
Aza-Amino Acids Disrupt β-Sheet Secondary Structures


Department of Chemistry, North Carolina State University, Raleigh, NC 27695-8204, USA
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Molecules 2019, 24(10), 1919; https://doi.org/10.3390/molecules24101919
Submission received: 1 May 2019
/
Revised: 15 May 2019
/
Accepted: 17 May 2019
/
Published: 18 May 2019
(This article belongs to the Special Issue Advances in Peptide and Peptidomimetic Design Inspiring Basic Science and Drug Discovery: A Themed Issue Honoring Professor Victor J. Hruby on the Occasion of His 80th Birthday)
Abstract
:Cα to N substitution in aza-amino acids imposes local conformational constraints, changes in hydrogen bonding properties, and leads to adaptive chirality at the nitrogen atom. These properties can be exploited in mimicry and stabilization of peptide secondary structures and self-assembly. Here, the effect of a single aza-amino acid incorporation located in the upper β-strand at a hydrogen-bonded (HB) site of a β-hairpin model peptide (H-Arg-Tyr-Val-Glu-Val-d-Pro-Gly-Orn-Lys-Ile-Leu-Gln-NH2) is reported. Specifically, analogs in which valine3 was substituted for aza-valine3 or aza-glycine3 were synthesized, and their β-hairpin stabilities were examined using Nuclear Magnetic Resonance (NMR) spectroscopy. The azapeptide analogs were found to destabilize β-hairpin formation compared to the parent peptide. The aza-valine3 residue was more disruptive of β-hairpin geometry than its aza-glycine3 counterpart.
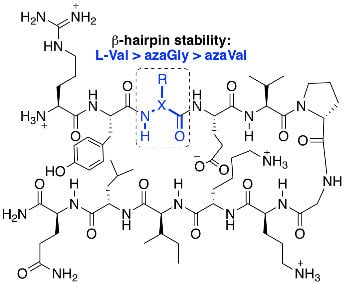
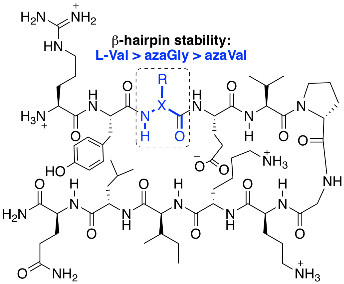
1. Introduction
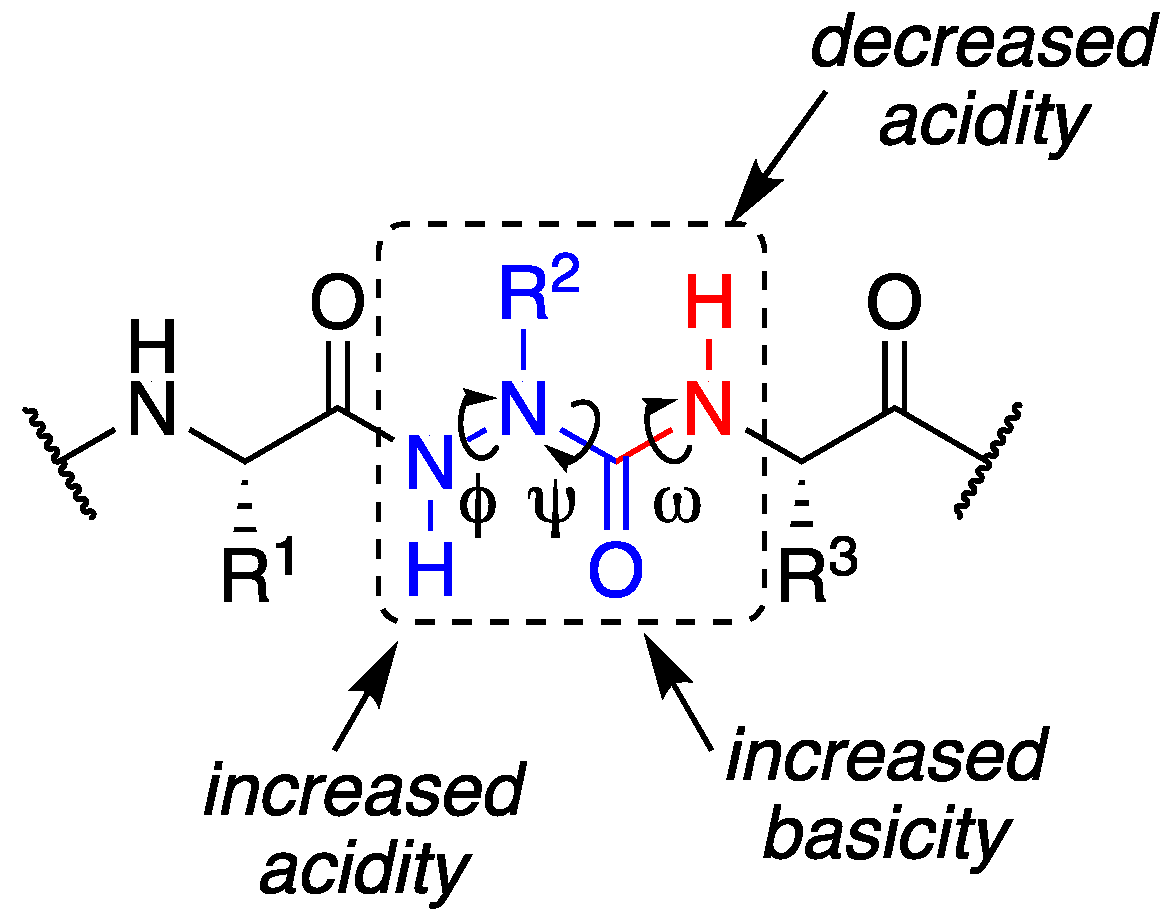
β-sheets are common protein secondary structures that are often involved in protein-protein interactions and protein aggregation [1,2]. To understand factors governing folding, stability, and molecular recognition events involving β-sheets, water soluble β-hairpin model systems that are partially folded have been developed, consisting of two antiparallel β-strands connected by a reverse turn unit [3]. In the pursuit of protein-protein interaction (PPI) inhibitors, β-strand and β-sheet peptidomimetics have been explored using a variety of unnatural scaffolds [4]. When mimicking β-strands in particular, the hydrogen bonding pattern of a natural peptide is ideally replicated by the artificial templates to maintain cross-strand interactions, yet surface exposed hydrogen bonding sites should be minimized to prevent uncontrolled aggregation, creating a so-called “blocking strand” [5,6,7]. Here, we describe the effect of a single Cα substitution for a nitrogen at the Val3 position of a model β-hairpin peptide. The aza-amino acid residue was envisioned to provide two distinct faces with divergent hydrogen bonding capabilities, while retaining side chain position and chemistry (Figure 1).
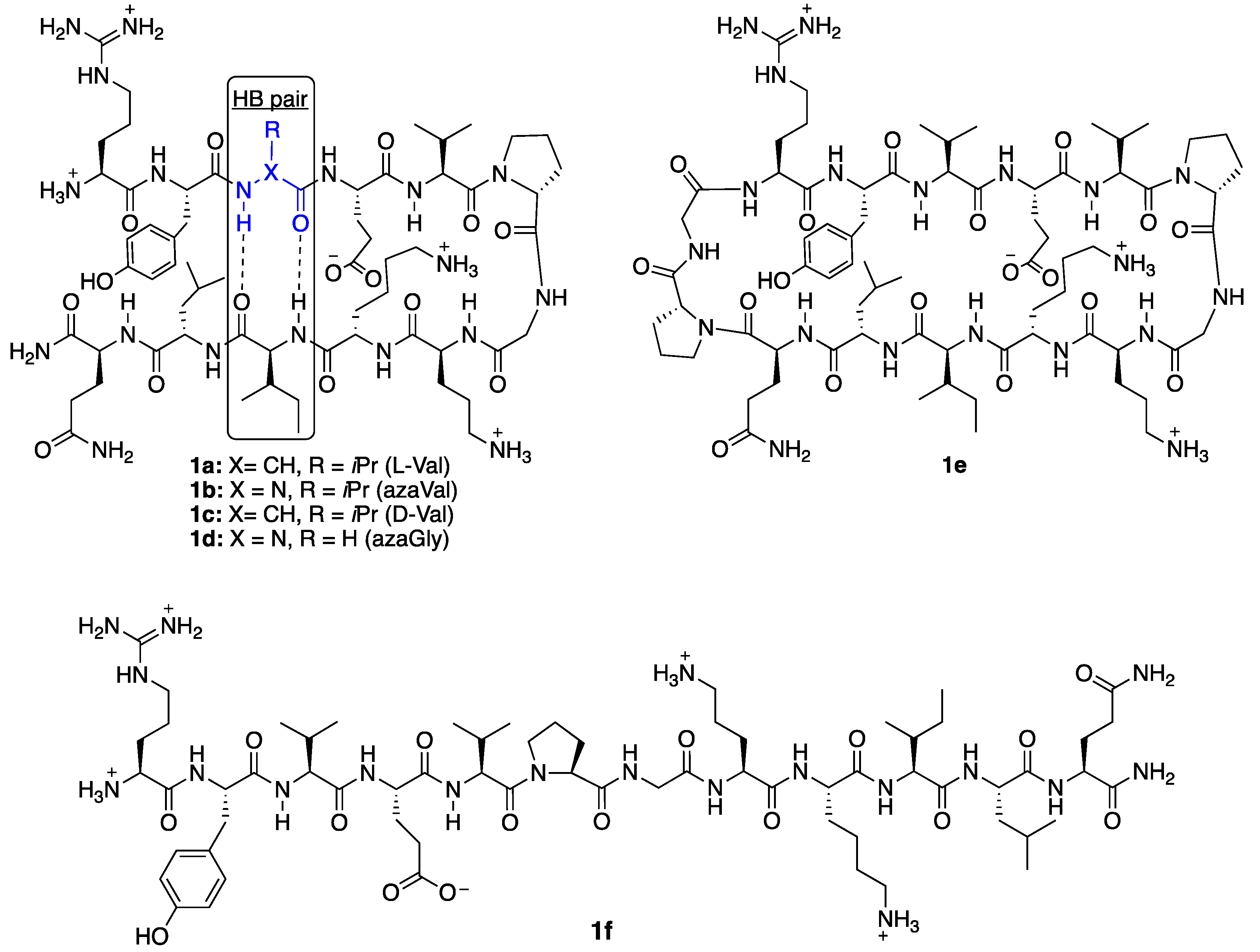
Aza-amino acids [8] have been used to promote β-turn secondary structures and hyperstable collagen peptide mimics [9,10] via a combination of hydrogen bonding and backbone dihedral angle modulation. Their effect on β-hairpin stability has yet to be quantified within β-strand regions. In aza-amino acids, perpendicular lone pair orientation between adjacent nitrogen and urea planarity typically reinforce backbone dihedral angles that fall within the range of β-turns and polyproline type II secondary structures [8]. Although this may prevent adoption of extended β-strand geometry [11], computational studies have set the energy barrier for rotation from the twisted conformer (φ = 90°) to the extended conformer (φ = 180°) to be < 1 kcal/mol within 1,2-diformylhydrazine in the Z,Z configuration as an azaGly model [12]. Two favorable intramolecular hydrogen bonds are proposed to stabilize the 180° orientation. Although addition of side chain chemistry in aza-amino acids removes an intramolecular hydrogen bond with a neighboring residue, cross-strand hydrogen bonding and side chain; side chain interactions in a β-hairpin may provide added stability to the extended conformer. Substitution of Val3 for aza-valine3 was thus examined in a well-studied β-hairpin model system for which Nuclear Magnetic Resonance (NMR)-based quantification of β-hairpin folding have been established, first reported by Gellman et al. [13,14]: H-Arg-Tyr-Val-Glu-Val-d-Pro-Gly-Orn-Lys-Ile-Leu-Gln-NH2 (1a). Valine3 is situated in the middle of the upper β-strand at a hydrogen bonded (HB) site (Figure 2, in blue), equally removed from the d-Pro-Gly turn region and the N-terminus. In addition to azaVal3 analog 1b, d-Val3 control 1c, azaGly3 analog 1d, and previously described [14] positive control cyclic peptide 1e, and negative control l-Pro6-Gly7 peptide 1f, all were synthesized and studied (Figure 2). Since the first incorporation of azaVal to give an azapeptide angiotensin analog [15], considerable interest has been generated in the synthesis and conformational properties of this aza-amino acid.
2. Results and Discussion
2.1. Synthesis
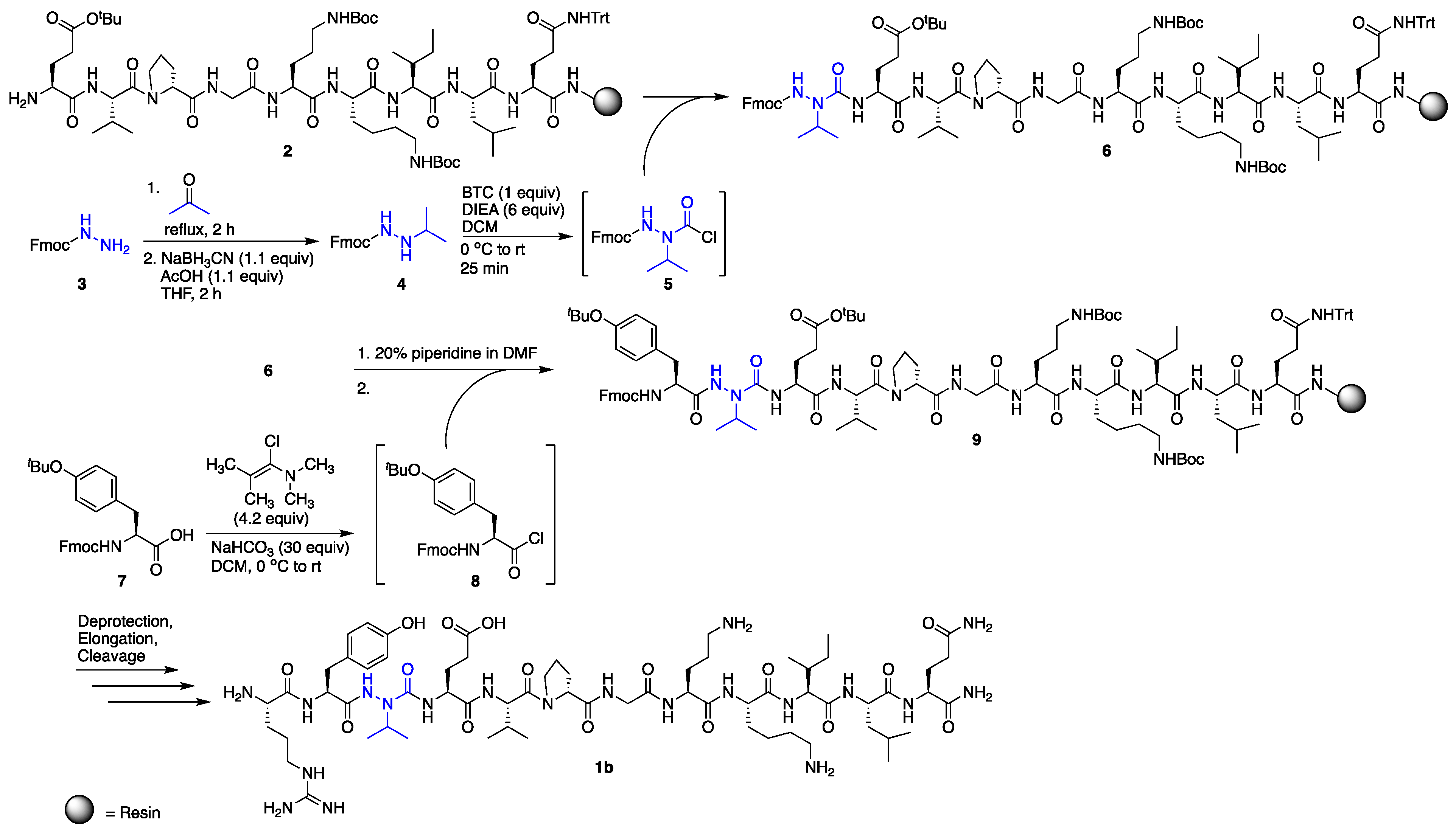
Solid-phase peptide synthesis was performed on Rink amide resin, using standard Fmoc procedures for all l- and d-amino acid coupling and deprotection cycles [16]. Incorporation of azaVal3 for the synthesis of 1b was accomplished via activation of N’-isopropyl-fluorenylmethyl carbazate [17] using bis-(trichloromethyl)carbonate (BTC) as the activating agent (Scheme 1) and as a safer alternative to phosgene. Although submonomer azapeptide synthesis protocols [18] were considered for the installation of aza-valine to circumvent the solution-phase synthesis and activation of hydrazine building blocks, N-alkylation of resin-bound semicarbazone intermediates with sterically hindered secondary alkyl halides has typically resulted in lower conversions [19]. In our hands, incorporation of aza-valine via N’-alkyl fluoren-9-ylmethyl carbazate activation with triphosgene afforded azapeptide 6 as the major product in 77% crude purity after a single overnight coupling reaction. In contrast, in accordance with previous literature, coupling to the aza-amino acid to give 9 was challenging due to the decreased nucleophilicity of semicarbazides [8] and steric hindrance from the branched aza-valine side chain. In the past, coupling to aza-valine and other aza-amino acids with branched side chains has been accomplished using N,N’-dicyclohexylcarbodiimide (DCC) [15], diisoproylcarbodiimide (DIC) [19,20], and BTC [21]. Here, the coupling of Fmoc-Tyr(tBu) to resin-bound azaV-EVpGOKILQ using BTC and 2,4,6-collidine in tetrahydrofuran (THF) provided 9 in only 33% conversion by liquid chromatography-mass spectrometry (LC-MS) analysis, with 21% uncoupled semicarbazide and 28% isocyanate or hydantoin byproduct [22] (Table 1, entry 1). In the context of β-hairpin synthesis, the already difficult coupling to aza-Val may be rendered even more challenging by folding or aggregation on-resin [23]. Attempts employing a LiCl salt additive to the BTC coupling solution to disrupt possible on-resin aggregation [24] were less successful than efforts to optimize the coupling reagent (Table 1, entries 2–5).
Recent successes using benzotriazol-1-yloxytris(pyrrolidinophosphonium-hexafluorophosphate (PyBOP)/N,N-diisopropylethylamine (DIEA) [25] and (1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylaminomorpholino-carbenium hexafluorophosphate (COMU)/DIEA [26,27] in difficult couplings to aza-amino acids led us to try these activating agents; however, 9 was obtained in low conversions in both cases (6–13%, Table 1, entries 2 and 3). We next investigated N,N,N′,N′-tetramethylchloroformamidinium hexafluorophosphate (TCFH)/N-methylimidazole (NMI) (Table 1, entry 4), which was recently developed [28] to couple sterically hindered carboxylic acids with non-nucleophilic aniline derivatives and sterically hindered amino acids, including valine. Gratifyingly, these conditions provided 9 in high conversion (89%) with little byproduct formation and no detectable starting material (Table 1, entry 4), although appearance of two peaks with identical masses by LC-MS suggested possible epimerization. Alternatively, the use of 1-chloro-N,N,2-trimethyl-1-propenylamine (Ghosez’s reagent) [29] (Table 1, entry 5) provided the desired elongated azapeptide 9 as a single peak by LC-MS analysis in 67% conversion, with no uncoupled semicarbazide detected. Using this procedure for coupling onto azaVal, azapeptide 1b was obtained in 53% crude purity, following deprotection, elongation, and cleavage from the resin. Purification of the crude azapeptide by reverse phase – high performance liquid chromatography (RP-HPLC) afforded 1b in 8% yield (see Supporting Information).
Activation of N-Fmoc-hydrazine with phosgene equivalents was previously shown to lead to the formation of a cyclic oxadiazolone [30]. Although ring-opening of this heterocycle to afford the desired Fmoc-protected azaGly-terminated peptides may be possible [30], activation and coupling of aza-glycine using these conditions has been reported to be problematic in certain cases [31]. Here, aza-glycine installation was accomplished by activating fluoren-9-methyl carbazate with N,N′-disuccinimidyl carbonate (DSC) as the activating agent instead of BTC, which afforded the desired azapeptide in higher yields [32]. Fmoc-Tyr(tBu)-OH activation and coupling to the resin-bound aza-glycine-terminated peptide was accomplished using BTC/2,4,6-collidine in THF and required no further optimization.
2.2. Conformational Analysis
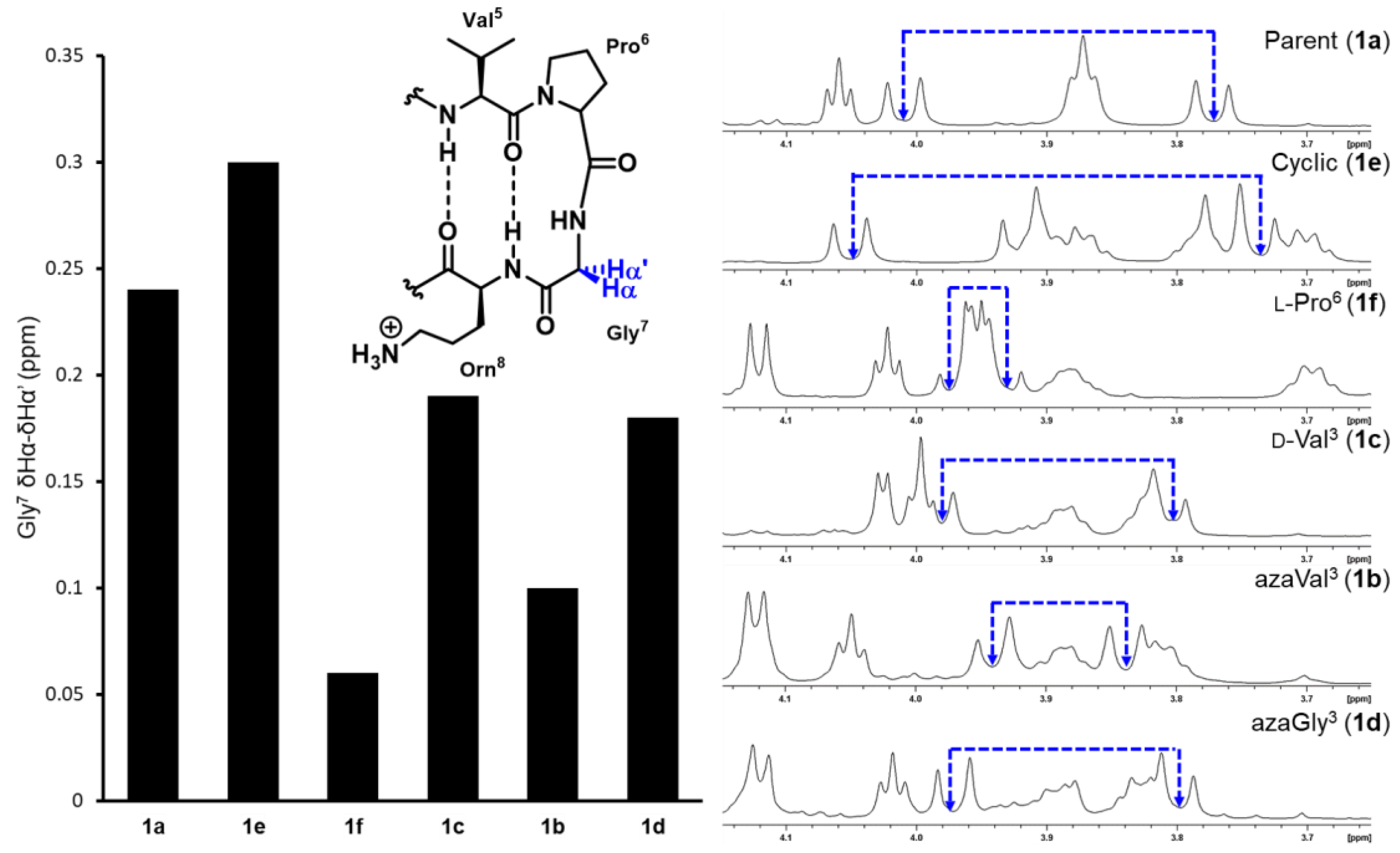
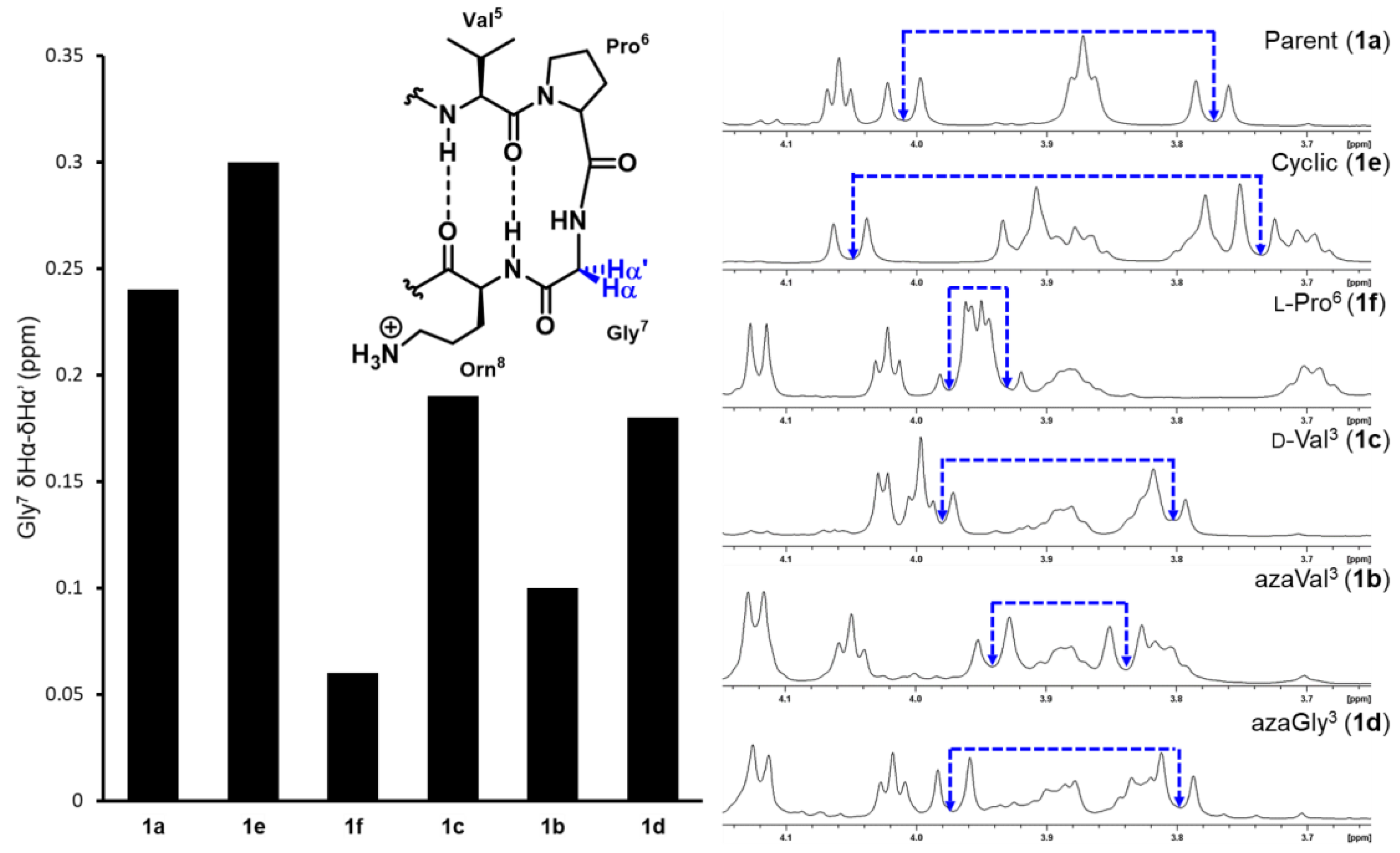
β-Hairpin folding was first assessed by measuring the extent of diastereotopic Gly7 Hα splitting in the turn region for each analog and comparing it to the fully folded cyclic control (Equation (1)), as previously described [33]. Using this method, the relative stability of each β-hairpin analog can be quickly compared as the glycine signals lie in a distinct region of the spectra (Figure 3, Table 2).
Fraction folded = (∆δGly Obs)/(∆δGly 100)
Replacing valine3 for an aza-valine residue in analog 1b caused a significant decrease in β-hairpin stability, reflected by the much smaller Gly7 δHα–δHα’ value (Figure 3). Potential cross-strand hydrogen bonding and side chain - side chain interactions between azaVal3 and Ile10 appear to be insufficient to stabilize the extended conformation in aza-valine3, which likely adopts a twisted conformation (φ = 90°) that disrupts β-hairpin formation. Because aza-amino acids may exhibit adaptive chirality at Nα, the aza-valine residue could be achiral or exhibit either l or d-like chirality [34], which could in turn affect β-hairpin stability. We synthesized d-Val3 analog 1c as a control and found its Gly7 δHα–δHα’ value to be larger than that of aza-Val3 analog 1b, yet smaller than the parent peptide 1a. While changing the stereochemistry of the valine3 residue from l to d decreased β-hairpin stability overall, this substitution was found to be less disruptive than the backbone Cα to Nα substitution in the aza-valine residue. As such, the destabilizing effect of the aza-valine residue cannot be solely attributed to the loss of a defined stereocenter.
Given that the extended conformation in aza-glycine vs. aza-valine residues may be more favorable due to additional intramolecular hydrogen-bonding interactions (vide supra), the synthesis and conformational analysis of azaGly3 analog 1d was also pursued. Despite the absence of an isopropyl side chain, the aza-glycine substitution was found to be less detrimental to β-hairpin stability compared to the aza-Val3 analog 1b; however, β-hairpin folding was still decreased relative to the parent peptide 1a. The disruptive effects of the azaGly3 substitution are similar those observed in the d-Val3 analog using Gly7 diastereotopic Hα splitting measurements (Figure 3) [35].
It should be noted that glycine splitting measurements have been found to overestimate β-hairpin folding in sequences that use d-Pro-Gly turn units [36]. Downfield chemical shifts in CHα protons relative to unfolded peptide for residues away from the turn region, especially those situated at HB sites, can be used to quantify the degree of β-hairpin folding more reliably according to Equation (2), where δ0 and δ100 are the chemical shift values for the unfolded analog 1f and the cyclic control 1e, respectively.
Fraction folded = (δObs − δ0)/(δ100 − δ0)
Specifically, the downfield chemical shifts of Val3, Ile10, and Orn8 have been found to be more reliable and are typically used to measure folded fractions for this β-hairpin model peptide and its derivatives [14]. Here, because the Cα of Val3 is substituted by a nitrogen in our azapeptide analogs, we omitted this residue from our studies (Table 2). The CHα chemical shift values for Ile10 are particularly informative because this residue sits in the middle of the lower strand and is positioned to form hydrogen bonds with the variable residue in the third position; Orn8 is situated closer to the turn region. Based on the chemical shift values for Ile10, aza-Val3 peptide 1b is completely unfolded, in contrast to the partially folded structure predicted based on glycine7 Hα splitting measurements (Figure 3, Table 2). Considering that diastereotopic splitting of Gly7 Hα overestimate folding in β-hairpin model peptides that have d-Pro-Gly turn units, incorporation of azaVal3 is likely completely disrupting the β-hairpin fold. In contrast, the azaGly3 analog 1d which lacks side chain functionality was ~50% folded based on the Ile10 downfield Hα chemical shifts, and exhibited a greater percentage of folding relative to both the aza-valine (1a) and d-valine (1c) analogs. The additional potential for azaGly to intra- and/or inter-strand hydrogen-bond may stabilize the extended conformer. The % folding values calculated from Orn8 chemical shifts reinforced the conclusion that the aza-Valine analog 1b was the least folded of all analogs, yet suggested similar degrees of folding for the d-Val (1c) and the azaGly (1d) analogs.
3. Materials and Methods
3.1. General
Polystyrene Rink Amide resin (0.78 mmol/g) and Fmoc-glycine-2-chlorotrityl resin (200–400 mesh, 0.493 mmol/g) were purchased from Protein Technology Inc™ and Chem-Impex Int’l, Inc, respectively, and the manufacturer’s reported loading of the resin was used in the calculation of the yields of the final products. Solid phase peptide synthesis was performed using an automated Biotage Syro WaveTM peptide synthesizer in 10 mL parallel reactors with polytetrafluoroethylene (PTFE) frits. Incorporation of aza-amino acids and further elongations were performed manually in disposable filter columns with 20 µM polyethylene (PE) frit filters and caps purchased from Applied Separations (cat # 2413 and 2416 for 3 mL and 6 mL filter columns, respectively) with gentle agitation on a Thermo Fisher vortex mixer equipped with a microplate tray. Solution draining and washing of the resin was accomplished by connecting the filter columns to a water aspirator vacuum via a waste trap. Analytical LC-MS analyses were performed using an Agilent Technologies 1260 Infinity II series LC-MS Single Quad instrument with ESI ion-source and positive mode ionization, equipped with a 5 µM, 150 × 4.6 mm C18 Vydac column purchased from Mac-Mod Analytical, Inc (cat # 218TP5415). A flow rate of 0.5 mL/min and 5–95%, 20–80%, or 12–60% gradients of CH3CN [0.1% trifluoroacetic acid (TFA)] in water (0.1% TFA) over 12 min (total run time = 22 min) were used for all LC-MS analyses. Peptides were purified on a preparative HPLC (Agilent 218 purification system) using a preparative column (10–20 µM, 250 mm × 22 mm, C18 Vydac column, cat # 218TP101522) at a flow rate of 10 mL/min with gradients of CH3CN [0.1% trifluoroacetic acid (TFA)] in water (0.1% TFA) over 30 min (total run time = 60 min).
3.2. Reagents
Amino acids, N,N′-diisopropylethylamine (DIEA), N,N′-Disuccinimidyl carbonate (DSC), Fmoc chloride, (1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate (COMU), chloro-N,N,N′,N′-tetramethylformamidinium hexafluorophospate (TCFH) and triphosgene (BTC) were purchased from Chem Impex Int’l, Inc. Reagents such as piperidine, lithium chloride, sodium cyanoborohydride (NaBH3CN), triisopropylsilane (TIPS), sodium acetate-d3, acetic acid-d4, and D2O were purchased from Sigma Aldrich. Reagents such as hydrazine monohydrate and 2,4,6-collidine were purchased from Alfa Aesar. Trifluoroacetic acid, glacial acetic acid, sodium bicarbonate, and solvents were purchased from Fisher. Reagents including HBTU and PyBOP were purchased from Oakwood Chemical. 1-Methylimidazole (NMI) and 1-chloro-N,N,2-trimethylpropenylamine (Ghosez’s Reagent) were purchased from Acros Organics.
3.3. 9-Fluorenylmethoxycarbonyl (Fmoc)-Based Solid Phase Peptide Synthesis (SPPS): Fmoc Deprotection and HBTU Couplings
Peptide syntheses were performed under standard manual conditions on an automated shaker or using a Biotage Syro WaveTM peptide synthesizer using Polystyrene Rink Amide resin (0.78 mmol/g). Couplings of amino acids (3 equiv) were performed in DMF using HBTU (3 equiv) as coupling reagent and DIEA (6 equiv) as base. Fmoc deprotections were performed initially by treating the resin with 20% piperidine in DMF (v/v) for 5 min, followed by treatment with a fresh solution of 20% piperidine in DMF (v/v) for 15 min. Resin was washed after each coupling with DMF (3 × 1 mL) and deprotection reaction with DMF, MeOH, and CH2Cl2 (2 × 1 mL). Prior to cleavage from the resin or storage, resin was washed with CH2Cl2 (3 × 1 mL).
3.4. Test Cleavages of Resin-Bound Peptides
A small amount of resin (1–5 mg) was washed with CH2Cl2 to remove traces of DMF, drained, and treated with a freshly made solution of TFA/H2O/TIPS (90:5:5, v/v/v, 0.5 mL) for 2 h at room temperature. The cleavage solution was collected by filtering the resin through a disposable, PE fritted cartridge. The filtrate was evaporated to dryness and the crude peptide was precipitated twice with cold ether (4 mL) followed by decanting. The pellet (crude peptide sample) was dissolved in 1:1 MeCN/H2O v/v (1 mg/mL) and subjected to LC-MS analysis.
3.5. N′-Isopropyl-fluoren-9-ylmethyl Carbazate (4)
Synthesized according to literature procedures [12,16]. Briefly, a solution of Fmoc-Cl (2.00 g, 7.73 mmol) in 120 mL of CH3CN was added dropwise over 2 h at 0 °C to a solution of excess hydrazine hydrate (3.8 mL, 78.0 mmol, 10 equiv) in 26 mL of CH3CN/H2O (1:1, v/v). The solution was warmed to room temperature and left stirring for 12 h, prior to being concentrated in vacuo and filtered. The resulting solid was washed with water, followed by hexanes, to give 1.92 g of white solid in 98% yield. The resulting 9H-fluoren-9-ylmethyl hydrazinecarboxylate 3 (1.92 g, 7.55 mmol) was suspended in acetone (50 mL) and heated at reflux for 2 h. Acetone was evaporated in vacuo and the hydrazone intermediate was dissolved in THF (50 mL), treated with acetic acid (0.48 mL, 8.39 mmol, 1.1 equiv) and NaBH3CN (521 mg, 8.31 mmol, 1.1 equiv), and stirred for 2 h. The volatiles were removed and the crude was dissolved in EtOAc (100 mL), washed with 1 M aqueous KHSO4 (4 × 50 mL) and brine (2 × 50 mL), dried over Na2SO4, and concentrated to give a white solid. The obtained product was dissolved in EtOH and heated for 1 h, followed by evaporation of EtOH to yield N’-isopropyl-fluoren-9-ylmethyl carbazate 4 as a white solid (1.32 g, 59% yield) after column chromatography using a 20–100% gradient of EtOAc in hexanes. NMR (CDCl3) spectra matched literature values [12].
3.6. Incorporation of Aza-Valine on the Solid Phase
N’-isopropyl-fluoren-9-ylmethyl carbazate 4 (138 mg, 0.466 mmol, 3 equiv) was dissolved in dry CH2Cl2 (1 mL) and cooled to 0 °C. A solution of bis(trichloromethyl)carbonate (BTC, 46.4 mg, 0.156 mmol, 1 equiv) dissolved in dry CH2Cl2 (1 mL) and cooled to 0 °C was added dropwise to the N’-isopropyl-fluoren-9-ylmethyl carbazate suspension. The reaction mixture was left stirring at 0 °C for 5 min, allowed to warm to room temperature, and stirred for another 25 min. DIEA (0.163 mL, 0.930 mmol, 6.0 equiv) was added to the solution and the solution was stirred for 5 min prior to being transferred to a SPPS cartridge containing resin-bound peptide 2 (200 mg, 0.156 mmol) swollen in CH2Cl2. The SPPS cartridge was left shaking for 12 h, after which the solution was drained, and the resin was washed with CH2Cl2 (3 × 2 mL).
3.7. Coupling to AzaV-EVpGOKILQ
3.7.1. BTC Coupling
Fmoc-Tyr(tBu)-OH (53.8 mg, 0.117 mmol, 3.0 equiv) was dissolved in dry THF (0.3 mL), cooled to 0 °C, and treated dropwise with a cold solution of BTC (11.5 mg, 0.039 mmol, 1.0 equiv) in dry THF (0.3 mL). The reaction mixture was left stirring at 0 °C for 15 min, before adding 2,4,6-collidine (0.052 mL, 0.39 mmol, 10.0 equiv). The solution was stirred at 0 °C for 2 min, allowed to warm to room temperature, and stirred another 5 min. The activated amino acid was then transferred to a SPPS cartridge containing the resin-bound azapeptide (50 mg, 0.039 mmol), previously swollen in dry THF. The resin was left shaking at room temperature overnight, then washed with DMF (2 × 0.5 mL), MeOH (2 × 0.5 mL), and THF (2 × 0.5 mL).
3.7.2. PyBOP Coupling
Fmoc-Tyr(tBu)-OH (53.8 mg, 0.117 mmol, 3.0 equiv) and PyBOP (60.9 mg, 0.117 mmol, 3.0 equiv) were each dissolved in DMF (0.3 mL) and cooled to 0 °C separately. The PyBOP solution was then added dropwise to the solution of Fmoc-Tyr(tBu)-OH and the reaction mixture was stirred at 0 °C for 5 min. DIEA (0.0408 mL, 0.234 mmol, 6.0 equiv) was added to the solution and the reaction was allowed to warm to room temperature before being transferred to a SPPS cartridge containing the resin-bound azapeptide (50 mg, 0.039 mmol), previously swollen in DMF. The resin was shaken at room temperature overnight, then washed with DMF (2 × 0.5 mL), MeOH (2 × 0.5 mL), and CH2Cl2 (2 × 0.5 mL).
3.7.3. COMU Coupling
Fmoc-Tyr(tBu)-OH (89.6 mg, 0.195 mmol, 5.0 equiv) and COMU (83.5 mg, 0.195 mmol, 5.0 equiv) were each dissolved in 0.3 mL of DMF and cooled to 0 °C separately. The COMU solution was added dropwise to the solution of Fmoc-Tyr(tBu)-OH and the reaction mixture was stirred at 0 °C for 5 min. DIEA (0.0504 mL, 0.39 mmol, 10.0 equiv) was added and the reaction was warmed to room temperature before being transferred to a SPPS cartridge containing the resin-bound azapeptide (50 mg, 0.039 mmol), previously swollen in DMF. The resin was shaken at room temperature overnight, then washed with DMF (2 × 0.5 mL), MeOH (2 × 0.5 mL), and CH2Cl2 (2 × 0.5 mL).
3.7.4. NMI/TCFH Coupling
Fmoc-Tyr(tBu)-OH (53.8 mg, 0.117 mmol, 3.0 equiv) was dissolved in dry CH2Cl2 (0.3 mL), to which N-methyl imidazole (0.028 mL, 0.351 mmol, 9 equiv) was added before cooling the solution to 0 °C. A solution of TCFH (32.8 mg, 0.117 mmol, 3 equiv) in dry CH2Cl2 (0.3 mL), cooled to 0 °C, was added to the Fmoc-Tyr(tBu)-OH and NMI solution in a single portion and the reaction was immediately transferred to a SPPS cartridge containing the resin-bound azapeptide (50 mg, 0.039 mmol), previously swollen in dry CH2Cl2. The resin was shaken at room temperature overnight, then washed with DMF (2 × 0.5 mL), MeOH (2 × 0.5 mL), and CH2Cl2 (2 × 0.5 mL).
3.7.5. Ghosez Coupling
NaHCO3 (98.3 mg, 1.17 mmol, 30 equiv) was added to the resin-bound azapeptide (50 mg, 0.039 mmol) pre-swelled in dry CH2Cl2 (0.2 mL) and shaken for 15 min. During that time, Fmoc-Tyr(tBu)-OH (53.8 mg, 0.117 mmol, 3.0 equiv) was dissolved in dry CH2Cl2 (0.4 mL) and cooled to 0 °C. Ghosez’s reagent (0.0217 mL, 0.164 mmol, 4.2 equiv) was added dropwise to the cooled solution of Fmoc-Tyr(tBu)-OH and stirred for 5 min. The reaction was allowed to warm to room temperature for 5 min, prior to being transferred to the SPPS cartridge containing NaHCO3 in CH2Cl2. The resin was shaken at room temperature overnight. The resin was washed with DMF (2 × 0.5 mL), MeOH (2 × 0.5 mL), and CH2Cl2 (2 × 0.5 mL).
3.8. Incorporation of Aza-Glycine on the Solid Phase
N,N′-Disuccinimidyl carbonate (DSC) (120 mg, 0.466 mmol, 3.0 equiv) was added to a solution of 9H-fluoren-9-ylmethyl hydrazinecarboxylate 3 (123 mg, 0.484 mmol, 3.1 equiv) in DMF and stirred for 5 min. The solution was then transferred to resin-bound peptide 2 (200 mg, 0.156 mmol) swollen in DMF and left shaking for 12 h. The solution was drained and the resin was washed with DMF (3 × 2 mL).
3.9. Coupling to azaG-EVpGOKILQ
Fmoc-Tyr(tBu)-OH (22.2 mg, 0.0484 mmol, 3.1 equiv) was dissolved in dry THF (0.3 mL), cooled to 0 °C, and treated dropwise with a cold solution of BTC (4.63 mg, 0.0156 mmol, 1.0 equiv) in dry THF (0.2 mL). The reaction mixture was left stirring at 0 °C for 15 min before adding 2,4,6-collidine (0.0206 mL, 0.156 mmol, 10.0 equiv). The solution was stirred at 0 °C for 5 min. The reaction was allowed to warm to room temperature prior to transferring the activated amino acid to a SPPS cartridge containing the resin-bound azapeptide (20 mg, 0.0156 mmol) previously swollen with dry THF. The reaction was left shaking at room temperature overnight, then washed with DMF (2 × 0.5 mL), MeOH (2 × 0.5 mL), and THF (2 × 0.5 mL).
3.10. Synthesis of Cyclic Peptide 1e
The linear precursor H2N-Arg(Pbf)-Tyr(tBu)-Val-Glu(OtBu)-Val-pro-Gly-Orn(Boc)-Lys(Boc)-Ile-Leu-Gln(Trt)-pro-Gly-OH was synthesized on Fmoc-glycine-2-chlorotrityl resin (200–400 mesh, 0.493 mmol/g loading) according to standard SPPS protocols described above. Liberation of the side chain protected peptide from solid support was conducted using a freshly prepared solution of hexafluoroisopropanol in CH2Cl2 (20% v/v), the procedure was repeated, solutions were combined, and volatiles were removed in vacuo. The protected linear peptide was dissolved in DMF (15 mL) and a solution of HBTU (41.2 mg, 0.109 mmol, 6 equiv) and HOBt●H2O (16.6 mg, 0.109 mmol, 6 equiv) in DMF (1 mL) was added dropwise, followed by an addition of DIEA (0.0494 mL, 0.283 mmol, 15 equiv) in DMF (1 mL). The reaction was allowed to proceed overnight under N2 atmosphere. DMF was removed in vacuo and the cyclic side chain protected peptide was carried forward to global deprotection. A fresh 90:5:5 (v/v/v) solution of TFA: H2O: TIPS was prepared and ~10 mL were added to the dried peptide. The reaction was stirred for 2 h at room temperature, followed by the removal of all volatiles in vacuo. Cold ether precipitation was performed followed by dissolution of the cyclic peptide in 20% MeCN in water (3 mL). The crude peptide sample was purified by RP-HPLC according to protocols described in General Methods.
3.11. Full Cleavage and Purification of (Aza)Peptides
The resin-bound (aza)peptide was washed with CH2Cl2 to remove traces of DMF, drained, transferred into a 20 mL scintillation glass vial, and treated a freshly made solution of TFA/H2O/TIPS (90:5:5, v/v/v, 5 mL). The vial was capped and agitated for 2 h at room temperature on an orbital shaker. The cleavage mixture was filtered through a disposable, PP fritted cartridge into a 50 mL falcon tube containing cold ether (~20 mL). The ether was decanted following centrifugation at 8000 rpm for 2 min. The peptide pellet was suspended in 1:9 MeCN/H2O v/v (5 mL). The sample was frozen and lyophilized. The crude (aza)peptpide was redissolved in a MeCN/H2O solvent mixture and purified using reverse-phase HPLC. See Supporting Information for characterization data.
3.12. NMR Spectroscopy
Lyophilized (aza)peptides were diluted to concentrations of 3–4 mM in D2O buffered with 50 mM NaOAc-d3 buffer at pH 4.2 (uncorrected), pH adjusted with AcOH-d3 [36]. All NMR spectra were collected at the Molecular Education, Technology, and Research Innovation Center (METRIC) at NC State University on a Bruker NEO 700 MHz instrument with a TCI cryoprobe at 5 °C. 1D spectra were collected with 32K data points and at least 32 scans and water suppression with a pre-saturation pulse or with a 1D-1H NOESY. 2D-experiments were collected on the same instrumentation, using the standard Bruker ROESY, COSY, and TOCSY pulse sequences with presaturation. All 2D experiments were done with eight scans, 2048 points in the first dimension, and 256 or 512 in the indirect dimension. TOCSY spectra were collected with 80 ms spin-lock and ROESY spectra were collected with a mixing time of 200 ms. Spectral analysis was performed with TopSpin 4.0.5 software.
4. Conclusions
Aza-amino acids have previously been shown to stabilize β-turn secondary structures and impart hyperstability to self-assembling collagen peptide mimics. Here, we expand the folding rules for aza-amino acids beyond turns and polyproline type II helices and characterize their effect on β-hairpin stability when incorporated into the β-strand region. Using NMR spectroscopy, we demonstrate that a valine to aza-valine substitution significantly disrupts β-hairpin folding, based on both glycine splitting measurements and CHα proton shifts at the Orn8 and Ile10 residues relative to cyclic and unfolded controls. Aza-glycine incorporation at the same position was found to retain some folding, despite loss of side chain chemistry, which may be due to additional hydrogen bonding. Overall, our findings support earlier computational hypothesis that aza-amino acids are not well tolerated within β-sheet secondary structures.
Supplementary Materials
The following are available online: peptide and aza(peptide) characterization data; LC-MS chromatograms; tabulated 1H NMR data for all peptide and aza(peptide) analogs.
Author Contributions
Conceptualization, C.P. and M.A.M.; Methodology and Analysis, M.A.M., E.L.W., P.C.G.; Writing-Original Draft Preparation, C.P.; Writing-Review and Editing, C.P., M.A.M., E.L.W., P.C.G.; Supervision, C.P.; Project Administration, C.P.
Funding
This research received no external funding.
Acknowledgments
The authors thank Peter Thompson for assistance with NMR spectroscopy and North Carolina State University for startup support. All NMR measurements were made in the Molecular Education, Technology, and Research Innovation Center (METRIC) at North Carolina State University.
Conflicts of Interest
The authors declare no conflict of interest.
References and Notes
- Cheng, P.N.; Pham, J.D.; Nowick, J.S. The Supramolecular Chemistry of beta-Sheets. J. Am. Chem. Soc. 2013, 135, 5477–5492. [Google Scholar] [CrossRef] [PubMed]
- Watkins, A.M.; Arora, P.S. Anatomy of beta-Strands at Protein-Protein Interfaces. ACS Chem. Biol. 2014, 9, 1747–1754. [Google Scholar] [CrossRef]
- Hughes, R.M.; Waters, M.L. Model systems for beta-hairpins and beta-sheets. Curr. Opin. Struct. Biol. 2006, 16, 514–524. [Google Scholar] [CrossRef]
- Loughlin, W.A.; Tyndall, J.D.A.; Glenn, M.P.; Fairlie, D.P. Beta-strand mimetics. Chem. Rev. 2004, 104, 6085–6117. [Google Scholar] [CrossRef]
- Doig, A.J. A three stranded beta-sheet peptide in aqueous solution containing N-methyl amino acids to prevent aggregation. Chem. Commun. 1997, 22, 2153–2154. [Google Scholar] [CrossRef]
- Nowick, J.S.; Chung, D.M.; Maitra, K.; Maitra, S.; Stigers, K.D.; Sun, Y. An unnatural amino acid that mimics a tripeptide beta-strand and forms beta-sheet like hydrogen-bonded dimers. J. Am. Chem. Soc. 2000, 122, 7654–7661. [Google Scholar] [CrossRef]
- Sarnowski, M.P.; Kang, C.W.; Elbatrawi, Y.M.; Wojtas, L.; Del Valle, J.R. Peptide N-Amination Supports beta-Sheet Conformations. Angew. Chem. Int. Ed. 2017, 56, 2083–2086. [Google Scholar] [CrossRef]
- Proulx, C.; Sabatino, D.; Hopewell, R.; Spiegel, J.; Ramos, Y.G.; Lubell, W.D. Azapeptides and their therapeutic potential. Future Med. Chem. 2011, 3, 1139–1164. [Google Scholar] [CrossRef]
- Zhang, Y.T.; Malamakal, R.M.; Chenoweth, D.M. Aza-Glycine Induces Collagen Hyperstability. J. Am. Chem. Soc. 2015, 137, 12422–12425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.T.; Herling, M.; Chenoweth, D.M. General Solution for Stabilizing Triple Helical Collagen. J. Am. Chem. Soc. 2016, 138, 9751–9754. [Google Scholar] [CrossRef]
- Thormann, M.; Hofmann, H.J. Conformational properties of azapeptides. J. Mol. Struct. THEOCHEM 1999, 469, 63–76. [Google Scholar] [CrossRef]
- Reynolds, C.H.; Hormann, R.E. Theoretical study of the structure and rotational flexibility of diacylhydrazines: Implications for the structure of nonsteroidal ecdysone agonists and azapeptides. J. Am. Chem. Soc. 1996, 118, 9395–9401. [Google Scholar] [CrossRef]
- Stanger, H.E.; Gellman, S.H. Rules for antiparallel beta-sheet design: D-Pro-Gly is superior to L-Asn-Gly for beta-hairpin nucleation. J. Am. Chem. Soc. 1998, 120, 4236–4237. [Google Scholar] [CrossRef]
- Syud, F.A.; Espinosa, J.F.; Gellman, S.H. NMR-based quantification of beta-sheet populations in aqueous solution through use of reference peptides for the folded and unfolded states. J. Am. Chem. Soc. 1999, 121, 11577–11578. [Google Scholar] [CrossRef]
- Hess, H.J.; Moreland, W.T.; Laubach, G.D. N- [2-Isopropyl-3-(L-Aspartyl-L-Arginyl)-Carbazoyl]-L-Tyrosyl-L-Valyl-L-Histidyl-L-Prolyl-L-Phenylalanine, an Isostere of Bovine Angiotensin Ii. J. Am. Chem. Soc. 1963, 85, 4040–4041. [Google Scholar] [CrossRef]
- Meienhofer, J.; Waki, M.; Heimer, E.P.; Lambros, T.J.; Makofske, R.C.; Chang, C.D. Solid-Phase Synthesis without Repetitive Acidolysis. Int. J. Pept. Prot. Res. 1979, 13, 35–42. [Google Scholar] [CrossRef]
- Boeglin, D.; Lubell, W.D. Aza-amino acid scanning of secondary structure suited for solid-phase peptide synthesis with Fmoc chemistry and aza-amino acids with heteroatomic side chains. J. Comb. Chem. 2005, 7, 864–878. [Google Scholar] [CrossRef]
- Chingle, R.; Proulx, C.; Lubell, W.D. Azapeptide Synthesis Methods for Expanding Side-Chain Diversity for Biomedical Applications. Acc. Chem. Res. 2017, 50, 1541–1556. [Google Scholar] [CrossRef]
- Sabatino, D.; Proulx, C.; Klocek, S.; Bourguet, C.B.; Boeglin, D.; Ong, H.; Lubell, W.D. Exploring Side-Chain Diversity by Submonomer Solid-Phase Aza-Peptide Synthesis. Org. Lett. 2009, 11, 3650–3653. [Google Scholar] [CrossRef]
- Kurian, L.A.; Silva, T.A.; Sabatino, D. Submonomer synthesis of azapeptide ligands of the Insulin Receptor Tyrosine Kinase domain. Bioorg. Med. Chem. Lett. 2014, 24, 4176–4180. [Google Scholar] [CrossRef]
- Spiegel, J.; Mas-Moruno, C.; Kessler, H.; Lubell, W.D. Cyclic Aza-peptide Integrin Ligand Synthesis and Biological Activity. J. Org. Chem. 2012, 77, 5271–5278. [Google Scholar] [CrossRef] [PubMed]
- Isocyanate and/or hydantoin byproducts likely arise from activation of the resin-bound semicarbazide with the carbonyl donor, followed by intramolecular attack by the nitrogen of the preceding amino acid residue. See: Quibell, M.; Turnell, W.G.; Johnson, T., Synthesis of Azapeptides by the Fmoc Tert-Butyl Polyamide Technique. J. Chem. Soc. Perk. Trans 1 1993, 22, 2843–2849. [CrossRef]
- Paradis-Bas, M.; Tulla-Puche, J.; Albericio, F. The road to the synthesis of “difficult peptides”. Chem. Soc. Rev. 2016, 45, 631–654. [Google Scholar] [CrossRef]
- Murray, J.K.; Farooqi, B.; Sadowsky, J.D.; Scalf, M.; Freund, W.A.; Smith, L.M.; Chen, J.D.; Gellman, S.H. Efficient synthesis of a beta-peptide combinatorial library with microwave irradiation. J. Am. Chem. Soc. 2005, 127, 13271–13280. [Google Scholar] [CrossRef]
- Chingle, R.; Ratni, S.; Claing, A.; Lubell, W.D. Application of Constrained aza-Valine Analogs for Smac Mimicry. Biopolymers 2016, 106, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Arujoe, M.; Ploom, A.; Mastitski, A.; Jarv, J. Comparison of various coupling reagents in solid-phase aza-peptide synthesis. Tetrahedron Lett. 2017, 58, 3421–3425. [Google Scholar] [CrossRef]
- Arujoe, M.; Ploom, A.; Mastitski, A.; Jarv, J. Influence of steric effects in solid-phase aza-peptide synthesis. Tetrahedron Lett. 2018, 59, 2010–2013. [Google Scholar] [CrossRef]
- Beutner, G.L.; Young, I.S.; Davies, M.L.; Hickey, M.R.; Park, H.; Stevens, J.M.; Ye, Q.M. TCFH-NMI: Direct Access to N-Acyl Imidazoliums for Challenging Amide Bond Formations. Org. Lett. 2018, 20, 4218–4222. [Google Scholar] [CrossRef] [PubMed]
- Devos, A.; Remion, J.; Frisquehesbain, A.M.; Colens, A.; Ghosez, L. Synthesis of Acyl Halides under Very Mild Conditions. J. Chem. Soc. Chem. Comm. 1979, 24, 1180–1181. [Google Scholar] [CrossRef]
- Gibson, C.; Goodman, S.L.; Hahn, D.; Ho¨lzemann, G.; Kessler, H. Novel Solid-Phase Synthesis of Azapeptides and Azapeptoides via Fmoc-Strategy and Its Application in the Synthesis of RGD-Mimetics. J. Org. Chem. 1999, 64, 7388–7394. [Google Scholar] [CrossRef]
- Bourguet, C.B.; Sabatino, D.; Lubell, W.D. Benzophenone Semicarbazone Protection Strategy for Synthesis of Aza-Glycine Containing Aza-Peptides. Biopolymers 2008, 90, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Chingle, R.; Mulumba, M.; Chung, N.N.; Nguyen, T.M.; Ong, H.; Ballet, S.; Schiller, P.W.; Lubell, W.D. Solid-Phase Azopeptide Diels-Alder Chemistry for Aza-Pipecolyl Residue Synthesis to Study Peptide Conformation. J. Org. Chem. 2019, 84, 6006–6016. [Google Scholar] [CrossRef] [PubMed]
- Searle, M.S.; Griffiths-Jones, S.R.; Skinner-Smith, H. Energetics of weak interactions in a beta-hairpin peptide: Electrostatic and hydrophobic contributions to stability from lysine salt bridges. J. Am. Chem. Soc. 1999, 121, 11615–11620. [Google Scholar] [CrossRef]
- Bouayad-Gervais, S.H.; Lubell, W.D. Examination of the potential for adaptive chirality of the nitrogen chiral center in aza-aspartame. Molecules 2013, 18, 14739. [Google Scholar] [CrossRef]
- It should be noted that replacement of valine for an aza-glycine residue results in an overall decrease in chiral environment, which may affect diastereotopic glycine splitting.
- Kiehna, S.E.; Waters, M.L. Sequence dependence of beta-hairpin structure: Comparison of a salt bridge and an aromatic interaction. Protein Sci. 2003, 12, 2657–2667. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |
Figure 1.
Aza-amino acid (in blue) creates one face with increased hydrogen bonding properties and one with reduced hydrogen bonding capacity.
Figure 1.
Aza-amino acid (in blue) creates one face with increased hydrogen bonding properties and one with reduced hydrogen bonding capacity.


Scheme 1.
Synthesis of azapeptide 1b.
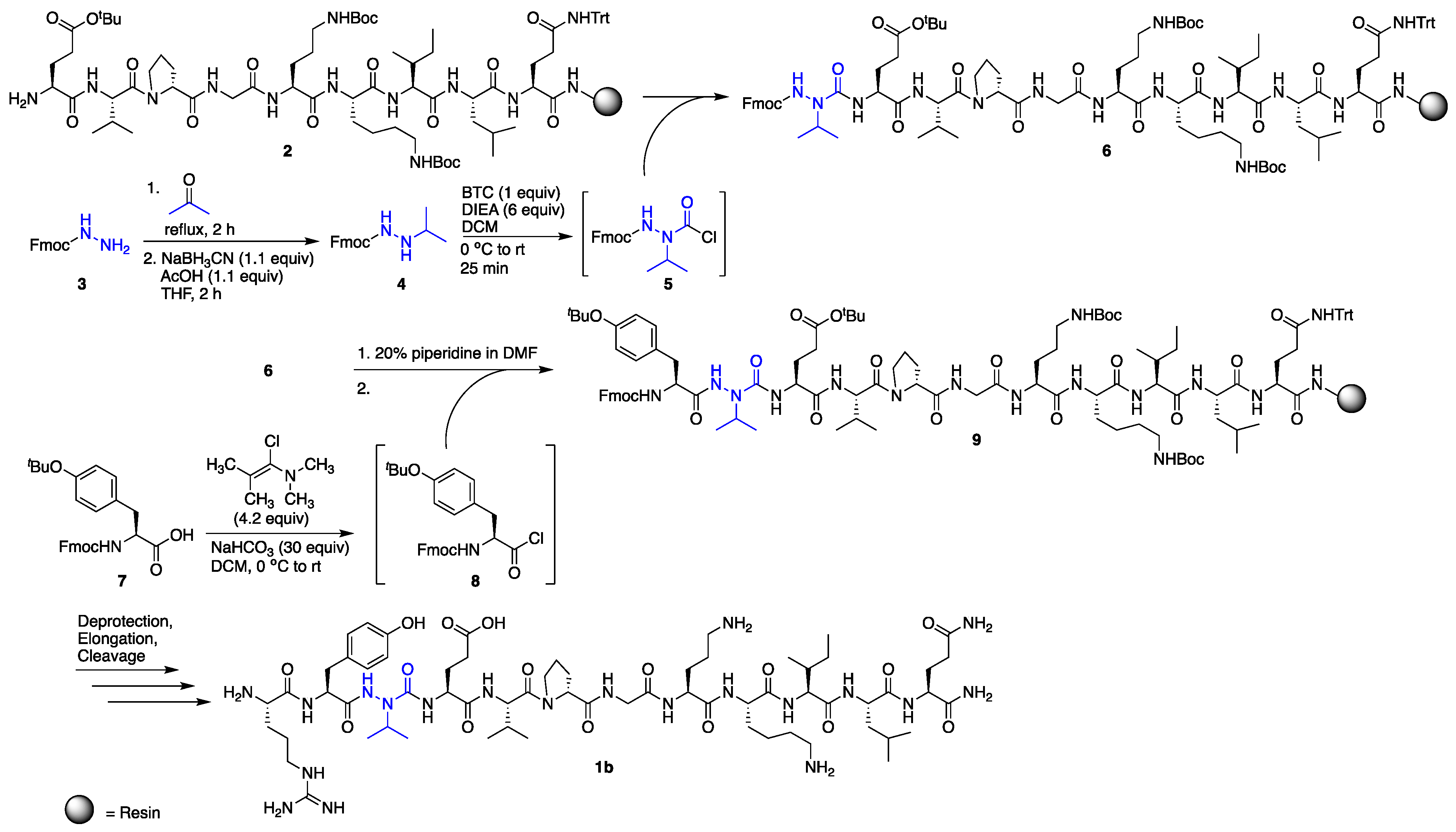
Figure 3.
Diastereotopic splitting of Gly7 Hα chemical shifts.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Optimization of coupling to resin-bound aza-VEVpGOKILQ.
| Entry | Coupling Reagent | Equiv. | Base | Equiv. | Solvent | % Coupled | % Uncoupled | % Isocyanate/Hydantoin Byproduct [22] |
|---|---|---|---|---|---|---|---|---|
| 1 | BTC | 1 | 2,4,6-collidine | 10 | THF | 33 | 21 | 28 |
| 2 | PyBOP | 3 | DIEA | 6 | DMF | 6 | 35 | 41 |
| 3 | COMU | 5 | DIEA | 10 | DMF | 13 | 17 | 53 |
| 4 | TCFHa | 3 | NMI | 9 | CH2Cl2 | 89a | 0 | 10 |
| 5 | Ghosez | 4.2 | NaHCO3 | 30 | CH2Cl2 | 67 | 0 | 22 |
a Possible epimerization was detected by LC-MS analysis.
Table 2.
% folding based on glycine7 splitting and Hα chemical shifts for Orn8 and Ile10.
| Peptide | % Folding | |||
|---|---|---|---|---|
| Gly7 δHα–δHα’ | Orn8 | Ile10 | Average from Orn8/Ile10 | |
| 1a | 75 | 72 | 64 | 68 |
| 1b | 17 | 36 | 0 | 18 |
| 1c | 54 | 56 | 15 | 35.5 |
| 1d | 50 | 50 | 49 | 49.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
McMechen, M.A.; Willis, E.L.; Gourville, P.C.; Proulx, C. Aza-Amino Acids Disrupt β-Sheet Secondary Structures. Molecules 2019, 24, 1919. https://doi.org/10.3390/molecules24101919
AMA Style
McMechen MA, Willis EL, Gourville PC, Proulx C. Aza-Amino Acids Disrupt β-Sheet Secondary Structures. Molecules. 2019; 24(10):1919. https://doi.org/10.3390/molecules24101919
Chicago/Turabian StyleMcMechen, Michael A., Evan L. Willis, Preston C. Gourville, and Caroline Proulx. 2019. "Aza-Amino Acids Disrupt β-Sheet Secondary Structures" Molecules 24, no. 10: 1919. https://doi.org/10.3390/molecules24101919