Author Contributions
F.Z. wrote the manuscript; P.L. and X.L. collected the literature references; X.J. and J.W. drew the schemes and contributed to the editing; H.L. conceived this review and made major revisions and edits.
Funding
This research was funded by National Natural Science Foundation of China (grant 21602022, 81620108027 and 21632008), Sichuan Science and Technology Program (grant 2018JY0345) and Chengdu University New Faculty Start-up Funding (grant 2081915037).
Acknowledgments
F.Z. gratefully acknowledge the support from 1000 Talents Program of Sichuan Province and Chengdu Talents Program.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Irvine, G.J.; Lesley, M.J.G.; Marder, T.B.; Norman, N.C.; Rice, C.R.; Robins, E.G.; Roper, W.R.; Whittell, G.R.; Wright, L.J. Transition metal-boryl compounds: Synthesis, reactivity, and structure. Chem. Rev. 1998, 98, 2685–2722. [Google Scholar] [CrossRef] [PubMed]
- Marder, T.B.; Norman, N.C. Transition metal catalysed diboration. Top. Catal. 1998, 5, 63–73. [Google Scholar] [CrossRef]
- Han, L.B.; Tanaka, M. Transition metal-catalysed addition reactions of H-heteroatom and inter-heteroatom bonds to carbon-carbon unsaturated linkages via oxidative additions. Chem. Commun. 1999, 395–402. [Google Scholar] [CrossRef]
- Beletskaya, I. Element-element addition to alkynes catalyzed by the group 10 metals. Chem. Rev. 1999, 99, 3435–3461. [Google Scholar] [CrossRef] [PubMed]
- Suginome, M.; Ito, Y. Transition-metal-catalyzed additions of silicon-silicon and silicon-heteroatom bonds to unsaturated organic molecules. Chem. Rev. 2000, 100, 3221–3256. [Google Scholar] [CrossRef] [PubMed]
- Ishiyama, T.; Miyaura, N. Chemistry of group 13 element-transition metal linkage—The platinum- and palladium-catalyzed reactions of (alkoxo)diborons. J. Organomet. Chem. 2000, 611, 392–402. [Google Scholar] [CrossRef]
- Suginome, M.; Ito, Y. Stereoselective accesses to enantioenriched allyl-, allenyl-, and propargyl-silanes via Si-Si bond activation by palladium-isocyanide catalysts. J. Organomet. Chem. 2003, 685, 218–229. [Google Scholar] [CrossRef]
- Dembitsky, V.M.; Ali, H.A.; Srebnik, M. Recent developments in bisdiborane chemistry: B–C–B, B–C–C–B, B–C=C–B and B–C≡C–B compounds. Appl. Organomet. Chem. 2003, 17, 327–345. [Google Scholar] [CrossRef]
- Ishiyama, T.; Miyaura, N. Metal-catalyzed reactions of diborons for synthesis of organoboron compounds. Chem. Rec. 2004, 3, 271–280. [Google Scholar] [CrossRef]
- Beletskaya, I. Element-element additions to unsaturated carbon-carbon bonds catalyzed by transition metal complexes. Chem. Rev. 2006, 106, 2320–2354. [Google Scholar] [CrossRef]
- Shimizu, M.; Hiyama, T. Polyborylated reagents for modern organic synthesis. Proc. Jpn. Acad. Ser. B 2008, 84, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Miyaura, N. Metal-catalyzed reactions of organoboronic acids and esters. Bull. Chem. Soc. Jpn. 2008, 81, 1535–1553. [Google Scholar] [CrossRef]
- Takaya, J.; Iwasawa, N. Catalytic, direct synthesis of bis(boronate) compounds. ACS Catal. 2012, 2, 1993–2006. [Google Scholar] [CrossRef]
- Zhao, F.; Jia, X.; Wang, D.; Fei, C.; Wu, C.; Wang, J.; Liu, H. Research progress in metal-catalyzed addition of carbon-hetero bonds to alkynes. Chin. J. Org. Chem. 2017, 37, 284–300. [Google Scholar] [CrossRef]
- Zhao, F.; Jia, X.; Li, P.; Zhao, J.; Zhou, Y.; Wang, J.; Liu, H. Catalytic and catalyst-free diboration of alkynes. Org. Chem. Front. 2017, 4, 2235–2255. [Google Scholar] [CrossRef]
- Miyabe, H. Transition-metal-free activation of amide bond by arynes. Molecules 2018, 23, 2145. [Google Scholar] [CrossRef] [PubMed]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The current state of peptide drug discovery: Back to the future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef]
- Eğe, S.N.; Carter, M.L.C.; Spencer, R.L.; Nordman, C.E.; Friedman, H.Z. Formation and acid-catalyzed rearrangement of 1,2-diazepin-5-ones. J. Chem. Soc. Perkin Trans. 1976, 1, 868–876. [Google Scholar] [CrossRef]
- Turk, C.; Svete, J.; Stanovnik, B.; Golič, L.; Golobič, A.; Selič, L. Unusual reactions of 5,5-dimethyl-2-(indenyl-2)-3-pyrazolidinone with acetylenedicarboxylates. Org. Lett. 2000, 2, 423–424. [Google Scholar] [CrossRef] [PubMed]
- Hanack, M.; Wilhelrn, B. Addition of carboxamides to alkynyl trifluoromethyl sulfones. Angew. Chem. Int. Ed. Engl. 1989, 28, 1057–1059. [Google Scholar] [CrossRef]
- Knölker, H.J.; El-Ahl, A.A. Imidazole derivatives, part VIII. Stereoselective formation of 1-[(E) 3-(1-imidazolyl)-2-alkenoyl]imidazoles. Heterocycles 1993, 36, 1381–1385. [Google Scholar] [CrossRef]
- Suzuki, K.; Ohkuma, T.; Tsuchihashi, G. Preparation of enaminones by two-carbon homologation of amides with lithium (triphenylsilyl)acetylide. J. Org. Chem. 1987, 52, 2930–2932. [Google Scholar] [CrossRef]
- Jeong, I.H.; Jeon, S.L.; Min, Y.K.; Kim, B.T. A novel approach to β-trifluoromethyl enaminones. Tetrahedron Lett. 2002, 43, 7171–7174. [Google Scholar] [CrossRef]
- Jeong, I.H.; Jeon, S.L.; Kim, M.S.; Kim, B.T. New approaches to β-trifluoromethylated enone derivatives. J. Fluorine Chem. 2004, 125, 1629–1638. [Google Scholar] [CrossRef]
- Jeon, S.L.; Kim, J.K.; Son, J.B.; Kim, B.T.; Jeong, I.H. One pot synthesis of novel α,β-dichloro-β-trifluoromethylated enones and their application to the synthesis of trifluoromethylated heterocycles. J. Fluorine Chem. 2007, 128, 153–157. [Google Scholar] [CrossRef]
- Persson, T.; Nielsen, J. Synthesis of N-methoxy-N-methyl-β-enaminoketoesters: New synthetic precursors for the regioselective synthesis of heterocyclic compounds. Org. Lett. 2006, 8, 3129–3222. [Google Scholar] [CrossRef]
- Choudhury, A.; Breslav, M.; Grimm, J.S.; Xiao, T.; Xu, D.; Sorgi, K.L. A facile one-pot synthesis of acyclic β-enamino ketones, an important class of versatile synthetic intermediates. Tetrahedron Lett. 2007, 48, 3069–3072. [Google Scholar] [CrossRef]
- Zheng, Z.; Wang, Y.; Xu, M.; Kong, L.; Wang, M.; Li, Y. Transition-metal-free insertion reactions of alkynes into the C–N r-bonds of imides: Synthesis of substituted enamides or chromones. Chem. Commun. 2018, 54, 6192–6195. [Google Scholar] [CrossRef]
- Zheng, Z.; Tao, Q.; Ao, Y.; Xu, M.; Li, Y. Transition-metal-free aminoacylation of ynones with amides: Synthesis of 3-carbonyl-4-quinolinones or functionalized enaminones. Org. Lett. 2018, 20, 3907–3910. [Google Scholar] [CrossRef]
- Shimada, T.; Nakamura, I.; Yamamoto, Y. Intramolecular C-N bond addition of amides to alkynes using platinum catalyst. J. Am. Chem. Soc. 2004, 126, 10546–10547. [Google Scholar] [CrossRef]
- Nakamura, I.; Sato, Y.; Konta, S.; Terada, M. Platinum-catalyzed consecutive C-N bond formation-[1,3] shift of carbamoyl and ester groups. Tetrahedron Lett. 2009, 50, 2075–2077. [Google Scholar] [CrossRef]
- Nakamura, I.; Mizushima, Y.; Yamagishi, U.; Yamamoto, Y. Synthesis of 2,3-disubstituted benzofurans and indoles by π-Lewis acidic transition metal-catalyzed cyclization of ortho-alkynylphenyl O,O- and N,O-acetals. Tetrahedron 2007, 63, 8670–8676. [Google Scholar] [CrossRef]
- Zhao, F.; Zhang, D.; Nian, Y.; Zhang, L.; Yang, W.; Liu, H. Palladium-catalyzed difunctionalization of alkynes via C–N and S–N cleavages: A versatile approach to highly functional indoles. Org. Lett. 2014, 16, 5124–5127. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhao, F.; Shu, S.; Wang, J.; Liu, H. Palladium-catalyzed intramolecular addition of C–N bond to alkynes: A novel approach to 3-diketoindoles. RSC Adv. 2015, 5, 90396–90399. [Google Scholar] [CrossRef]
- Okauchi, T.; Itonaga, M.; Minami, T.; Owa, T.; Kitoh, K.; Yoshino, H. A general method for acylation of indoles at the 3-position with acyl chlorides in the presence of dialkylaluminum chloride. Org. Lett. 2000, 2, 1485–1487. [Google Scholar] [CrossRef] [PubMed]
- Merkul, E.; Dohe, J.; Gers, C.; Rominger, F.; Müller, T.J.J. Three-component synthesis of ynediones by a Glyoxylation/Stephens-Castro coupling sequence. Angew. Chem. Int. Ed. 2011, 50, 2966–2969. [Google Scholar] [CrossRef]
- Wu, J.C.; Song, R.J.; Wang, Z.Q.; Huang, X.C.; Xie, Y.X.; Li, J.H. Copper-catalyzed C-H oxidation/cross-coupling of α-amino carbonyl compounds. Angew. Chem. Int. Ed. 2012, 51, 3453–3457. [Google Scholar] [CrossRef]
- Tang, R.Y.; Guo, X.K.; Xiang, J.N.; Li, J.H. Palladium-catalyzed synthesis of 3-acylated indoles involving oxidative cross-coupling of indoles with α-amino carbonyl compounds. J. Org. Chem. 2013, 78, 11163–11171. [Google Scholar] [CrossRef]
- Gao, Q.; Zhang, J.; Wu, X.; Liu, S.; Wu, A. Direct regioselective oxidative cross-coupling of indoles with methyl ketones: A novel route to C3-dicarbonylation of indoles. Org. Lett. 2015, 17, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.; Hu, M.; Liu, Y.; Song, R.J; Lei, Y.; Tang, B.X.; Li, R.J.; Li, J.H. Ruthenium-catalyzed annulation of alkynes with amides via formyl translocation. Chem. Commun. 2012, 48, 3197–3199. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.; Bess, M.; Huber, S.M.; Heinemann, F.W. Nucleophilic β-Oniovinylation: Concept, mechanism, scope, and applications. J. Am. Chem. Soc. 2008, 130, 4610–4617. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Yoshida, M.; Uekusa, H.; Doi, T. Facile synthesis of pyrrolyl 4-quinolinone alkaloid quinolactacide by 9-AJ-catalyzed tandem acyl transfer-cyclization of o-alkynoylaniline derivatives. ACS Omega. 2017, 2, 4370–4381. [Google Scholar] [CrossRef]
- Nakamura, I.; Yamagishi, U.; Song, D.; Konta, S.; Yamamoto, Y. Gold- and indium-catalyzed synthesis of 3- and 6-sulfonylindoles from ortho-alkynyl-N-sulfonylanilines. Angew. Chem. Int. Ed. 2007, 46, 2284–2287. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, I.; Yamagishi, U.; Song, D.; Konta, S.; Yamamoto, Y. Synthesis of 3- and 6-sulfonylindoles from ortho-alkynyl-N-sulfonylanilines by the use of Lewis acidic transition-metal catalysts. Chem. Asian J. 2008, 3, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.M.; Ciccarone, T.M.; MacTough, S.C.; Rooney, C.S.; Balani, S.K.; Condra, J.H.; Emini, E.A.; Goldman, M.E.; Greenlee, W.J.; Kauffman, L.R.; et al. 5-Chloro-3-(phenylsulfonyl)indole-2-carboxamide: A novel, non-nucleoside inhibitor of HIV-1 reverse transcriptase. J. Med. Chem. 1993, 36, 1291–1294. [Google Scholar] [CrossRef]
- Silvestri, R.; Martino, G.D.; Regina, G.L.; Artico, M.; Massa, S.; Vargiu, L.; Mura, M.; Loi, A.G.; Marceddu, T.; Colla, P.L. Novel Indolyl Aryl Sulfones Active against HIV-1 Carrying NNRTI Resistance Mutations: Synthesis and SAR Studies. J. Med. Chem. 2003, 46, 2482–2493. [Google Scholar] [CrossRef]
- Teo, W.T.; Rao, W.; Koh, M.J.; Chan, P.W.H. Gold-catalyzed domino aminocyclization/1,3-sulfonyl migration of N-substituted N-sulfonyl-aminobut-3-yn-2-ols to 1-substituted 3-sulfonyl-1H-pyrroles. J. Org. Chem. 2013, 78, 7508–7517. [Google Scholar] [CrossRef]
- Wu, C.; Zhao, F.; Du, Y.; Zhao, L.; Chen, L.; Wang, J.; Liu, H. Highly selective intramolecular addition of C–N and S–N bonds to alkynes catalyzed by palladium: A practical access to two distinct functional indoles. RSC Adv. 2016, 6, 70682–70690. [Google Scholar] [CrossRef]
Scheme 1.
The addition of amide/sulfonamide bonds to alkynes.
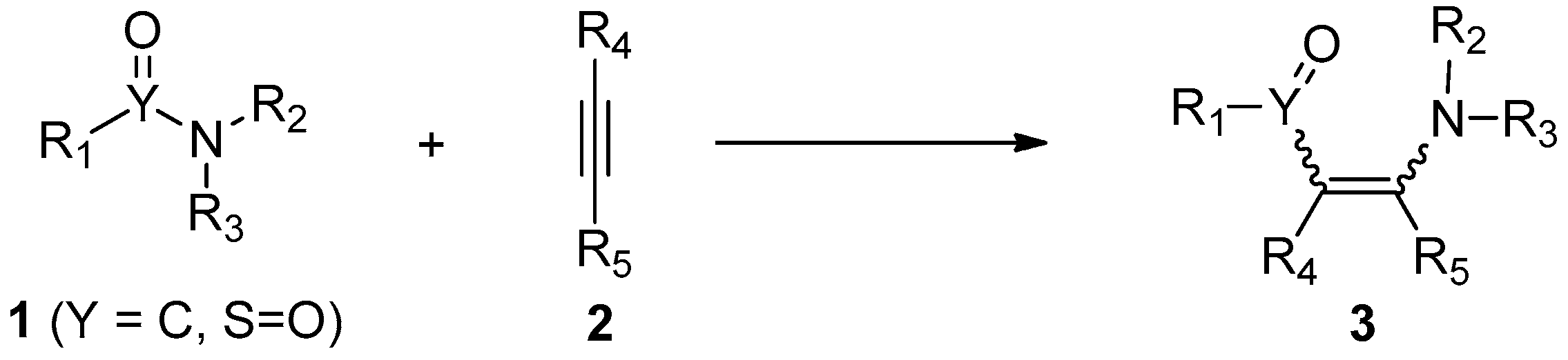
Scheme 2.
Insertion of 2-phenylpyrazolidin-3-ones into amide bonds.

Scheme 3.
Catalyst- and additive-free amide bond addition to alkynes.
Scheme 4.
Proposed mechanism for the insertion of 2-phenylpyrazolidin-3-ones into amide bonds.

Scheme 5.
Direct aminoacylation of alkynyl trifluoromethyl sulfones.

Scheme 6.
Proposed mechanism for the direct aminoacylation of alkynyl trifluoromethyl sulfones.

Scheme 7.
Direct addition of 1,1′-carbonyldiimidazole to alkynes.

Scheme 8.
The insertion of lithium (triphenylsilyl)acetylide into amides. Method A: THF, −45 °C, 5 h, then quenching with MeOH. Method B: BF3∙OEt2 (1.2 equiv.), THF:Hexane = 1:5 (v/v), −45 °C, 5 h, then quenching with 50% aqueous TFA.
Scheme 8.
The insertion of lithium (triphenylsilyl)acetylide into amides. Method A: THF, −45 °C, 5 h, then quenching with MeOH. Method B: BF3∙OEt2 (1.2 equiv.), THF:Hexane = 1:5 (v/v), −45 °C, 5 h, then quenching with 50% aqueous TFA.

Scheme 9.
Addition of Weinreb amides to trifluoropropynyl lithium.

Scheme 10.
(a) Insertion of sodium acetylide of ethyl propynoate into Weinreb amides. (b) Synthesis of pyrazoles employing β-enaminoketoesters.
Scheme 10.
(a) Insertion of sodium acetylide of ethyl propynoate into Weinreb amides. (b) Synthesis of pyrazoles employing β-enaminoketoesters.

Scheme 11.
One-pot sequential transformation of Weinreb amides to enamino ketones.

Scheme 12.
Addition of imides to alkynones promoted by K2CO3 to synthesize enamides.

Scheme 13.
Addition of imides to alkynones promoted by K2CO3 to synthesize chromones.

Scheme 14.
Proposed catalytic cycle for the addition of imides to alkynones promoted by K2CO3.

Scheme 15.
Insertion of alkynones into amides to synthesize enaminones promoted by Cs2CO3.

Scheme 16.
Insertion of alkynones into amides to synthesize quinolinones promoted by Cs2CO3.

Scheme 17.
Proposed reaction mechanism for the insertion of alkynones into amides promoted by Cs2CO3.
Scheme 17.
Proposed reaction mechanism for the insertion of alkynones into amides promoted by Cs2CO3.
Scheme 18.
PtCl2-catalyzed intramolecular aminoacylation of alkynes.

Scheme 19.
Proposed catalytic cycle of PtCl2-catalyzed intramolecular aminoacylation of alkynes.

Scheme 20.
Platinum-catalyzed intramolecular carboamination.

Scheme 21.
Proposed pathway for the generation of 3-protonated byproducts.
Scheme 22.
PdBr2-catalyzed intramolecular aminoacylation of alkynes.

Scheme 23.
PdCl2(CH3CN)2-catalyzed intramolecular aminoacylation of alkynes.
Scheme 24.
PdCl2(CH3CN)2-catalyzed synthesis of 3-diketoindoles via the intramolecular amide bond addition to alkynes.
Scheme 24.
PdCl2(CH3CN)2-catalyzed synthesis of 3-diketoindoles via the intramolecular amide bond addition to alkynes.

Scheme 25.
PdCl2(CH3CN)2-catalyzed synthesis of 3-diketoindole dimer via the intramolecular amide bond addition to alkynes.
Scheme 25.
PdCl2(CH3CN)2-catalyzed synthesis of 3-diketoindole dimer via the intramolecular amide bond addition to alkynes.
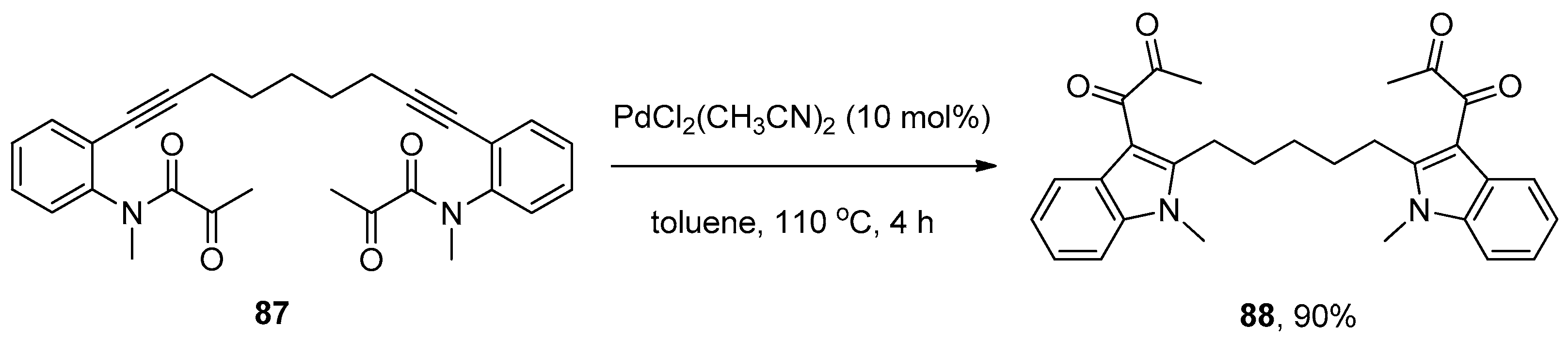
Scheme 26.
[RuCl2(p-cym)]2 catalyzed intramolecular aminoacylation of alkynes.
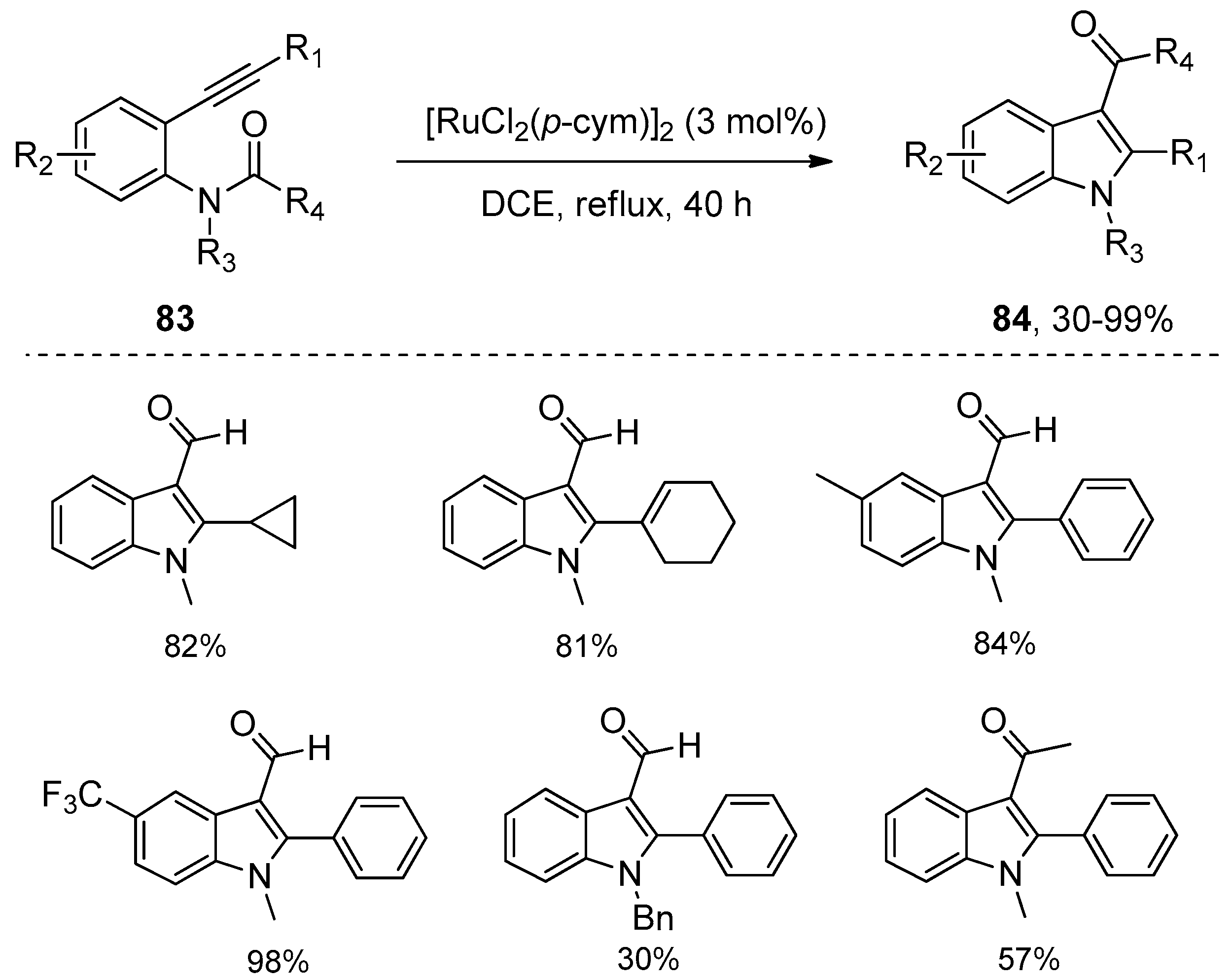
Scheme 27.
Possible mechanism of [RuCl2(p-cym)]2 catalyzed intramolecular aminoacylation of alkynes.
Scheme 27.
Possible mechanism of [RuCl2(p-cym)]2 catalyzed intramolecular aminoacylation of alkynes.

Scheme 28.
The addition of amide bonds of acyl-onio salts to alkynes through PPh3 or 4-dimethylaminopyridine (DMAP) organocatalysis.
Scheme 28.
The addition of amide bonds of acyl-onio salts to alkynes through PPh3 or 4-dimethylaminopyridine (DMAP) organocatalysis.

Scheme 29.
Proposed catalytic cycle for the addition of amide bond of acyl-onio salts to alkynes.
Scheme 30.
The 9-azajulolidine-catalyzed intramolecular amide addition to alkynes.

Scheme 31.
Proposed mechanism for the 9-azajulolidine-catalyzed intramolecular amide addition to alkynes.
Scheme 31.
Proposed mechanism for the 9-azajulolidine-catalyzed intramolecular amide addition to alkynes.

Scheme 32.
AuBr3-catalyzed intramolecular aminosulfonylation of alkynes.

Scheme 33.
InBr3-catalyzed intramolecular aminosulfonylation of alkynes.

Scheme 34.
Proposed mechanism for AuBr3/InBr3-catalyzed intramolecular aminosulfonylation of alkynes.
Scheme 34.
Proposed mechanism for AuBr3/InBr3-catalyzed intramolecular aminosulfonylation of alkynes.
Scheme 35.
Gold-catalyzed intramolecular aminosulfonylation of N-substituted N-sulfonyl-aminobut-3-yn-2-ols.
Scheme 35.
Gold-catalyzed intramolecular aminosulfonylation of N-substituted N-sulfonyl-aminobut-3-yn-2-ols.
Scheme 36.
Possible reaction mechanism for gold-catalyzed intramolecular aminosulfonylation of N-substituted N-sulfonyl-aminobut-3-yn-2-ols.
Scheme 36.
Possible reaction mechanism for gold-catalyzed intramolecular aminosulfonylation of N-substituted N-sulfonyl-aminobut-3-yn-2-ols.
Scheme 37.
PdCl2(CH3CN)2-catalyzed intramolecular aminosulfonylation of alkynes.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhao, F.; Li, P.; Liu, X.; Jia, X.; Wang, J.; Liu, H. Recent Advances in the Addition of Amide/Sulfonamide Bonds to Alkynes. Molecules 2019, 24, 164. https://doi.org/10.3390/molecules24010164
AMA Style
Zhao F, Li P, Liu X, Jia X, Wang J, Liu H. Recent Advances in the Addition of Amide/Sulfonamide Bonds to Alkynes. Molecules. 2019; 24(1):164. https://doi.org/10.3390/molecules24010164
Chicago/Turabian StyleZhao, Fei, Pinyi Li, Xiaoyan Liu, Xiuwen Jia, Jiang Wang, and Hong Liu. 2019. "Recent Advances in the Addition of Amide/Sulfonamide Bonds to Alkynes" Molecules 24, no. 1: 164. https://doi.org/10.3390/molecules24010164