
Structural Changes of the Trinuclear Copper Center in Bilirubin Oxidase upon Reduction

 ,
,
Abstract
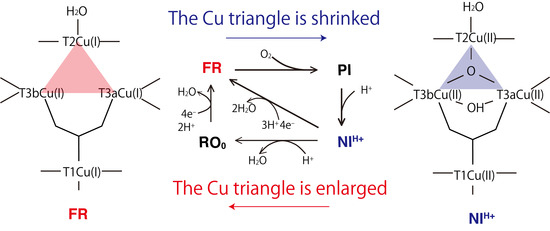
:
1. Introduction
2. Results and Discussions
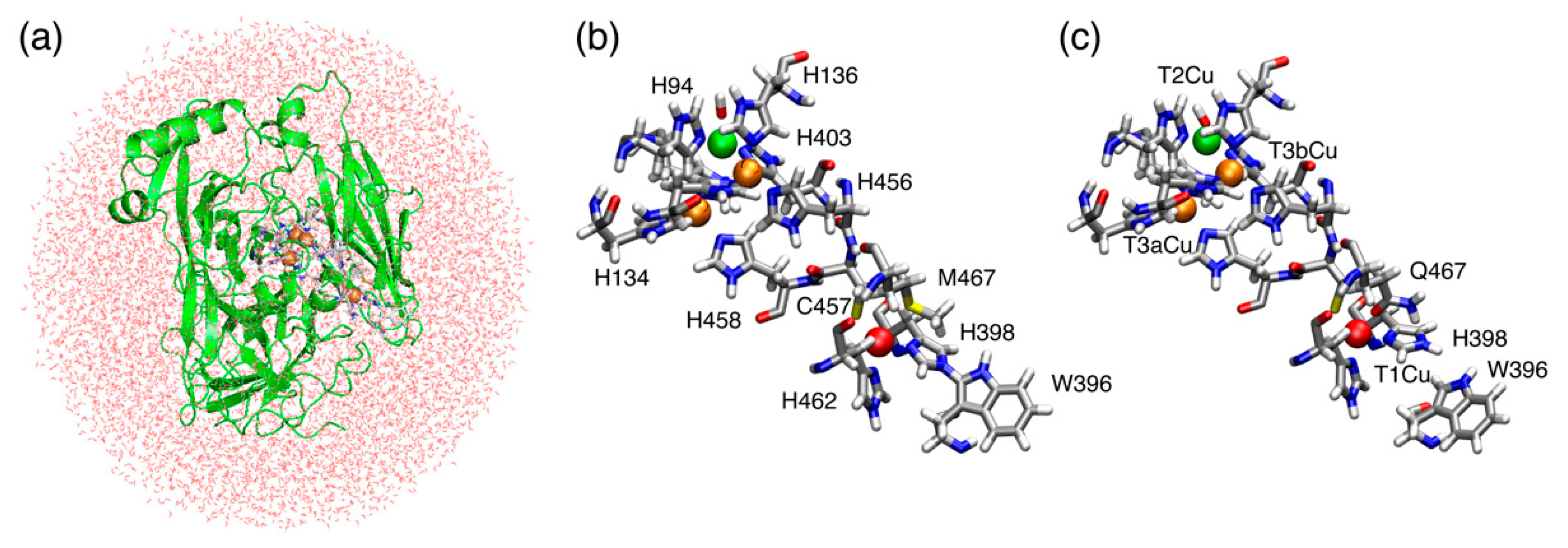
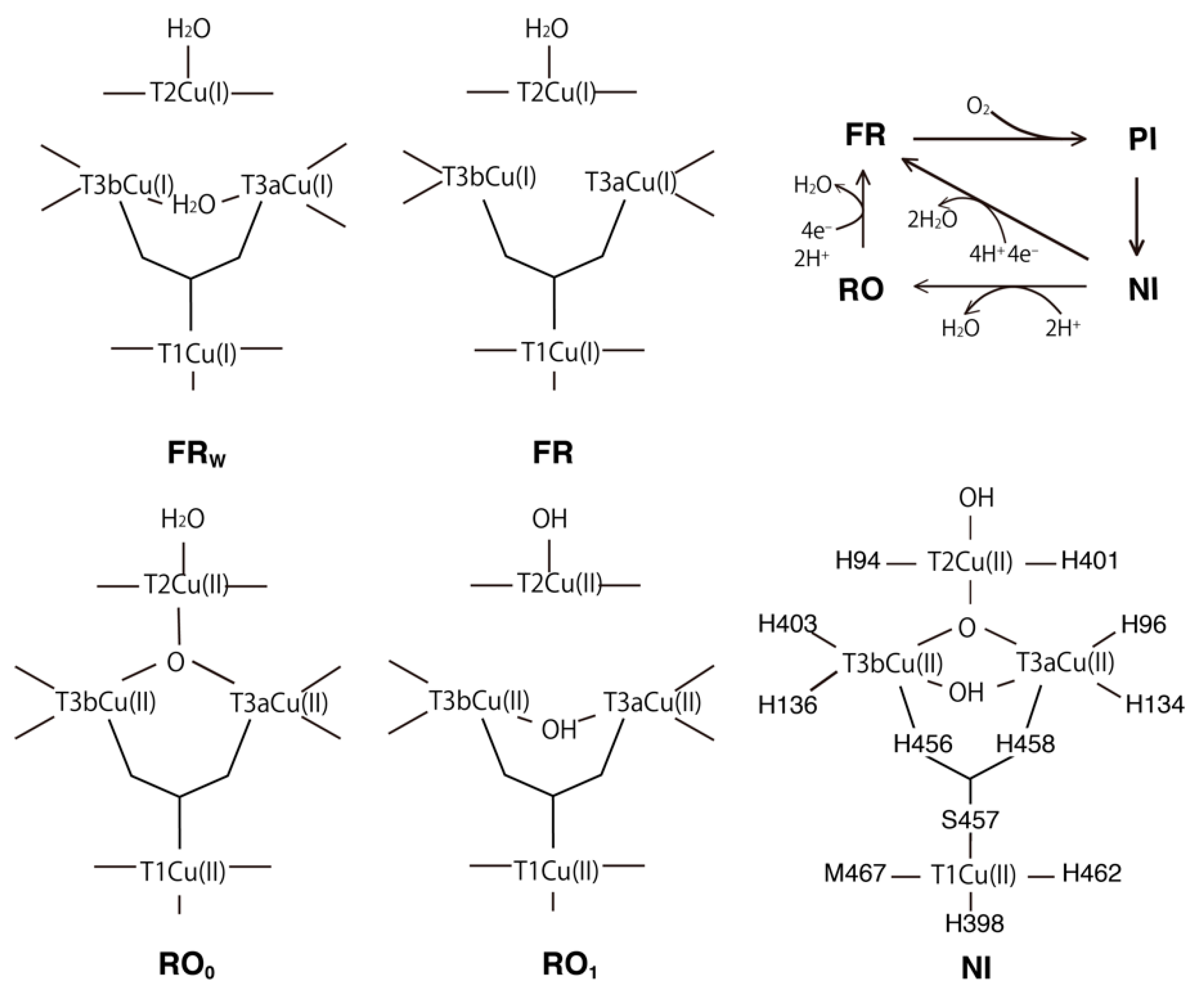
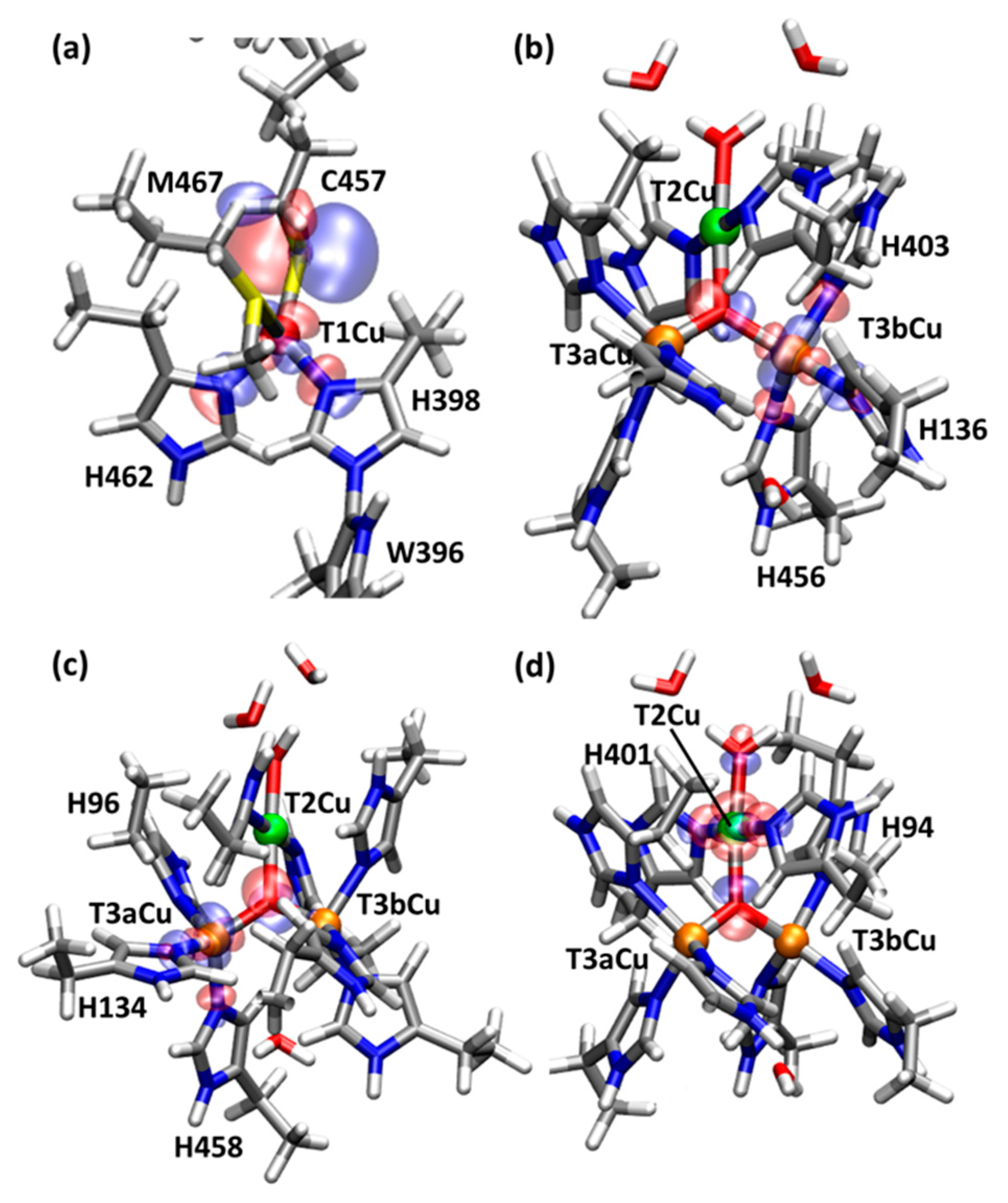
2.1. T1Cu
2.2. T2Cu and T3Cu
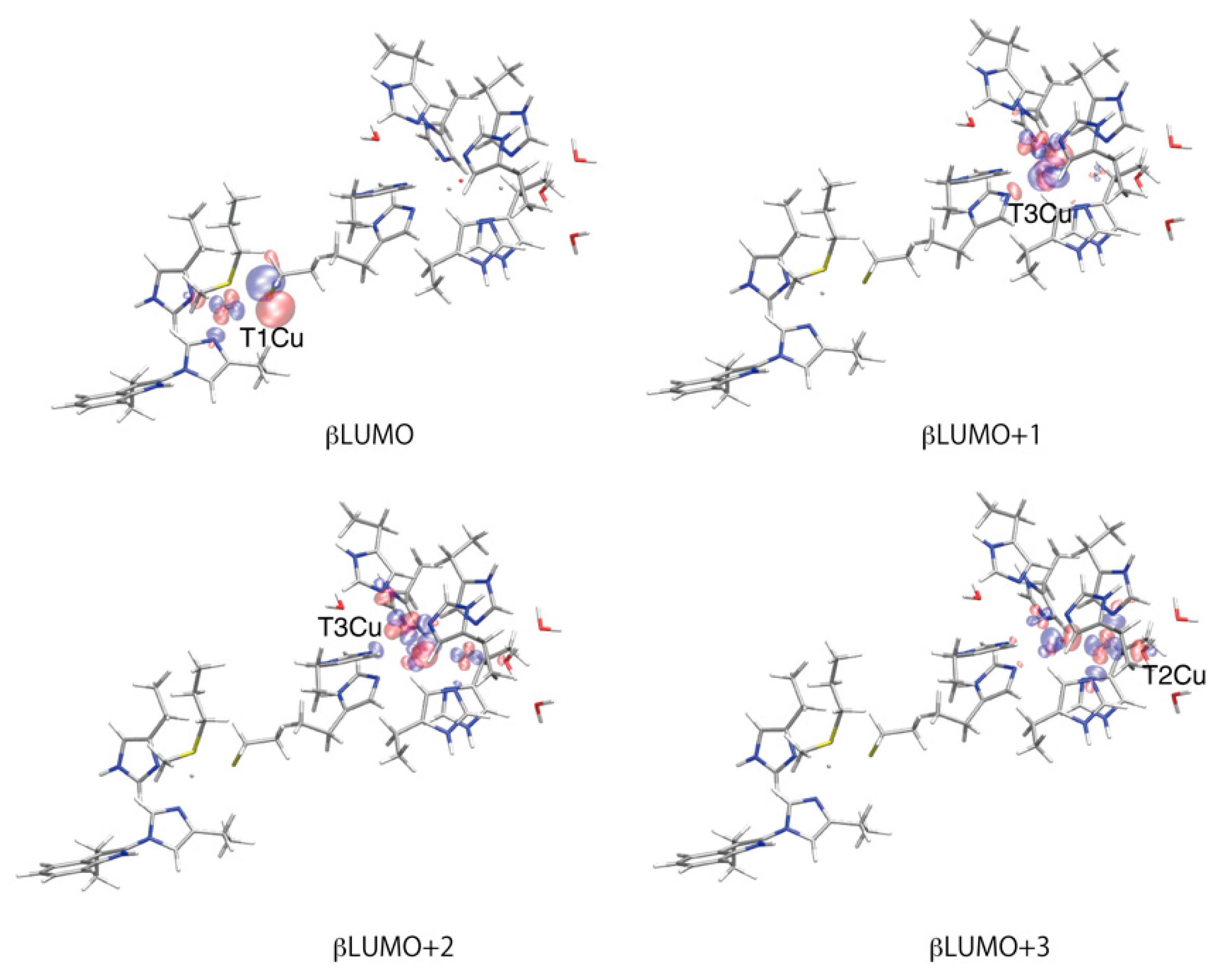
2.3. Molecular Orbital Analysis for the Reduction Process of WT and M467Q BOD
3. Materials and Methods
Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Permyakov, E.A. Metalloproteomics; John Wiley & Sons. Inc.: Hoboken, NJ, USA, 2009; pp. 339–388. ISBN 978-0-470-39248-5. [Google Scholar]
- Mizutani, K.; Toyoda, M.; Sagara, K.; Takahashi, N.; Sato, A.; Kamitaka, Y.; Tsujimura, S.; Nakanishi, Y.; Sugiura, T.; Yamguchi, S.; et al. X-ray analysis of bilirubin oxidase from Myrothecium verrucaria at 2.3 Å resolution using a twinned crystal. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2010, 66, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Cracknell, J.A.; McNamara, T.P.; Lowe, E.D.; Blanford, C.F. Bilirubin oxidase from Myrothecium verrucaria: X-ray determination of the complete crystal structure and a rational surface modification for enhanced electrocatalytic O2 reduction. Dalton. Trans. 2011, 40, 6668–6675. [Google Scholar] [CrossRef] [PubMed]
- Sakai, H.; Nakagawa, T.; Tokita, Y.; Hatazawa, T.; Ikeda, T.; Tsujimura, S.; Kano, K. A high-power glucose/oxygen biofuel cell operating under quiescent conditions. Energy Environ. Sci. 2009, 2, 133–138. [Google Scholar] [CrossRef]
- Mano, N.; de Poulpiquet, A. O2 Reduction in Enzymatic Biofuel Cells. Chem. Rev. 2018, 118, 2392–2468. [Google Scholar] [CrossRef] [PubMed]
- Solomon, E.I.; Augustine, A.J.; Yoon, J. O2 Reduction to H2O by the multicopper oxidases. Dalton Trans. 2008, 3921–3932. [Google Scholar] [CrossRef] [PubMed]
- Solomon, E.I.; Sundaram, U.M.; Machonkin, T.E. Multicopper Oxidases and Oxygenases. Chem. Rev. 1996, 96, 2563–2605. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Sugiyama, R.; Hirota, S.; Inoue, M.; Urata, K.; Minagawa, Y.; Seo, D.; Sakurai, T. Four-electron Reduction of Dioxygen by a Multicopper Oxidase, CueO, and Roles of Asp112 and Glu506 Located Adjacent to the Trinuclear. Copp. Cent. J. Biol. Chem. 2009, 284, 14405–14413. [Google Scholar] [PubMed]
- Tasca, F.; Farias, D.; Castro, C.; Acuna-Rougier, C.; Antiochia, R. Bilirubin Oxidase from Myrothecium verrucaria Physically Absorbed on Graphite Electrodes. Insights into the Alternative Resting Form and the Sources of Activity Loss. PLoS ONE 2015, 10, e0132181. [Google Scholar] [CrossRef]
- Akter, M.; Tokiwa, T.; Shoji, M.; Nishikawa, K.; Shigeta, Y.; Sakurai, T.; Higuchi, Y.; Kataoka, K.; Shibata, N. Redox Potential-Dependent Formation of an Unusual His–Trp Bond in Bilirubin Oxidase. Chem. Eur. J. 2018. [Google Scholar] [CrossRef]
- Shimizu, A.; Sasaki, T.; Kwon, J.H.; Odaka, A.; Satoh, T.; Sakurai, N.; Sakurai, T.; Yamaguchi, S.; Samejima, T. Site-Directed Mutagenesis of a Possible Type 1 Copper Ligand of Bilirubin Oxidase; a Met467Gln Mutant Shows Stellacyanin-Like Properties. J. Biochem. 1999, 125, 662–668. [Google Scholar] [CrossRef]
- Kataoka, K.; Kitagawa, R.; Inoue, M.; Naruse, D.; Sakurai, T.; Huang, H. Point Mutations at the Type I Cu Ligands, Cys457 and Met467, and at the Putative Proton Donor, Asp105, in Myrothecium verrucaria Bilirubin Oxidase and Reactions with Dioxygen. Biochemistry 2005, 44, 7004–7012. [Google Scholar] [CrossRef] [PubMed]
- Shoji, M.; Isobe, H.; Yamanaka, S.; Umena, Y.; Kawakami, K.; Kamiya, N.; Shen, J.-R.; Yamaguchi, K. Large-Scale QM/MM Calculations of Hydrogen Bonding Networks for Proton Transfer and Water Inlet Channels for Water Oxidation-Theoretical System Models of the Oxygen-Evolving Complex of Photosystem II. Adv. Quantum. Chem. 2014, 7, 325–413. [Google Scholar]
- Shoji, M.; Isobe, H.; Nakajima, T.; Yamaguchi, K. Full geometry optimizations of the CaMn4O4 model cluster for the oxygen evolving complex of photosystem II. Chem. Phys. Lett. 2015, 640, 23–30. [Google Scholar] [CrossRef]
- Shoji, M.; Isobe, H.; Shen, J.-R.; Yamaguchi, K. Geometric and electronic structures of the synthetic Mn4CaO4 model compound mimicking the photosynthetic oxygen-evolving complex. Phys. Chem. Chem. Phys. 2016, 18, 11330–11340. [Google Scholar] [CrossRef] [PubMed]
- Shoji, M.; Isobe, H.; Shigeta, Y.; Nakajima, T.; Yamaguchi, K. Concerted Mechanism of Water Insertion and O2 Release during the S4 to S0 Transition of the Oxygen-Evolving Complex in Photosystem II. J. Phys. Chem. B 2018, 122, 6491–6502. [Google Scholar] [CrossRef] [PubMed]
- Shoji, M.; Yoshioka, Y.; Yamaguchi, K. An efficient initial guess formation of broken-symmetry solutions by using localized natural orbitals. Chem. Phys. Lett. 2014, 608, 50–54. [Google Scholar] [CrossRef]
- Komori, H.; Sugiyama, R.; Kataoka, K.; Higuchi, Y.; Sakurai, T. An O-Centered Structure of the Trinuclear Copper Center in the Cys500Ser/Glu506Gln Mutant of CueO and Structural Changes in Low to High X-Ray Dose Conditions. Angew. Chem. Int. Ed. 2012, 51, 1861–1864. [Google Scholar] [CrossRef] [Green Version]
- Valiev, M.; Bylaska, E.J.; Govind, N.; Kowalski, K.; Straatsma, T.P.; van Dam, H.J.J.; Wang, D.; Nieplocha, J.; Apra, E.; Windus, T.L.; et al. NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 2010, 181, 1477–1489. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Wang, J.; Cieplak, P.; Kollman, P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta. 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Sondergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.M.; Sondergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System, version 1.8; DeLano Scientific LLC: Palo Alto, CA, USA, 2015. [Google Scholar]
- Tokiwa, T.; Nakano, S.; Yamamoto, Y.; Ishikawa, T.; Ito, S.; Sladek, V.; Fukuzawa, K.; Mochizuki, Y.; Tokiwa, H.; Misaizu, F.; et al. Development of an Analysis Toolkit, AnalysisFMO, to Visualize Interaction Energies Generated by Fragment Molecular Orbital Calculations. J. Chem. Inf. Model. 2018. [Google Scholar] [CrossRef] [PubMed]
- Humphery, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 27–28. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T1Cu | S (C457) | T2Cu | T3aCu | T3bCu | μ3-oxo | μ2-OH/W | |
|---|---|---|---|---|---|---|---|
| NI | 0.00 | 0.00 | 0.59 | 0.64 | 0.66 | 1.07 | 0.28 |
| NIH+ | 0.13 | 0.30 | 0.58 | 0.62 | 0.65 | 0.75 | 0.29 |
| ROW | 0.30 | 0.55 | 0.59 | 0.46 | 0.65 | 0.76 | 0.08 |
| RO0 | 0.31 | 0.57 | 0.62 | 0.53 | 0.65 | 0.63 | - |
| RO1 | 0.32 | 0.57 | 0.50 | 0.56 | 0.60 | - | 0.22 |
| FRW | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | - | 0.00 |
| FR | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | - | - |
| T2Cu-T3aCu | T2Cu-T3bCu | T3aCu-T3bCu | Cu- μ3-oxo (T2Cu, T3aCu, T3bCu) | Cu- μ2-OH/W (T3aCu, T3bCu) | |
|---|---|---|---|---|---|
| NI | 3.84 | 3.75 | 3.14 | 2.07, 2.18, 2.02 | 1.94, 1.90 (μ2OH) |
| NIH+ | 3.60 | 3.56 | 3.06 | 1.94, 1.99, 2.07 | 1.95, 1.90 (μ2OH) |
| ROW | 3.52 | 3.59 | 3.38 | 2.03, 1.94, 2.11 | 2.58, 1.99 (μ2W) |
| RO0 | 3.48 | 3.48 | 3.27 | 2.09, 1.94, 1.89 | – |
| RO1 | 4.28 | 4.21 | 3.66 | – | 1.99, 1.93 (μ2OH) |
| FRW | 4.34 | 4.52 | 4.79 | – | 2.45, 2.46 (μ2W) |
| FR | 4.27 | 4.45 | 4.60 | – | – |
| 2XLL | 4.06 | 4.07 | 4.87 | – | – |
| 6IQZ | 4.18 | 3.98 | 4.98 | – | 2.69, 2.29 |
| 3UAA | 3.65 | 3.61 | 3.22 | 1.83, 2.07, 2.27 | 2.06, 1.82 |
| 3UAE | 4.40 | 3.98 | 4.96 | – | 2.77, 2.27 |
| l | RMSD (2XLL) | RMSD (6IQZ) | RMSD (3UAA) | RMSD (3UAE) | |
|---|---|---|---|---|---|
| NI | 3.538 | 0.716 | 0.766 | 0.105 | 0.749 |
| NIH+ | 3.382 | 0.720 | 0.766 | 0.051 | 0.765 |
| ROW | 3.493 | 0.610 | 0.654 | 0.097 | 0.669 |
| RO0 | 3.405 | 0.658 | 0.702 | 0.088 | 0.713 |
| RO1 | 4.023 | 0.566 | 0.614 | 0.327 | 0.579 |
| FRW | 4.538 | 0.232 | 0.245 | 0.683 | 0.257 |
| FR | 4.433 | 0.245 | 0.270 | 0.606 | 0.282 |
| 2XLL | 4.278 | 0 | 0.073 | 0.674 | 0.147 |
| 6IQZ | 4.309 | 0.073 | 0 | 0.721 | 0.202 |
| 3UAA | 3.477 | 0.674 | 0.721 | 0 | 0.719 |
| 3UAE | 4.394 | 0.147 | 0.202 | 0.719 | 0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tokiwa, T.; Shoji, M.; Sladek, V.; Shibata, N.; Higuchi, Y.; Kataoka, K.; Sakurai, T.; Shigeta, Y.; Misaizu, F. Structural Changes of the Trinuclear Copper Center in Bilirubin Oxidase upon Reduction. Molecules 2019, 24, 76. https://doi.org/10.3390/molecules24010076
Tokiwa T, Shoji M, Sladek V, Shibata N, Higuchi Y, Kataoka K, Sakurai T, Shigeta Y, Misaizu F. Structural Changes of the Trinuclear Copper Center in Bilirubin Oxidase upon Reduction. Molecules. 2019; 24(1):76. https://doi.org/10.3390/molecules24010076
Chicago/Turabian StyleTokiwa, Takaki, Mitsuo Shoji, Vladimir Sladek, Naoki Shibata, Yoshiki Higuchi, Kunishige Kataoka, Takeshi Sakurai, Yasuteru Shigeta, and Fuminori Misaizu. 2019. "Structural Changes of the Trinuclear Copper Center in Bilirubin Oxidase upon Reduction" Molecules 24, no. 1: 76. https://doi.org/10.3390/molecules24010076