
Theoretical Study on the Difference in Electron Conductivity of a One-Dimensional Penta-Nickel(II) Complex between Anti-Ferromagnetic and Ferromagnetic States—Possibility of Molecular Switch with Open-Shell Molecules

 ,
,
Abstract
:
1. Introduction
2. Theoretical Background and Computational Details
2.1. Calculation of Electron Conductivity of Open-Shell Systems
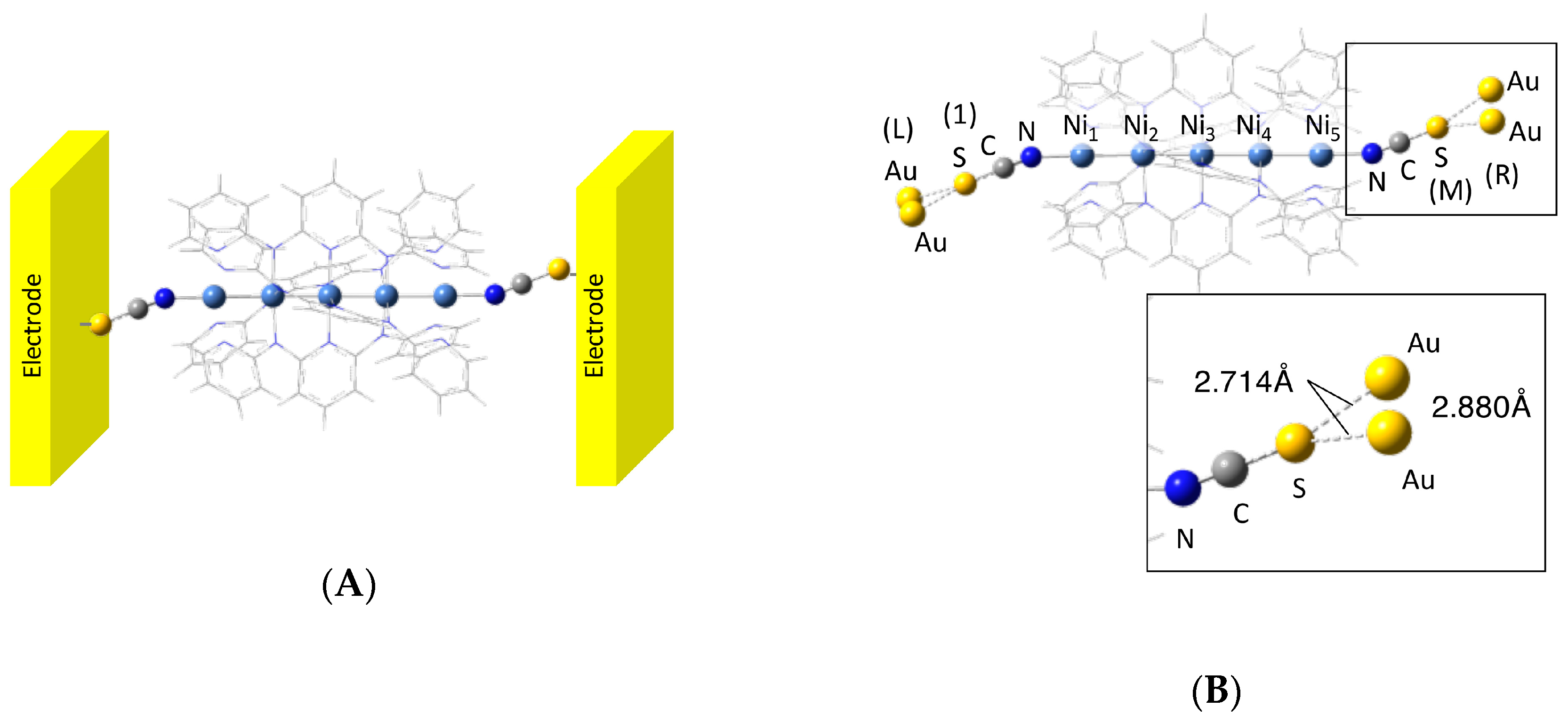
2.2. Computational Details
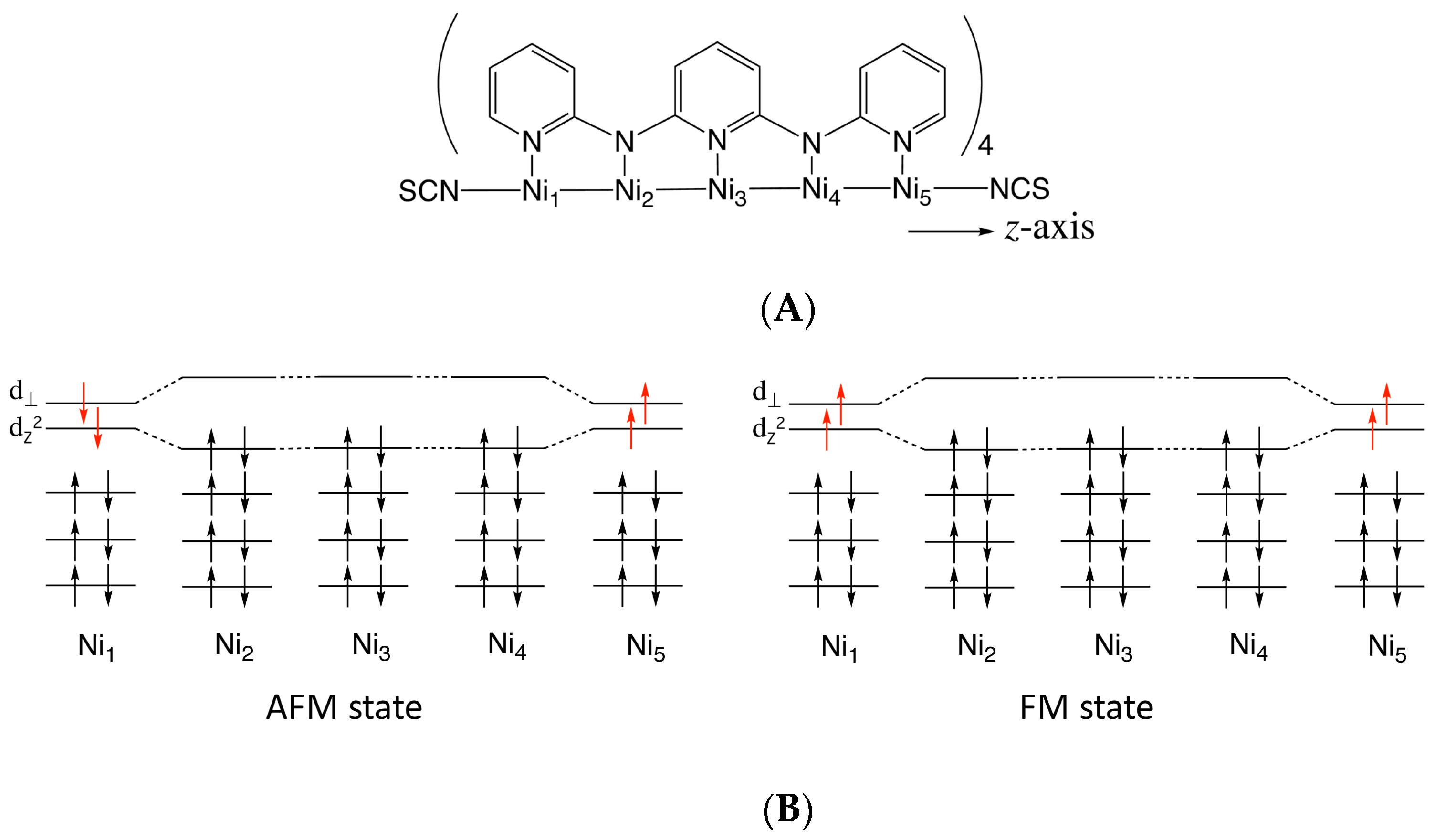
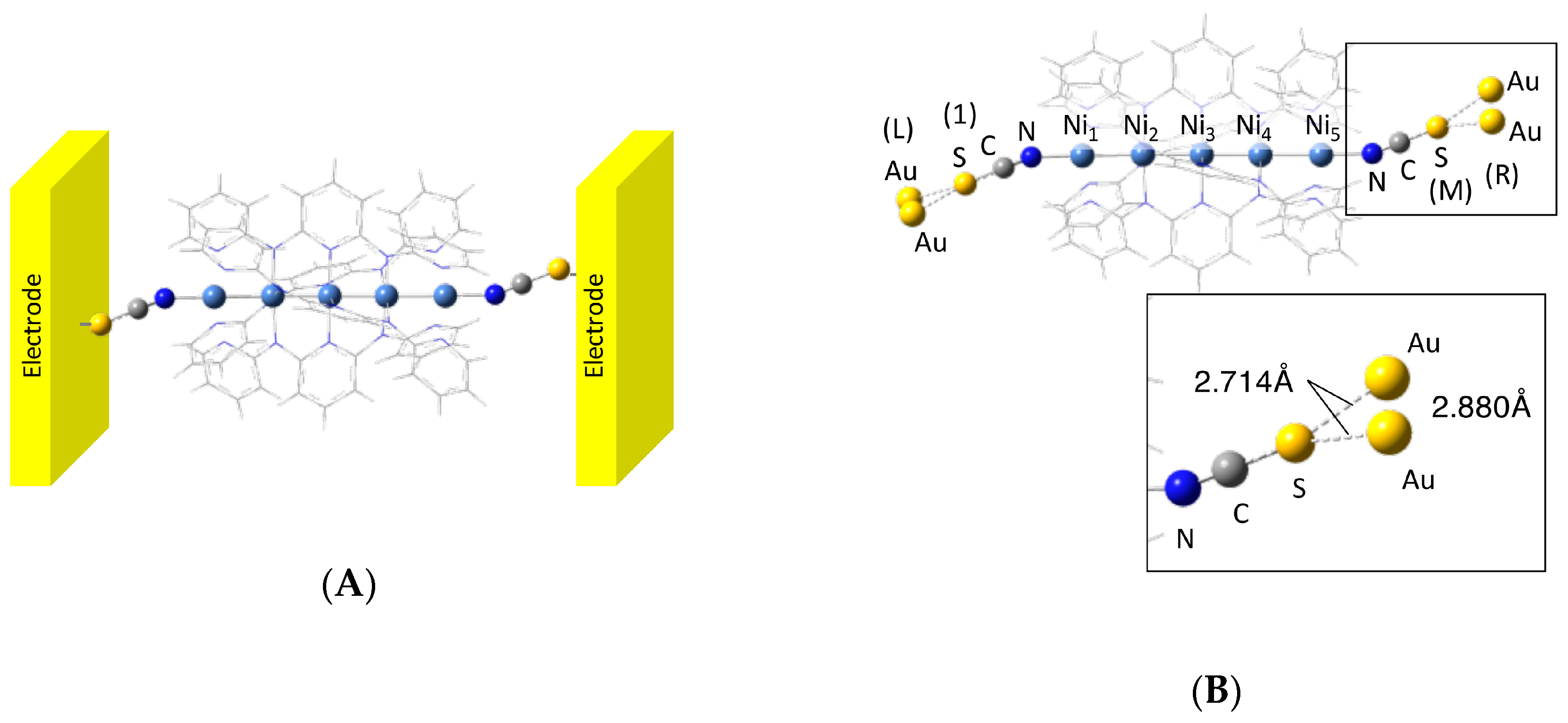
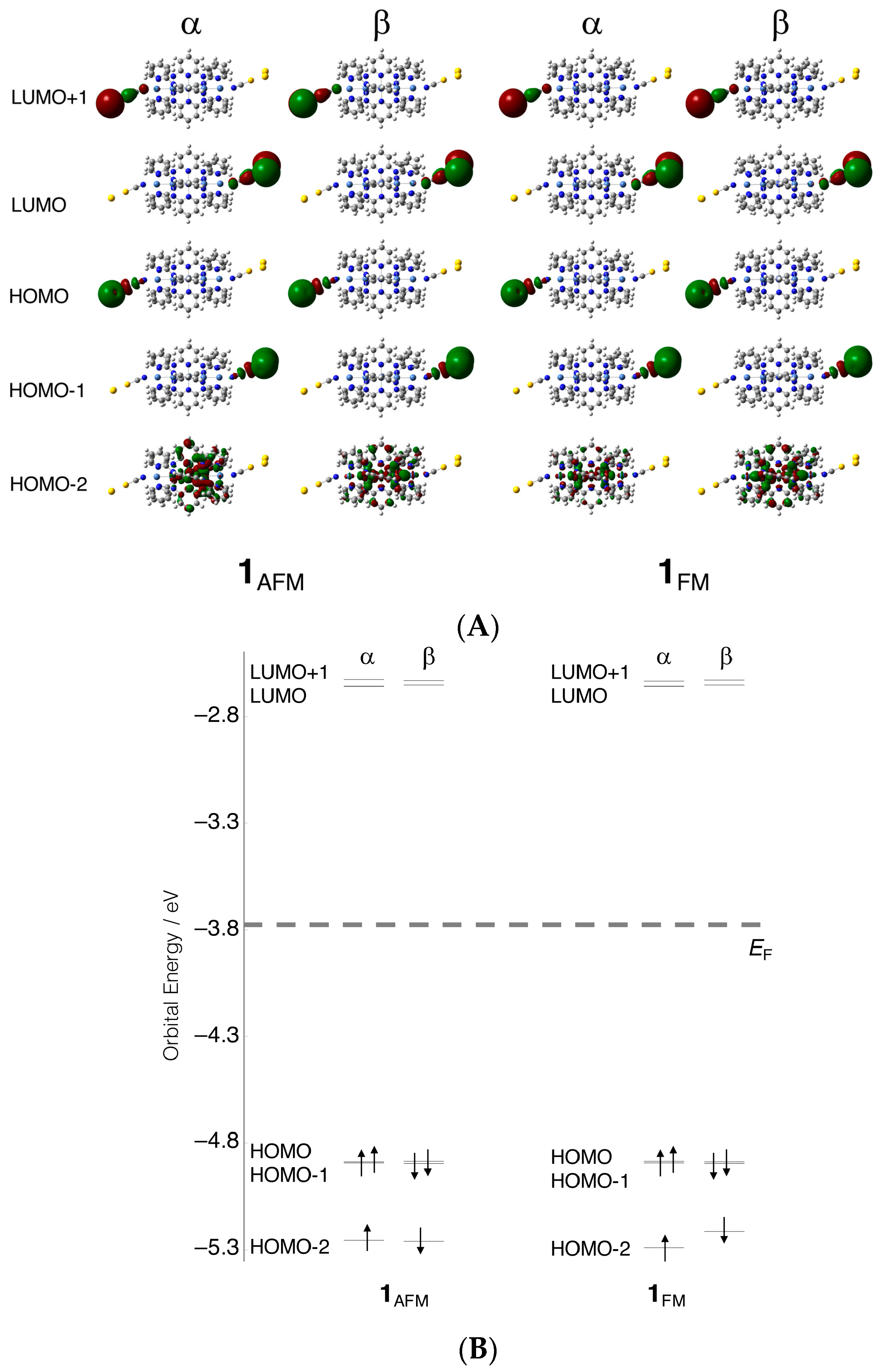
3. Calculated Results
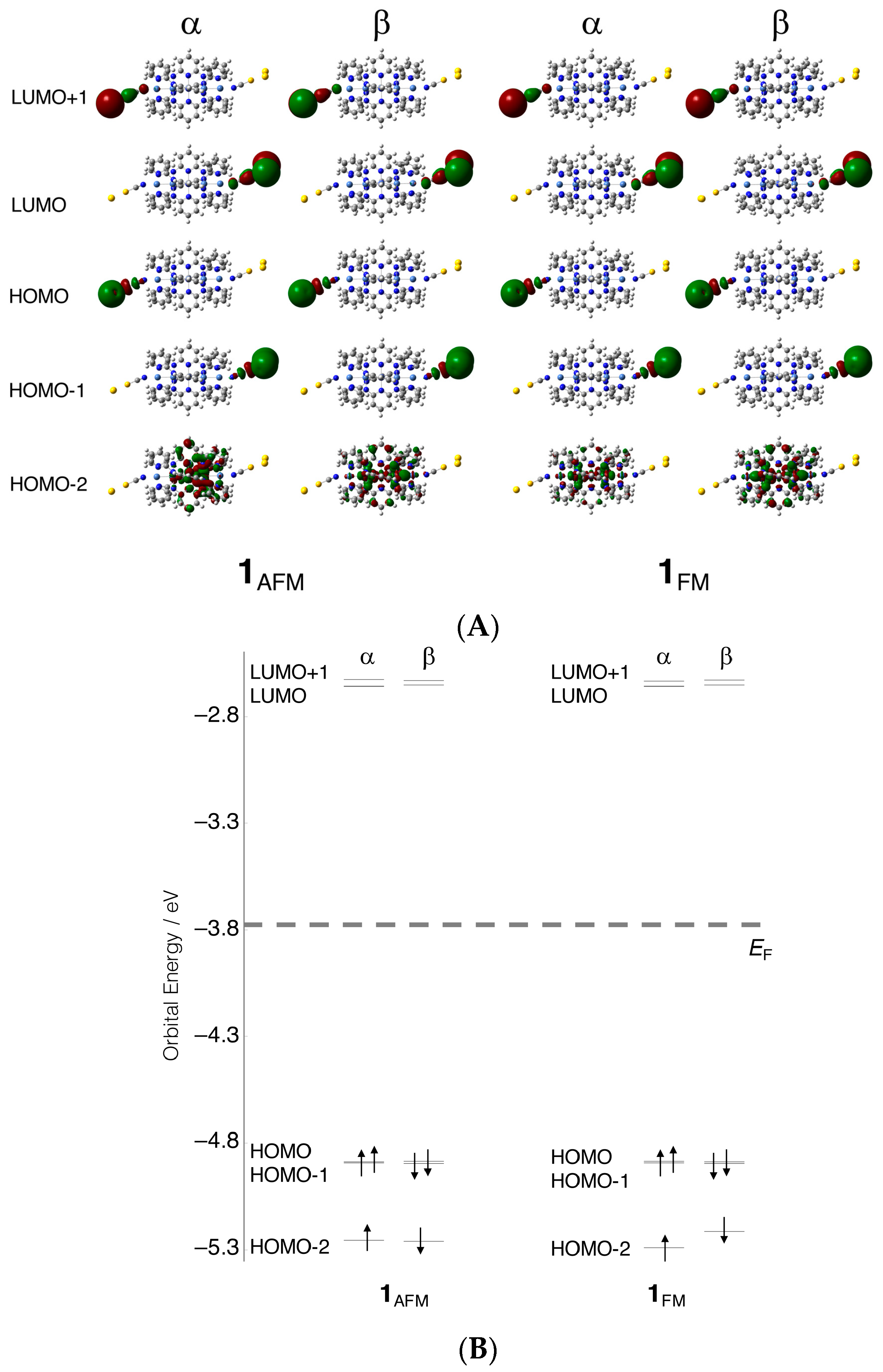
3.1. Electronic Structures
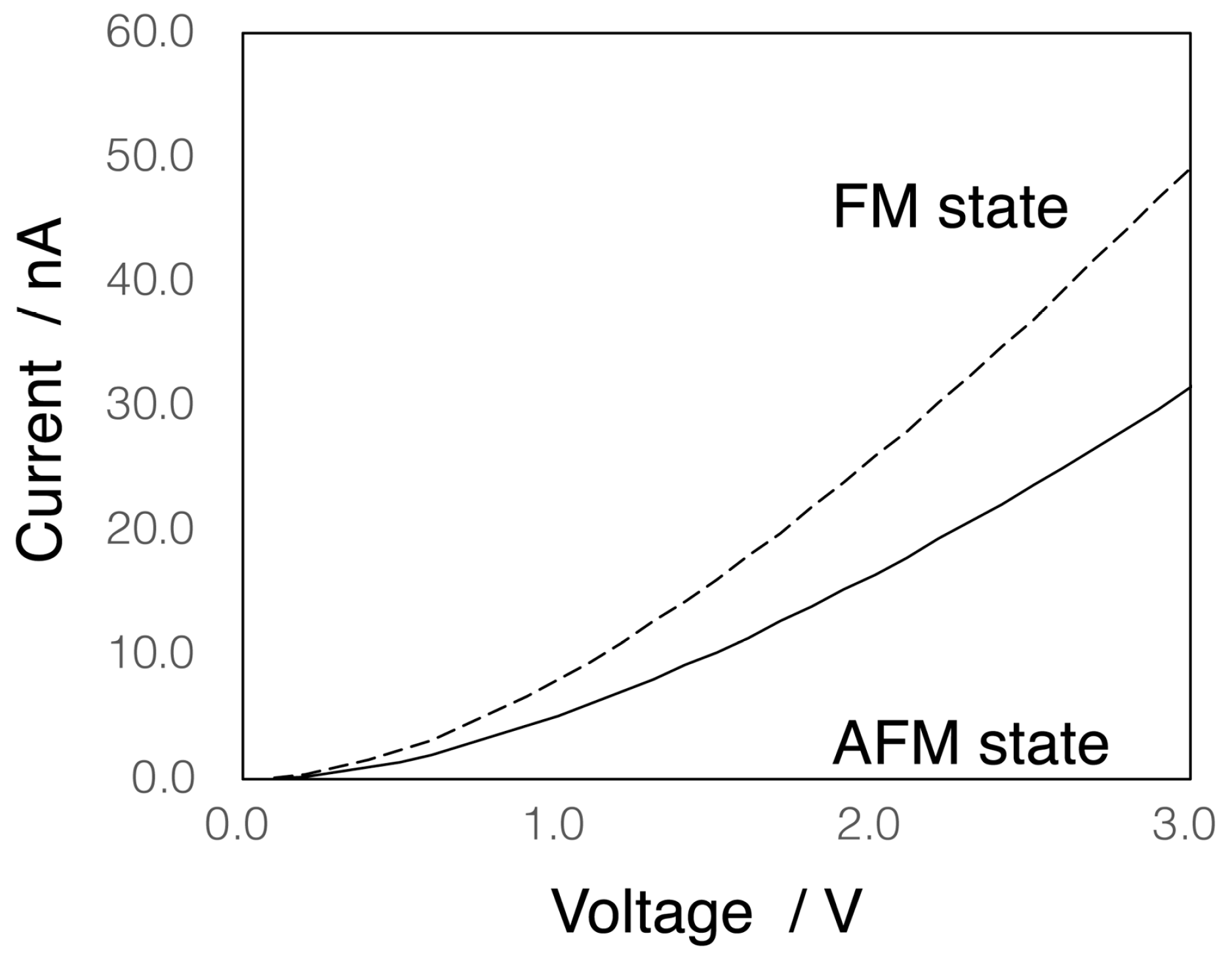
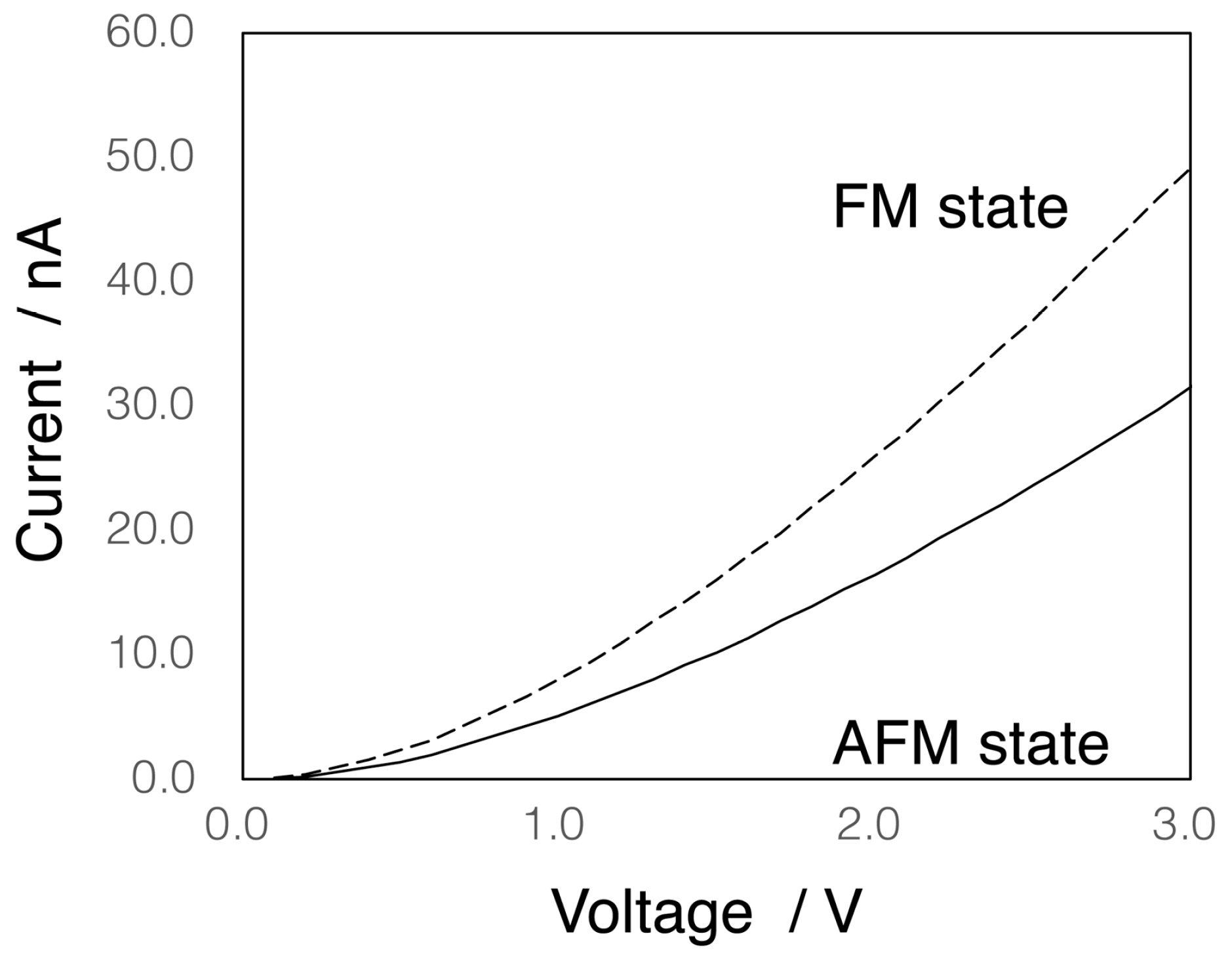
3.2. Electron Conductivity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tans, S.J.; Devoret, M.H.; Dai, H.; Thess, A.; Smalley, R.E.; Geerligs, L.J.; Dekker, C. Individual single-wall carbon nanotubes as quantum wires. Nature 1997, 386, 474–477. [Google Scholar] [CrossRef] [Green Version]
- Nitzan, A.; Ratner, M. Electron Transport in Molecular Wire Junctions. Science 2003, 300, 1384–1389. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, H. Coordination Programming: A New Concept for the Creation of Multifunctional Molecular Systems. Chem. Lett. 2014, 43, 388–395. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, Y.; Staykov, A.; Yoshizawa, K. Orbital view concept applied on photoswitching systems. Thin Solid Films 2009, 518, 444–447. [Google Scholar] [CrossRef]
- Ruiz, E. Charge transport properties of spin crossover systems. Phys. Chem. Chem. Phys. 2014, 16, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Ruben, M.; Wernsdorfer, W. Synthetic Hilbert Space Enginnering of Molecular Qudits: Isotopologue Chemistry. Adv. Mater. 2019, 1806687. [Google Scholar] [CrossRef]
- Cotton, F.A.; Daniels, L.M.; Jordan, G.T., IV. Efficient preparation of a linear, symmetrical, metal–metalbonded tricobalt compound; should we believe there is a bond stretchisomer? Chem. Commun. 1997, 421–422. [Google Scholar] [CrossRef]
- Cotton, F.A.; Daniels, L.M.; Murillo, C.A.; Pascual, I. Compounds with Linear, Bonded Trichromium Chains. J. Am. Chem. Soc. 1997, 119, 10223–10224. [Google Scholar] [CrossRef]
- Aduldecha, S.; Hathaway, B. Crystal structure and electronic properties of tetrakis [μ3-bis(2-pyridyl)amido]dichlorotrinickel(II)–water–acetone (1/0.23/0.5). J. Chem. Soc. Dalton Trans. 1991, 993–998. [Google Scholar] [CrossRef]
- Bera, J.K.; Dunbar, K.R. Chain Compounds Based on Transition Metal Backbones: New Life for an Old Topic. Angew. Chem. Int. Ed. 2002, 41, 4453–4457. [Google Scholar] [CrossRef]
- Mashima, K.; Tanaka, M.; Tani, K.; Nakamura, A.; Takeda, S.; Mori, W.; Yamaguchi, K. Elongation of the Quadruple CrII−CrII Bond Induced by Two PtMe2 Moieties in the Linearly Aligned Tetrametal System, PtMe2···Cr−Cr···PtMe2. J. Am. Chem. Soc. 1997, 119, 4307–4308. [Google Scholar] [CrossRef]
- Tatsumi, Y.; Murahashi, T.; Okada, M.; Ogoshi, S.; Kurosawa, H. A stable zerovalent palladium chain enveloped by a π-electron sheath of conjugated polyene ligands. Chem. Commun. 2008, 477–479. [Google Scholar] [CrossRef] [PubMed]
- Takemura, Y.; Takenaka, H.; Nakajima, T.; Tanase, T. Hexa- and Octagold Chains from Flexible Tetragold Molecular Units Supported by Linear Tetraphosphine Ligands. Angew. Chem. Int. Ed. 2009, 121, 2157–2161. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jagličić, Z.; Han, L.L.; Zhuang, G.L.; Luo, G.G.; Zeng, S.Y.; Tunga, C.H.; Sun, D. Octanuclear Ni(II) cubes based on halogen-substituted pyrazolates: synthesis, structure, electrochemistry and magnetism. Cryst. Eng. Commun. 2016, 18, 3462–3471. [Google Scholar] [CrossRef]
- Nakamae, K.; Takemura, T.; Kure, B.; Nakajima, T.; Kitagawa, Y.; Tanase, T. Self-Alignment of Low-Valent Octanuclear Palladium Atoms. Angew. Chem. Int. Ed. 2015, 54, 1016–1021. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Lo, W.C.; Chou, C.C.; Lee, G.H.; Chen, J.M.; Peng, S.M. Synthesis, Crystal Structures, and Magnetic Properties of a Series of Linear Pentanickel(II) Complexes: [Ni5(μ5-tpda)4X2] (X = Cl-, CN-, N3-, NCS-) and [Ni5(μ5-tpda)4(CH3CN)2]- (PF6)2 (tpda2- = the Tripyridyldiamido Dianion). Inorg. Chem. 1998, 37, 4059–4065. [Google Scholar] [CrossRef] [PubMed]
- Shieh, S.-J.; Chou, C.C.; Lee, G.H.; Wang, C.C.; Peng, S.M. Linear Pentanuclear Complexes Containing a Chain of Metal Atoms: [CoII5(μ5-tpda)4(NCS)2] and (Ni II5(μ5-tpda)4Cl2]. Angew. Chem. Int. Ed. 1997, 36, 56–59. [Google Scholar] [CrossRef]
- Lai, S.Y.; Lin, Z.W.; Chen, Y.H.; Wang, C.C.; Lee, G.H.; Yang, M.H.; Leung, M.-K.; Peng, S.M. Metal String Complexes: Synthesis and Crystal Structure of [Ni4(μ4-phdpda)4] and [Ni7(μ7-teptra)4Cl2] (H2phdpda = N-Phenyldipyridyldiamine and H3teptra = Tetrapyridyltriamine). J. Am Chem. Soc. 1999, 121, 250–251. [Google Scholar] [CrossRef]
- Ismayilov, R.H.; Wang, W.Z.; Lee, G.H.; Yeh, C.Y.; Hua, S.A.; Song, Y.; Rohmer, M.M.; Bénard, M.; Peng, S.M. Two Linear Undecanickel Mixed-Valence Complexes: Increasing the Size and the Scope of the Electronic Properties of Nickel Metal Strings. Angew. Chem. Int. Ed. 2011, 50, 2045–2048. [Google Scholar] [CrossRef]
- Peng, S.M.; Wang, C.C.; Jang, Y.L.; Chen, Y.H.; Li, F.Y.; Mou, C.Y.; Leung, M.K. One-dimensional metal string complexes. J. Magn. Magn. Mat. 2000, 209, 80–83. [Google Scholar] [CrossRef]
- Chen, I.W.; Fu, M.D.; Tseng, W.H.; Yu, J.Y.; Wu, S.H.; Ku, C.J.; Chen, C.h.; Peng, S.M. Conductance and Stochastic Switching of Ligand-Supported Linear Chains of Metal Atoms. Angew. Chem. Int. Ed. 2006, 45, 5814–5818. [Google Scholar] [CrossRef]
- Huang, M.J.; Hua, S.A.; Fu, M.D.; Huang, G.C.; Yin, C.; Ko, C.H.; Kuo, C.K.; Hsu, C.H.; Hsu, G.H.; Lee, G.H.; et al. The First Heteropentanuclear Extended Metal-Atom Chain: [Ni+–Ru25+–Ni2+–Ni2+(tripyridyldiamido)4(NCS)2]. Chem. Euro. J. 2014, 20, 4526–4531. [Google Scholar] [CrossRef]
- Tsai, T.W.; Huang, Q.R.; Peng, S.M.; Jin, B.Y. Smallest Electrical Wire Based on Extended Metal-Atom Chains. J. Phys. Chem. C 2010, 114, 3641–3644. [Google Scholar] [CrossRef]
- Georgiv, V.P.; McGrady, J.E. Influence of Low-Symmetry Distortions on Electron Transport through Metal Atom Chains: When Is a Molecular Wire Really “Broken”? J. Am. Chem. Soc. 2011, 133, 12590–12599. [Google Scholar] [CrossRef]
- Nakanishi, Y.; Matsui, T.; Kitagawa, Y.; Shigeta, Y.; Saito, T.; Kataoka, Y.; Kawakami, T.; Okumura, M.; Yamaguchi, K. Electron Conductivity in Modified Models of Artificial Metal–DNA Using Green’s Function-Based Elastic Scattering Theory. Bull. Chem. Soc. Jpn. 2011, 84, 366–375. [Google Scholar] [CrossRef]
- Kitagawa, Y.; Shoji, M.; Koizumi, K.; Kawakami, T.; Okumura, M.; Yamaguchi, K. Theoretical studies on magnetic interactions and charge-dope effects in one-dimensional Ni5 and Ni7 complexes. Polyhedron 2005, 24, 2751–2757. [Google Scholar] [CrossRef]
- Kitagawa, Y.; Matsui, T.; Nakanishi, Y.; Shigeta, Y.; Kawakami, T.; Okumura, M.; Yamaguchi, K. Theoretical studies of electronic structures, magnetic properties and electron conductivities of one-dimensional Nin (n = 3, 5, 7) complexes. Dalton Trans. 2013, 42, 16200–16208. [Google Scholar] [CrossRef]
- Kitagawa, Y.; Asaoka, M.; Natori, Y.; Miyagi, K.; Teramoto, R.; Matsui, T.; Shigeta, Y.; Okumura, M.; Nakano, M. Theoretical study on relationship between spin structure and electron conductivity of one-dimensional tri-nickel(II) complex. Polyhedron 2017, 136, 125–131. [Google Scholar] [CrossRef]
- Mujica, V.; Kemp, M.; Ratner, M.A. Electron conduction in molecular wires. I. A scattering formalism. J. Chem. Phys. 1994, 101, 6849–6855. [Google Scholar] [CrossRef]
- Mujica, V.; Kemp, M.; Ratner, M.A. Electron conduction in molecular wires. II. Application to scanning tunneling microscopy. J. Chem. Phys. 1994, 101, 6856–6864. [Google Scholar] [CrossRef]
- Wang, C.K.; Fu, Y.; Luo, Y. A quantum chemistry approach for current–voltage characterization of molecular junctions. Phys. Chem. Chem. Phys. 2001, 3, 5017–5023. [Google Scholar] [CrossRef]
- Luo, Y.; Wang, C.K.; Fu, Y. Effects of chemical and physical modifications on the electronic transport properties of molecular junctions. J. Chem. Phys. 2002, 117, 10283–10290. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Rev.C01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Kittel, C. Introduction to Solid State Physics; John Wiley & Sons, Inc.: New York, NY, USA, 2005. [Google Scholar]
- Yamaguchi, K.; Fukui, H.; Fueno, T. Molecular orbital (MO) theory for magnetically interacting organic compounds. Ab-initio MO calculations of the effective exchange integrals for cyclophane-type carbene dimers. Chem. Lett. 1986, 15, 625–628. [Google Scholar] [CrossRef]
- Soda, T.; Kitagawa, Y.; Onishi, T.; Takano, Y.; Shigeta, Y.; Nagao, H.; Yoshioka, Y.; Yamaguchi, K. Ab initio computations of effective exchange integrals for H–H, H–He–H and Mn2O2 complex: Comparison of broken-symmetry approaches. Chem. Phys. Lett. 2000, 319, 223–230. [Google Scholar] [CrossRef]
- Huang, P.J.; Natori, Y.; Kitagawa, Y.; Sekine, Y.; Kosaka, W.; Miyasaka, H. Strong electronic influence of equatorial ligands on frontier orbitals in paddlewheel dichromium(II,II) complexes. Dalton Trans. 2019, 48, 908–914. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1AFM | 1FM | |
|---|---|---|
| EF/eV | α −3.77/β −3.77 | α −3.77/β −3.77 |
| ΔH-L/eV | α 2.23/β 2.24 | α 2.23/β 2.24 |
| Atomic spin densities/atomic units b | ||
| Ni1 | −1.592 | 1.593 |
| Ni2 | −0.114 | 0.120 |
| Ni3 | 0.000 | 0.034 |
| Ni4 | 0.109 | 0.116 |
| Ni5 | 1.595 | 1.596 |
| Sum of NCS ligands | 0.001 | 0.104 |
| Sum of gold dimers | 0.000 | −0.008 |
| Other ligands | 0.001 | 0.445 |
| Total energy/atomic unit | −12470.68913 | −12470.68895 |
| 2.0201 | 6.024 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitagawa, Y.; Tada, H.; Era, I.; Fujii, T.; Ikenaga, K.; Nakano, M. Theoretical Study on the Difference in Electron Conductivity of a One-Dimensional Penta-Nickel(II) Complex between Anti-Ferromagnetic and Ferromagnetic States—Possibility of Molecular Switch with Open-Shell Molecules. Molecules 2019, 24, 1956. https://doi.org/10.3390/molecules24101956
Kitagawa Y, Tada H, Era I, Fujii T, Ikenaga K, Nakano M. Theoretical Study on the Difference in Electron Conductivity of a One-Dimensional Penta-Nickel(II) Complex between Anti-Ferromagnetic and Ferromagnetic States—Possibility of Molecular Switch with Open-Shell Molecules. Molecules. 2019; 24(10):1956. https://doi.org/10.3390/molecules24101956
Chicago/Turabian StyleKitagawa, Yasutaka, Hayato Tada, Iori Era, Takuya Fujii, Kazuki Ikenaga, and Masayoshi Nakano. 2019. "Theoretical Study on the Difference in Electron Conductivity of a One-Dimensional Penta-Nickel(II) Complex between Anti-Ferromagnetic and Ferromagnetic States—Possibility of Molecular Switch with Open-Shell Molecules" Molecules 24, no. 10: 1956. https://doi.org/10.3390/molecules24101956