Sorption, Structure and Dynamics of CO2 and Ethane in Silicalite at High Pressure: A Combined Monte Carlo and Molecular Dynamics Simulation Study



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
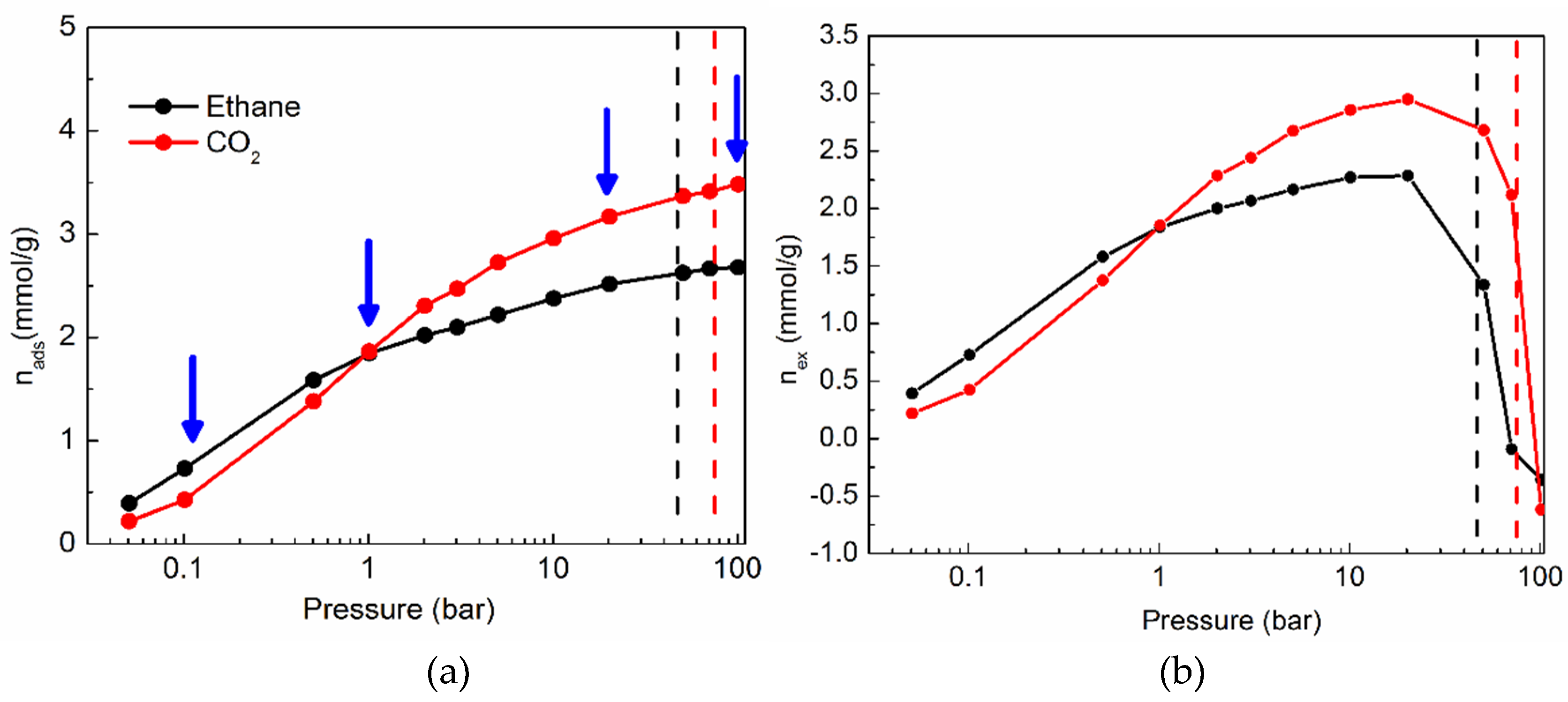
2.1. Sorption Isotherms of CO2 and Ethane in Silicalite
2.2. Structure of Ethane and CO2 in Silicalite
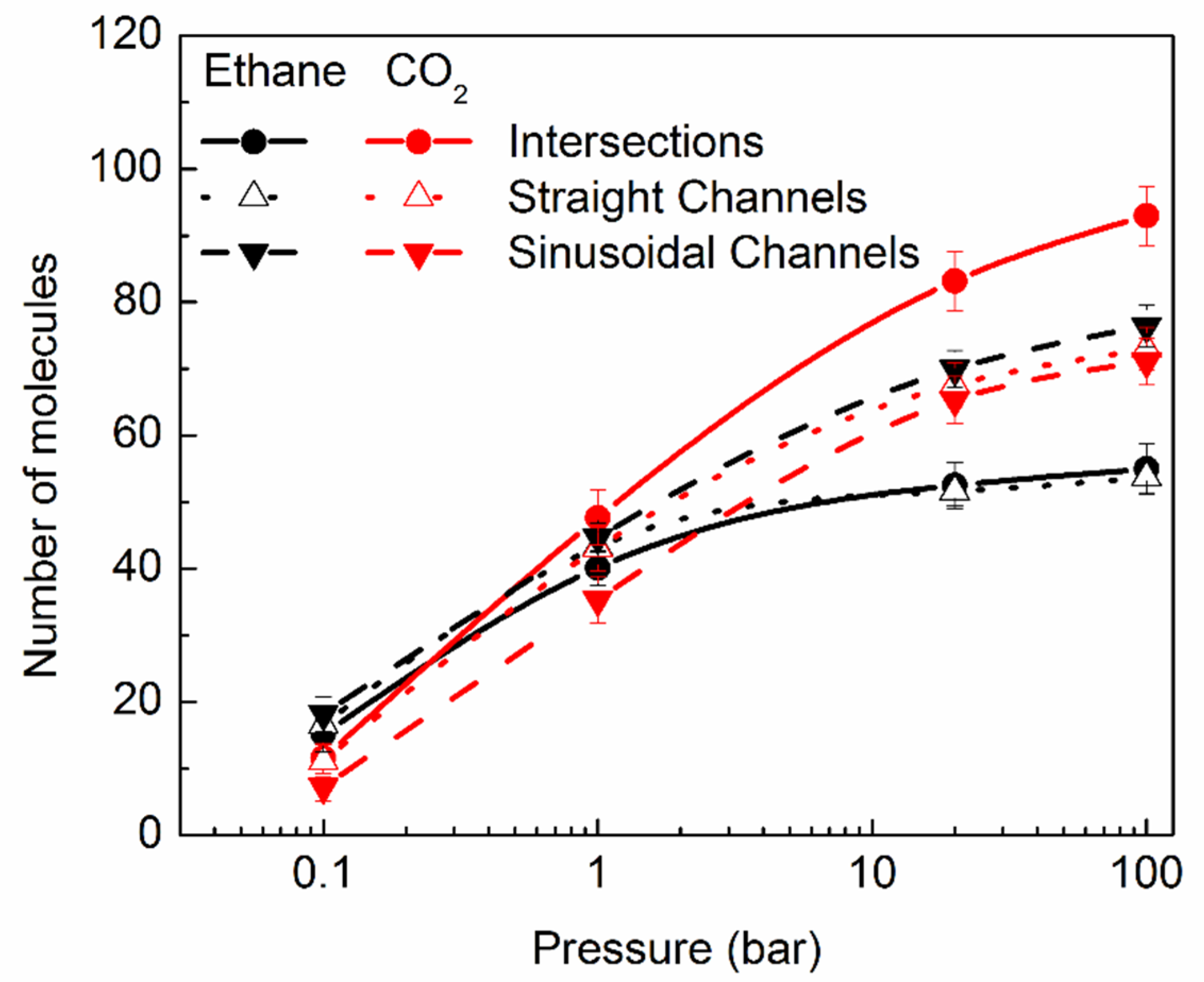
2.2.1. Distribution of the Molecules in Different Pores
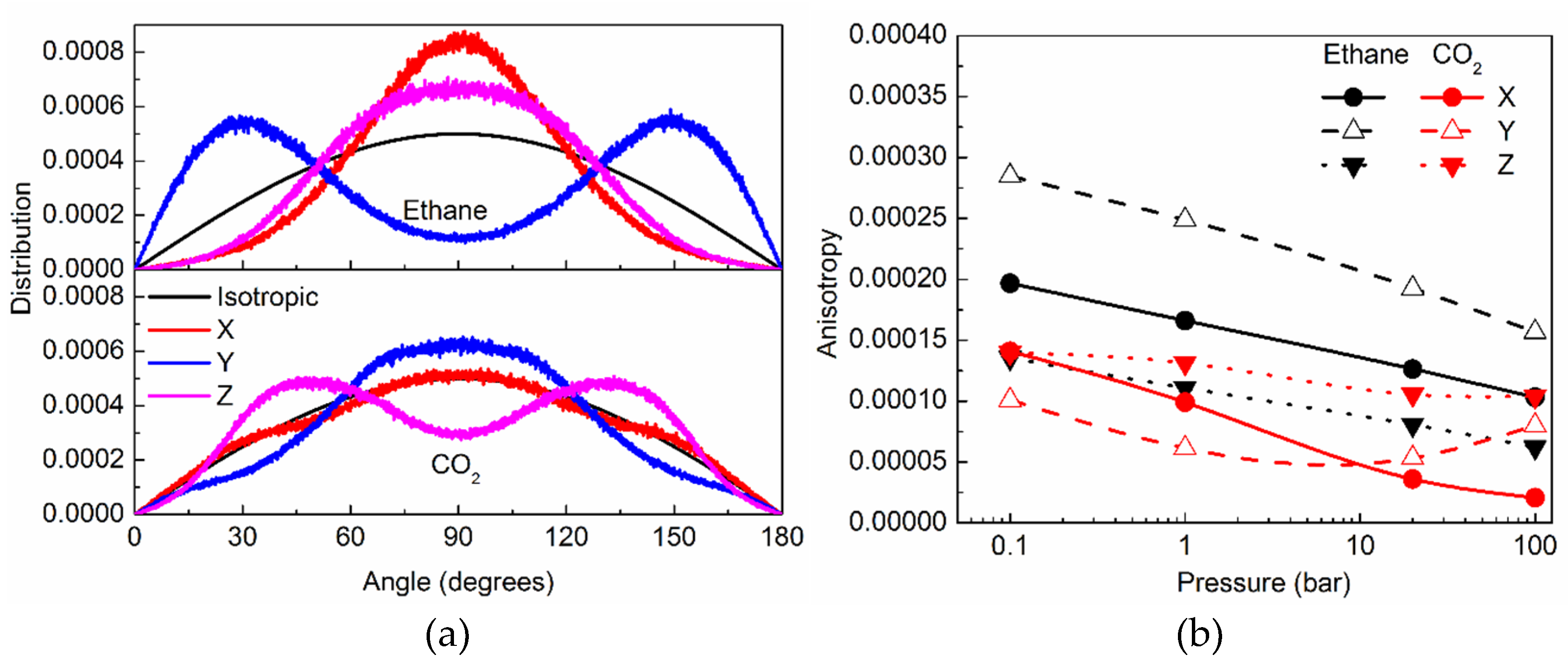
2.2.2. Orientational Distribution of Molecules in Silicalite and Anisotropy
2.3. Dynamics
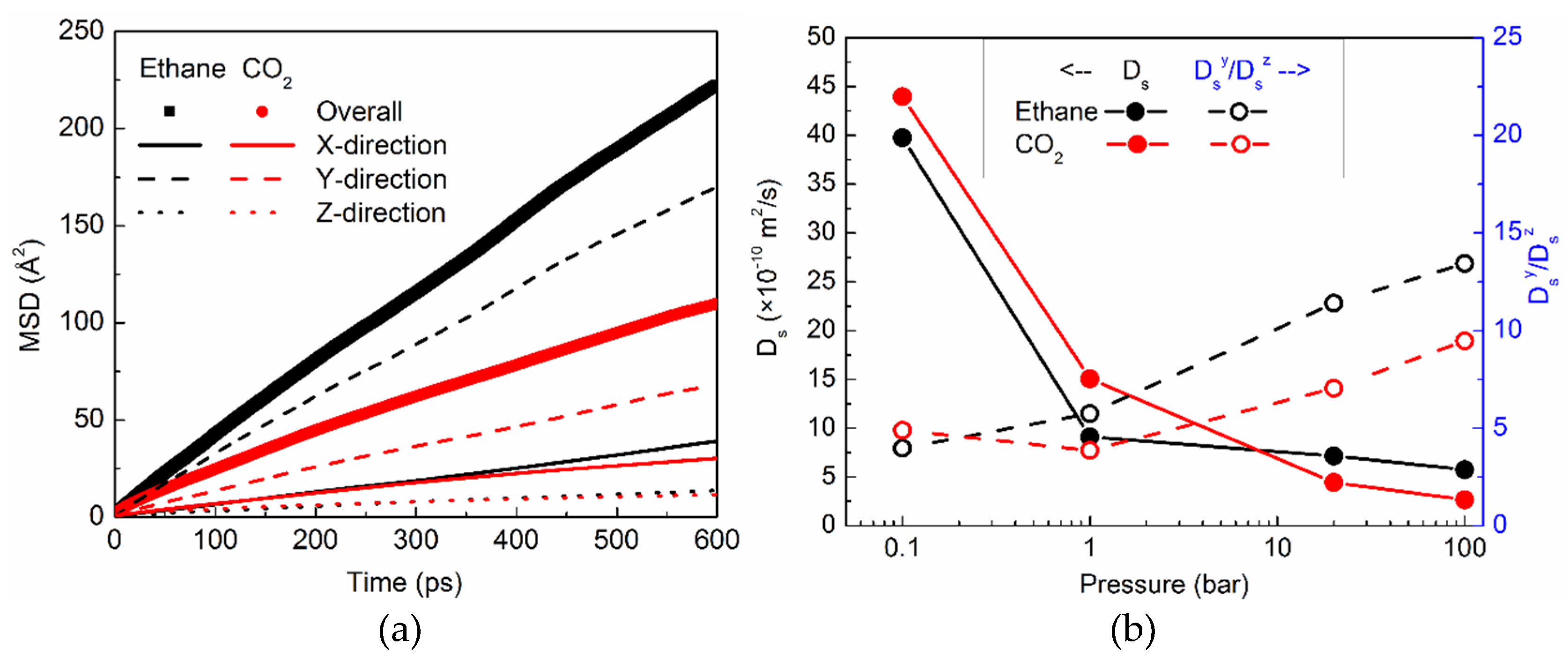
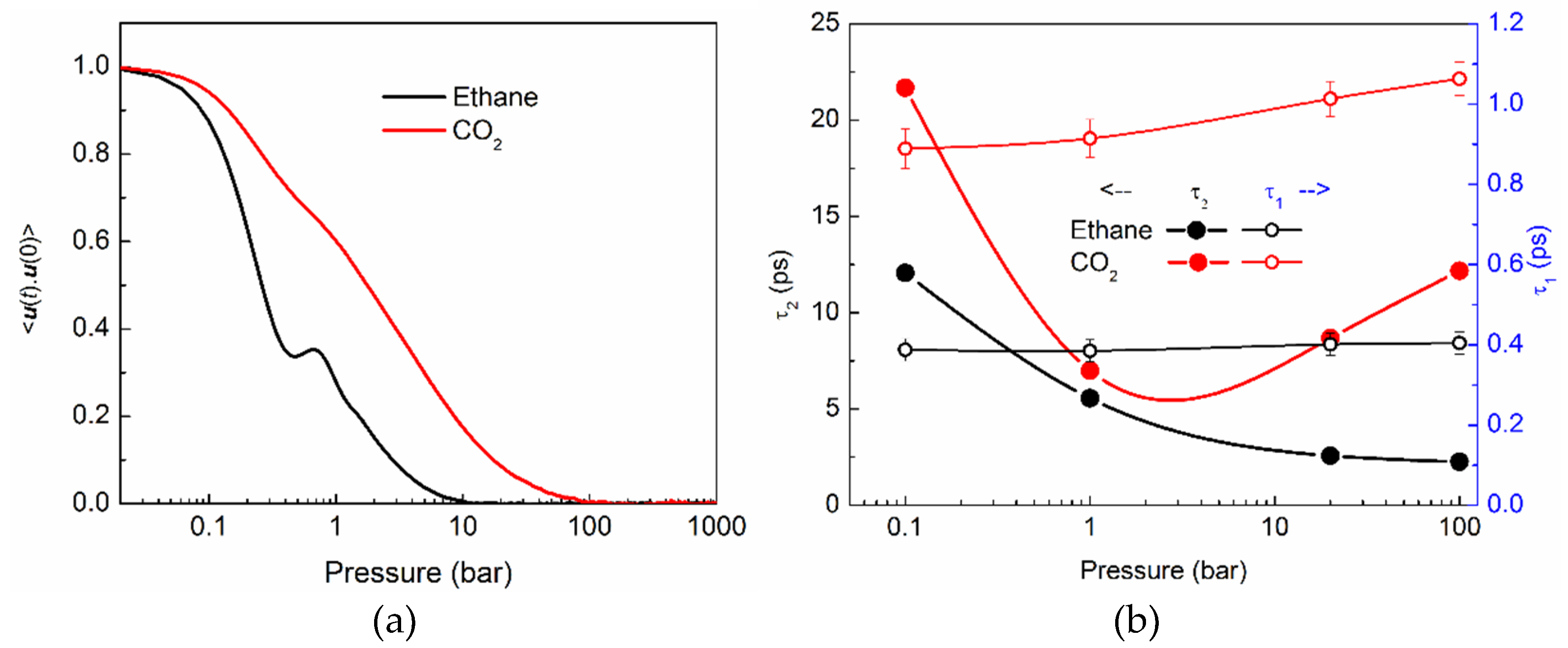
2.3.1. Translational Motion and its Anisotropy
2.3.2. Rotational Motion
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gautam, S.S.; Ok, S.; Cole, D.R. Structure and Dynamics of Confined C-O-H Fluids Relevant to the Subsurface: Application of Magnetic Resonance, Neutron Scattering, and Molecular Dynamics Simulations. Front. Earth Sci. 2017, 5, 1–19. [Google Scholar] [CrossRef]
- Kolesnikov, A.I.; Zanotti, J.-M.; Loong, C.-K.; Thiyagarajan, P.; Moravsky, A.P.; Loutfy, R.O.; Burnham, C.J. Anomalously soft dynamics of Water in a Nanotube: A Revelation of Nanoscale Confinement. Phys. Rev. Lett. 2004, 93, 035503. [Google Scholar] [CrossRef] [PubMed]
- Granick, S. Motions and Relaxations of Confined Liquids. Science 1991, 253, 1374–1379. [Google Scholar] [CrossRef]
- Pitakbunkate, T.; Balbuena, P.B.; Moridis, G.J.; Blasingame, T.A. Effect of Confinement on Pressure/Volume/Temperature Properties of Hydrocarbons in Shale Reservoirs. SPE J. 2016, 21, 621–634. [Google Scholar] [CrossRef]
- Cole, D.R.; Mamontov, E.; Rother, G. Structure and dynamics of fluids in microporous and mesoporous earth and engineered materials. In Neutron Applications in Earth, Energy and Environmental Sciences; Liang, L., Rinaldi, R., Schober, H., Eds.; Springer: Boston, MA, USA, 2009; pp. 547–570. ISBN 978-0-387-09416-8. [Google Scholar]
- Dhiman, I.; Bhowmik, D.; Shrestha, U.R.; Cole, D.R.; Gautam, S. Effect of molecular shape on rotation under severe confinement. Chem. Eng. Sci. 2018, 180, 33–41. [Google Scholar] [CrossRef]
- Dhiman, I.; Shrestha, U.R.; Bhowmik, D.; Cole, D.R.; Gautam, S. Influence of molecular shape on self-diffusion under severe confinement: A molecular dynamics study. Chem. Phys. 2019, 516, 92–102. [Google Scholar] [CrossRef]
- Le, T.; Striolo, A.; Cole, D.R. CO2-C4H10 Mixtures Simulated in Silica Slit Pores: Relation between Structure and Dynamics. J. Phys. Chem. C 2015, 119, 15274–15284. [Google Scholar] [CrossRef]
- Gautam, S.; Liu, T.; Rother, G.; Jalarvo, N.; Mamontov, E.; Welch, S.; Sheets, J.; Droege, M.; Cole, D.R. Dynamics of Propane in Nanoporous Silica Aerogel: A Quasielastic Neutron Scattering Study. J. Phys. Chem. C 2015, 119, 18188–18195. [Google Scholar] [CrossRef]
- Patankar, S.; Gautam, S.; Rother, G.; Podlesnyak, A.; Ehlers, G.; Liu, T.; Cole, D.R.; Tomasko, D.L. Role of Confinement on Adsorption and Dynamics of Ethane and an Ethane-CO2 Mixture in Mesoporous CPG Silica. J. Phys. Chem. C 2016, 120, 4843–4853. [Google Scholar] [CrossRef]
- Choudhary, V.R.; Mayadevi, S. Adsorption of methane, ethane, ethylene, and carbon dioxide on silicalite-I. Zeolites 1996, 17, 501–507. [Google Scholar] [CrossRef]
- Hirotani, A.; Mizukami, K.; Miura, R.; Takaba, H.; Miya, T.; Fahmi, A.; Stirling, A.; Kubo, M.; Miyamoto, A. Grand canonical Monte Carlo simulation of the adsorption of CO2 on silicalite and NaZSM-5. Appl. Surf. Sci. 1997, 120, 81–84. [Google Scholar] [CrossRef]
- Sun, M.S.; Shah, D.B.; Xu, H.H.; Talu, O. Adsorption Equilibria of C1 to C4 Alkanes, CO2, and SF6 on Silicalite. J. Phys. Chem. B 1998, 102, 1466–1473. [Google Scholar] [CrossRef]
- Pascual, P.; Ungerer, P.; Tavitian, B.; Pernot, P.; Boutin, A. Development of a transferable guest-host force field for adsorption of hydrocarbons in zeolites: I. Reinvestigation of alkane adsorption in silicalite by grand canonical Monte Carlo simulation. Phys. Chem. Chem. Phys. 2003, 5, 3684–3693. [Google Scholar] [CrossRef]
- Gautam, S.; Liu, T.; Patankar, S.; Tomasko, D.; Cole, D. Location dependent orientational structure and dynamics of ethane in ZSM5. Chem. Phys. Lett. 2016, 648, 130–136. [Google Scholar] [CrossRef] [Green Version]
- García-Pérez, E.; Parra, J.B.; Ania, C.O.; García-Sánchez, A.; van Baten, J.M.; Krishna, R.; Dubbeldam, D.; Calero, S. A computational study of CO2, N2, and CH4 adsorption in zeolites. Adsorption 2007, 13, 469–476. [Google Scholar] [CrossRef]
- Selassie, D.; Davis, D.; Dahlin, J.; Feise, E.; Haman, G.; Sholl, D.S.; Kohen, D. Atomistic Simulations of CO2 and N2 Diffusion in Silica Zeolites: The Impact of Pore Size and Shape. J. Phys. Chem. C 2008, 112, 16521–16531. [Google Scholar] [CrossRef]
- Razavian, M.; Fatemi, S.; Masoudi-Nejad, M. A Comparative Study of CO2 and CH4 Adsorption on Silicalite-1 Fabricated by Sonication and Conventional Method. Adsorpt. Sci. Technol. 2014, 32, 73–87. [Google Scholar] [CrossRef]
- Shi, X.; Xiao, H.; Liao, X.; Armstrong, M.; Chen, X.; Lackner, K.S. Humidity Effect on Ion Behaviors of Moisture-driven CO2 Sorbents. J. Chem. Phys. 2018, 149, 164708. [Google Scholar] [CrossRef]
- Shi, X.; Xiao, H.; Lackner, K.S.; Chen, X. Capture CO2 from Ambient Air Using Nanoconfined Ion Hydration. Angew. Chem. 2016, 128, 4094–4097. [Google Scholar] [CrossRef]
- Martin, M.G.; Siepmann, J.I. Transferable Potentials for Phase Equilibria. 1. United-Atom Description of n-Alkanes. J. Phys. Chem. B 1998, 102, 2569–2577. [Google Scholar] [CrossRef]
- Aguado, S.; Bergeret, G.; Daniel, C.; Farrusseng, D. Absolute Molecular Sieve Separation of Ethylene/Ethane Mixtures with Silver Zeolite A. J. Am. Chem. Soc. 2012, 134, 14635–14637. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhao, Q.; Xu, H.; Li, L.; Dong, J.; Li, J. Adsorption of CO2, CH4, and N2 on Gas Diameter Grade Ion-exchange Small Pore Zeolites. J. Chem. Eng. Data 2012, 57, 3701–3709. [Google Scholar] [CrossRef]
- Lemmon, E.W.; McLinden, M.O.; Friend, D.G. Thermophysical Properties of Fluid Systems in NIST Chemistry WebBook, NIST Standard Reference Database Number 69; Linstrom, P.J., Mallard, W.G., Eds.; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2017.
- Pini, R. Interpretation of net and excess adsorption isotherms in microporous adsorbents. Microporous Mesoporous Mater. 2014, 187, 40–52. [Google Scholar] [CrossRef]
- First, E.L.; Gounaris, C.E.; Wei, J.; Floudas, C.A. Computational characterization of zeolite porous networks: An automated approach. Phys. Chem. Chem. Phys. 2011, 13, 17339–17358. [Google Scholar] [CrossRef] [PubMed]
- Gautam, S.; Sharma, V.K.; Mitra, S.; Chaplot, S.L.; Mukhopadhyay, R. Rotational dynamics of propylene in ZSM-5 zeolitic frameworks. Chem. Phys. Lett. 2011, 501, 345–350. [Google Scholar] [CrossRef]
- Gautam, S.; Mitra, S.; Chaplot, S.L.; Mukhopadhyay, R. Dynamics of 1,3-butadiene adsorbed in Na-Y zeolite: A molecular dynamics simulation study. Phys. Rev. E 2008, 77, 061201. [Google Scholar] [CrossRef] [PubMed]
- Gautam, S.; Kolesnikov, A.I.; Rother, G.; Dai, S.; Qiao, Z.A.; Cole, D. Effects of Confinement and Pressure on the Vibrational Behavior of Nano-Confined Propane. J. Phys. Chem. A 2018, 122, 6736–6745. [Google Scholar] [CrossRef]
- Mitchell, P.C.H.; Parker, S.F.; Ramirez-Cuesta, A.J.; Tomkinson, J. Vibrational Spectroscopy with Neutrons: With Applications in Chemistry, Biology, Materials Science and Catalysis; World Scientific: Singapore, 2005; Volume 3, ISBN 9812567836. [Google Scholar]
- Tomkinson, J.; ISIS, Chilton, UK. Unpublished work.
- ISIS INS Database. Available online: https://www.isis.stfc.ac.uk/Pages/INS-database.aspx (accessed on 29 November 2018).
- Gautam, S.; Cole, D. Molecular dynamics simulation study of meso-confined propane in TiO2. Chem. Phys. 2015, 458, 68–76. [Google Scholar] [CrossRef]
- Gautam, S.; Le, T.; Striolo, A.; Cole, D. Molecular dynamics simulations of propane in slit shaped silica nano-pores: Direct comparison with quasielastic neutron scattering experiments. Phys. Chem. Chem. Phys. 2017, 19, 32320–32332. [Google Scholar] [CrossRef]
- Walton, K.S.; Millward, A.R.; Dubbeldam, D.; Frost, H.; Low, J.J.; Yaghi, O.M.; Snurr, R.Q. Understanding Inflections and Steps in Carbon Dioxide Adsorption Isotherms in Metal-Organic Frameworks. J. Am. Chem. Soc. 2008, 130, 406–407. [Google Scholar] [CrossRef]
- Vlugt, T.J.H.; Zhu, W.; Kapteijn, F.; Moulijn, J.A.; Smit, B.; Krishna, R. Adsorption of Linear and Branched Alkanes in the Zeolite Silicalite-1. J. Am. Chem. Soc. 1998, 120, 5599–5600. [Google Scholar] [CrossRef] [Green Version]
- Purton, J.A.; Crabtree, J.C.; Parker, S.C. DL_Monte: A general purpose program for parallel Monte Carlo simulation. Mol. Simul. 2013, 39, 1240–1252. [Google Scholar] [CrossRef]
- Todorov, I.T.; Smith, W.; Trachenko, K.; Dove, M.T. DL_POLY_3: New dimensions in molecular dynamics simulations via massive parallelism. J. Mater. Chem. 2006, 16, 1911–1918. [Google Scholar] [CrossRef]
- Gowers, R.J.; Farmahini, A.H.; Friedrich, D.; Sarkisov, L. Automated analysis and benchmarking of GCMC simulation programs in application to gas adsorption. Mol. Simul. 2018, 44, 309–321. [Google Scholar] [CrossRef]
- Van Koningsveld, H.; van Bekkum, H.; Jansen, J.C. On the Location and Disorder of the Tetrapropylammonium (TPA) Ion in zeolite ZSM-5 with Improved Framework Accuracy. Acta Crystallogr. Sect. B 1987, B43, 127–132. [Google Scholar] [CrossRef]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gautam, S.; Liu, T.; Cole, D. Sorption, Structure and Dynamics of CO2 and Ethane in Silicalite at High Pressure: A Combined Monte Carlo and Molecular Dynamics Simulation Study. Molecules 2019, 24, 99. https://doi.org/10.3390/molecules24010099
Gautam S, Liu T, Cole D. Sorption, Structure and Dynamics of CO2 and Ethane in Silicalite at High Pressure: A Combined Monte Carlo and Molecular Dynamics Simulation Study. Molecules. 2019; 24(1):99. https://doi.org/10.3390/molecules24010099
Chicago/Turabian StyleGautam, Siddharth, Tingting Liu, and David Cole. 2019. "Sorption, Structure and Dynamics of CO2 and Ethane in Silicalite at High Pressure: A Combined Monte Carlo and Molecular Dynamics Simulation Study" Molecules 24, no. 1: 99. https://doi.org/10.3390/molecules24010099