Structural Characterization of Arabidopsis thaliana NAP1-Related Protein 2 (AtNRP2) and Comparison with Its Homolog AtNRP1


Abstract
:1. Introduction
2. Results
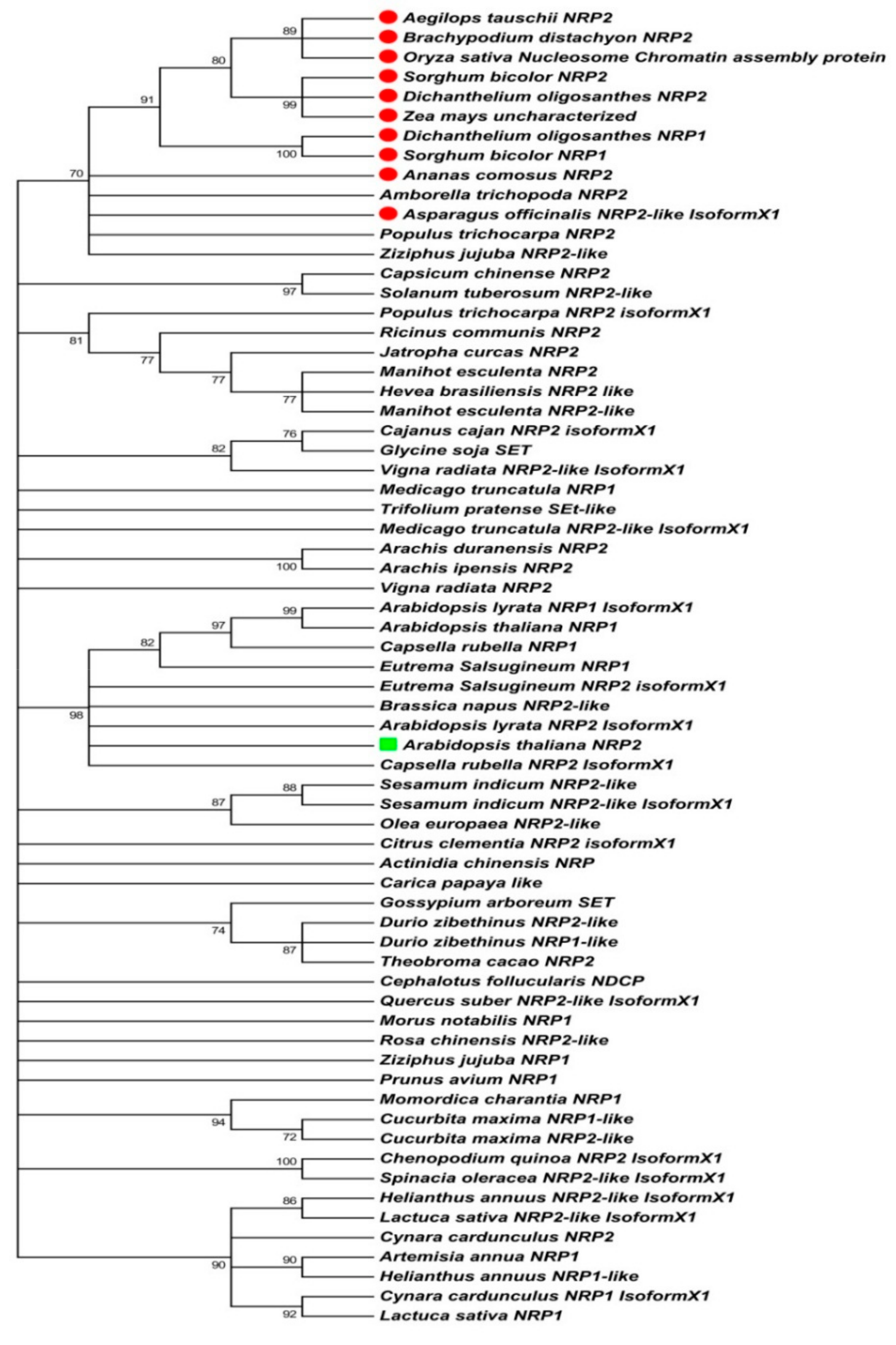
2.1. Sequence-Based Molecular Phylogenetic Studies on Plant NRP1 and NRP2
2.2. Expression, Purification, and Estimation of the Oligomeric Status of AtNRPs
2.3. Crystallization of AtNRP2
2.4. Crystal Structure of AtNRP2
2.5. AtNRP2 Structure vs. AtNRP1 Structure
2.6. Structural Comparison of AtNRP2 with Other NAP Family Proteins
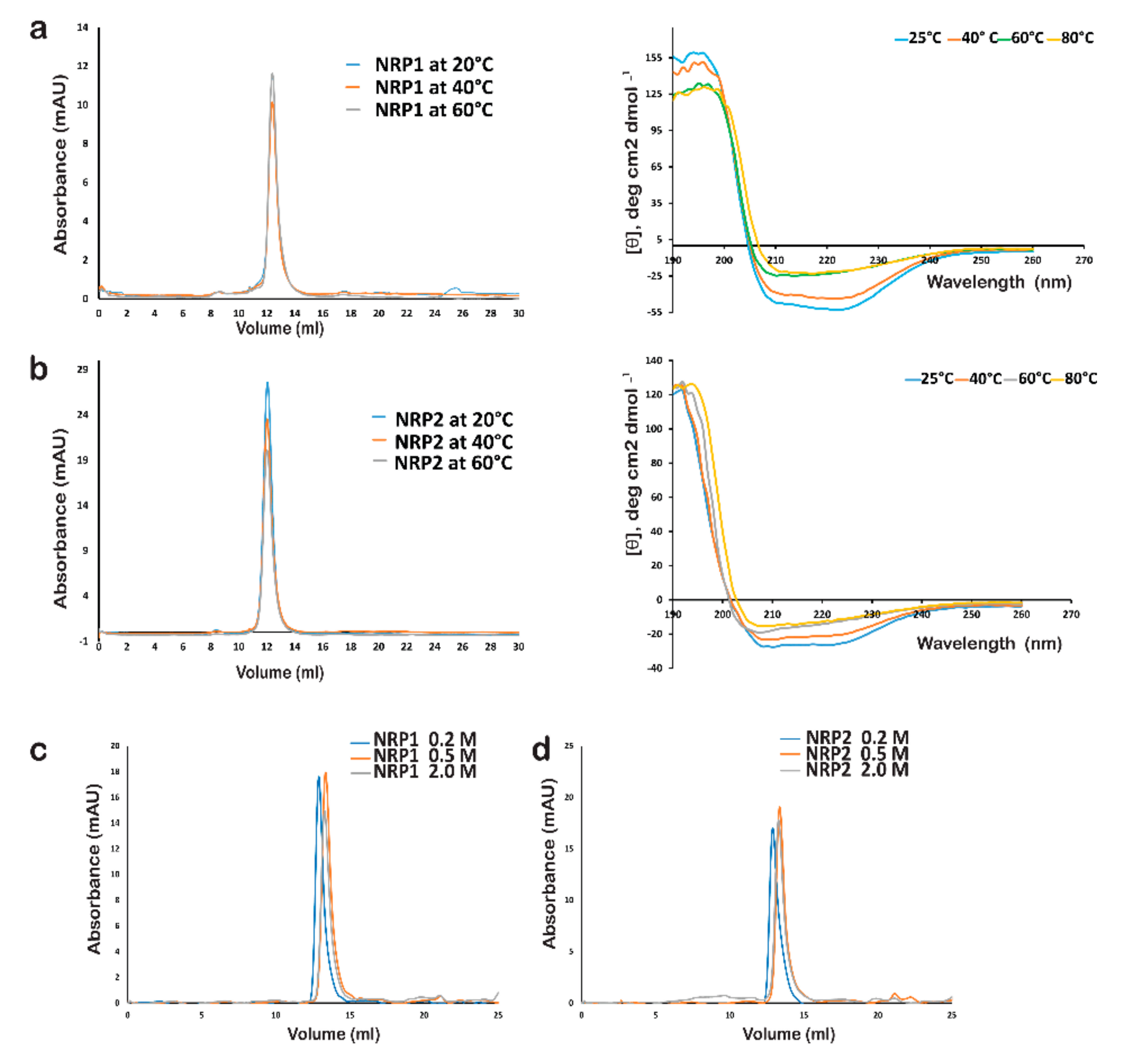
2.7. The In-Solution Architecture of AtNRP1 and AtNRP2
2.8. Stability of AtNRP1 and AtNRP2
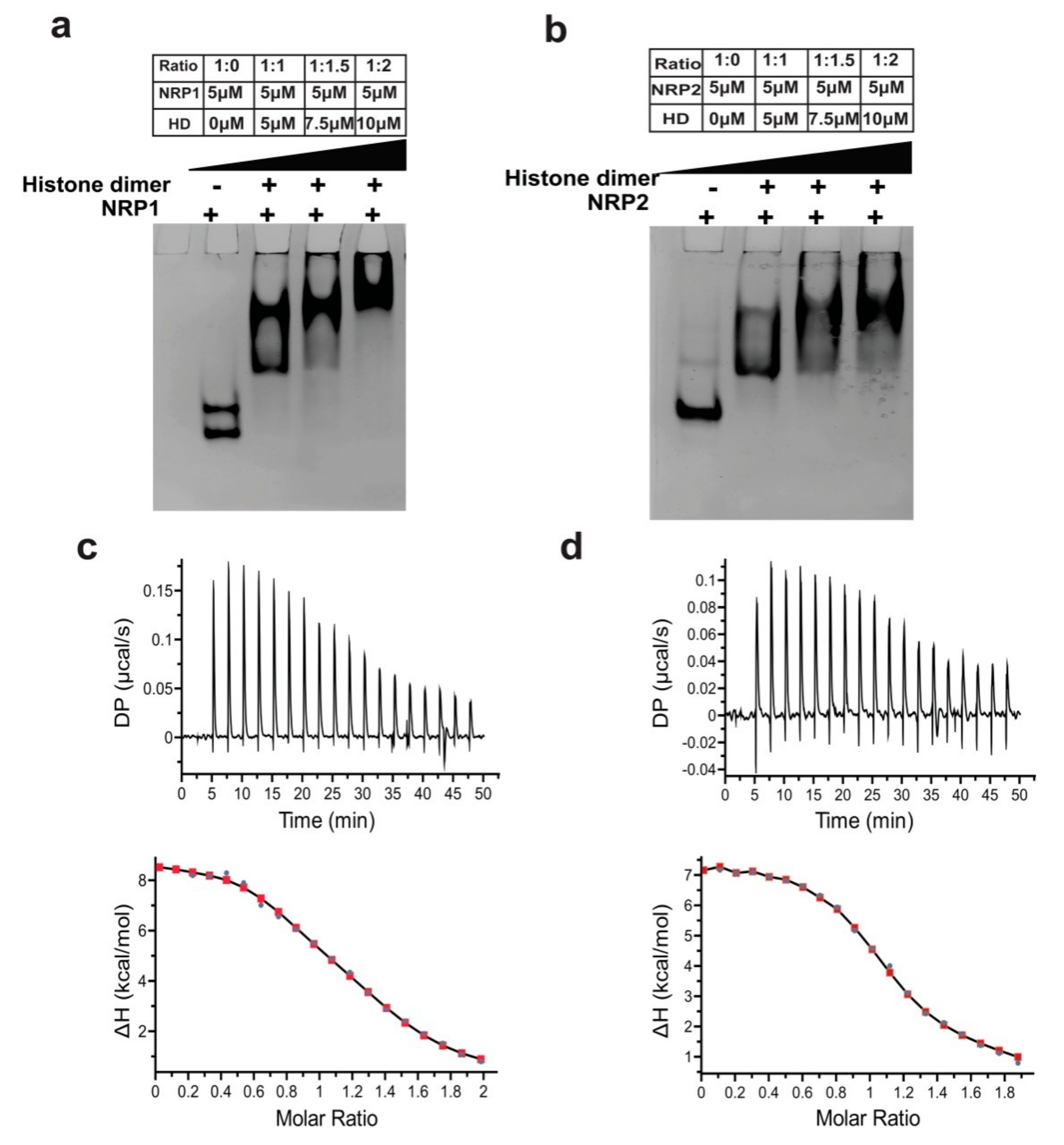
2.9. Histone-Binding and Chaperoning Activities of AtNRP1 and AtNRP2
3. Discussion
4. Materials and Methods
4.1. Phylogenetic Analysis
4.2. Construction of E. coli Expression Plasmids, AtNRP1/AtNRP2 Expression, and Purification
4.3. Histone Expression and Purification
4.4. Crystallization and Data Collection
4.5. Structure Determination
4.6. Analytical Gel-Filtration Chromatography
4.7. Sedimentation Velocity Analytical Ultra-Centrifugation
4.8. Circular Dichroism Spectroscopy
4.9. Small Angle X-ray Scattering Analysis
4.10. Isothermal Titration Calorimetry
4.11. DNA Supercoiling Assay
Accession Numbers
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aalfs, J.D.; Kingston, R.E. What does ‘chromatin remodeling’ mean? Trends Biochem. Sci. 2000, 25, 548–555. [Google Scholar] [CrossRef]
- Miyaji-Yamaguchi, M.; Kato, K.; Nakano, R.; Akashi, T.; Kikuchi, A.; Nagata, K. Involvement of nucleocytoplasmic shuttling of yeast Nap1 in mitotic progression. Mol. Cell. Biol. 2003, 23, 6672–6684. [Google Scholar] [CrossRef] [PubMed]
- McBryant, S.J.; Park, Y.J.; Abernathy, S.M.; Laybourn, P.J.; Nyborg, J.K.; Luger, K. Preferential binding of the histone (H3-H4)2 tetramer by NAP1 is mediated by the amino-terminal histone tails. J. Biol. Chem. 2003, 278, 44574–44583. [Google Scholar] [CrossRef] [PubMed]
- Avvakumov, N.; Nourani, A.; Cote, J. Histone chaperones: Modulators of chromatin marks. Mol. Cell 2011, 41, 502–514. [Google Scholar] [CrossRef]
- Zhu, Y.; Dong, A.; Meyer, D.; Pichon, O.; Renou, J.P.; Cao, K.; Shen, W.H. Arabidopsis NRP1 and NRP2 encode histone chaperones and are required for maintaining postembryonic root growth. Plant Cell 2006, 18, 2879–2892. [Google Scholar] [CrossRef] [PubMed]
- Hammond, C.M.; Stromme, C.B.; Huang, H.; Patel, D.J.; Groth, A. Histone chaperone networks shaping chromatin function. Nat. Rev. Mol. Cell Biol. 2017, 18, 141–158. [Google Scholar] [CrossRef] [Green Version]
- Das, C.; Tyler, J.K.; Churchill, M.E. The histone shuffle: Histone chaperones in an energetic dance. Trends Biochem. Sci. 2010, 35, 476–489. [Google Scholar] [CrossRef]
- Ohkuni, K.; Shirahige, K.; Kikuchi, A. Genome-wide expression analysis of NAP1 in Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 2003, 306, 5–9. [Google Scholar] [CrossRef]
- Park, Y.J.; Luger, K. The structure of nucleosome assembly protein 1. Proc. Natl. Acad. Sci. USA 2006, 103, 1248–1253. [Google Scholar] [CrossRef] [Green Version]
- Rogner, U.C.; Spyropoulos, D.D.; Le Novere, N.; Changeux, J.P.; Avner, P. Control of neurulation by the nucleosome assembly protein-1-like 2. Nat. Genet. 2000, 25, 431–435. [Google Scholar] [CrossRef]
- Lankenau, S.; Barnickel, T.; Marhold, J.; Lyko, F.; Mechler, B.M.; Lankenau, D.H. Knockout targeting of the Drosophila nap1 gene and examination of DNA repair tracts in the recombination products. Genetics 2003, 163, 611–623. [Google Scholar] [PubMed]
- Dong, A.; Zhu, Y.; Yu, Y.; Cao, K.; Sun, C.; Shen, W.H. Regulation of biosynthesis and intracellular localization of rice and tobacco homologues of nucleosome assembly protein 1. Planta 2003, 216, 561–570. [Google Scholar] [PubMed]
- Dong, A.; Liu, Z.; Zhu, Y.; Yu, F.; Li, Z.; Cao, K.; Shen, W.H. Interacting proteins and differences in nuclear transport reveal specific functions for the NAP1 family proteins in plants. Plant Physiol. 2005, 138, 1446–1456. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.K.; Singh, K.; Pareek, A.; Singla-Pareek, S.L. Histone chaperones in Arabidopsis and rice: Genome-wide identification, phylogeny, architecture and transcriptional regulation. BMC Plant Biol. 2015, 15, 42. [Google Scholar] [CrossRef]
- Tripathi, A.K.; Pareek, A.; Singla-Pareek, S.L. A NAP-Family Histone Chaperone Functions in Abiotic Stress Response and Adaptation. Plant Physiol. 2016, 171, 2854–2868. [Google Scholar] [Green Version]
- Barna, B.; Gemes, K.; Domoki, M.; Bernula, D.; Ferenc, G.; Balint, B.; Nagy, I.; Feher, A. Arabidopsis NAP-related proteins (NRPs) contribute to the coordination of plant growth, developmental rate, and age-related pathogen resistance under short days. Plant Sci. 2018, 267, 124–134. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.Q.; Gao, J.; Dong, A.W.; Shen, W.H. A truncated Arabidopsis NUCLEOSOME ASSEMBLY PROTEIN 1, AtNAP1;3T, alters plant growth responses to abscisic acid and salt in the Atnap1;3-2 mutant. Mol. Plant 2009, 2, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Rong, L.; Luo, Q.; Wang, B.; Zhou, N.; Yang, Y.; Zhang, C.; Feng, H.; Zheng, L.; Shen, W.H.; et al. The Histone Chaperone NRP1 Interacts with WEREWOLF to Activate GLABRA2 in Arabidopsis Root Hair Development. Plant Cell 2017, 29, 260–276. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Dong, A.; Shen, W.H. Chromatin remodeling in Arabidopsis root growth. Plant Signal. Behav. 2007, 2, 160–162. [Google Scholar] [CrossRef]
- Biro, J.; Farkas, I.; Domoki, M.; Otvos, K.; Bottka, S.; Dombradi, V.; Feher, A. The histone phosphatase inhibitory property of plant nucleosome assembly protein-related proteins (NRPs). Plant Physiol. Biochem. 2012, 52, 162–168. [Google Scholar] [CrossRef]
- Gao, J.; Zhu, Y.; Zhou, W.; Molinier, J.; Dong, A.; Shen, W.H. NAP1 family histone chaperones are required for somatic homologous recombination in Arabidopsis. Plant Cell 2012, 24, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Arzola, K.; Diaz-Quintana, A.; Rivero-Rodriguez, F.; Velazquez-Campoy, A.; De la Rosa, M.A.; Diaz-Moreno, I. Histone chaperone activity of Arabidopsis thaliana NRP1 is blocked by cytochrome c. Nucleic Acids Res. 2017, 45, 2150–2165. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Liu, Y.; Zhou, W.; Zhu, Y.; Dong, A.; Shen, W.H. Histone chaperones play crucial roles in maintenance of stem cell niche during plant root development. Plant J. 2018, 95, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felsenstein, J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Zuckerkandl, E.; Pauling, L. Evolutionary divergence and convergence in proteins. In Evolving Genes and Proteins; Bryson, V., Vogel, H.J., Eds.; Academic Press: New York, NY, USA, 1965; pp. 97–166. [Google Scholar]
- AmborellaGenome Project. The Amborella genome and the evolution of flowering plants. Science 2013, 342, 1241089. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Capella-Gutierrez, S.; Pryszcz, L.P.; Marcet-Houben, M.; Gabaldon, T. PhylomeDB v4: Zooming into the plurality of evolutionary histories of a genome. Nucleic Acids Res. 2014, 42, D897–D902. [Google Scholar] [CrossRef]
- Dale, G.E.; Oefner, C.; D’Arcy, A. The protein as a variable in protein crystallization. J. Struct. Biol. 2003, 142, 88–97. [Google Scholar] [CrossRef]
- Graslund, S.; Sagemark, J.; Berglund, H.; Dahlgren, L.G.; Flores, A.; Hammarstrom, M.; Johansson, I.; Kotenyova, T.; Nilsson, M.; Nordlund, P.; et al. The use of systematic N- and C-terminal deletions to promote production and structural studies of recombinant proteins. Protein Expr. Purif. 2008, 58, 210–221. [Google Scholar] [CrossRef]
- Mooij, W.T.; Mitsiki, E.; Perrakis, A. ProteinCCD: Enabling the design of protein truncation constructs for expression and crystallization experiments. Nucleic Acids Res. 2009, 37, W402–W405. [Google Scholar] [CrossRef]
- Derewenda, Z.S. Application of protein engineering to enhance crystallizability and improve crystal properties. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 5, 604–615. [Google Scholar] [CrossRef] [Green Version]
- Sagemark, J.; Kraulis, P.; Weigelt, J. A software tool to accelerate design of protein constructs for recombinant expression. Protein Expr. Purif. 2010, 72, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.D.O.; Marsden, B.D. N- and C-Terminal Truncations to Enhance Protein Solubility and Crystallization: Predicting Protein Domain Boundaries with Bioinformatics Tools. Methods Mol. Biol. 2017, 1586, 11–31. [Google Scholar]
- Dosztanyi, Z. Prediction of protein disorder based on IUPred. Protein Sci. 2018, 27, 331–340. [Google Scholar] [CrossRef]
- Dosztanyi, Z.; Csizmok, V.; Tompa, P.; Simon, I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J. Mol. Biol. 2005, 347, 827–839. [Google Scholar] [CrossRef]
- Dosztanyi, Z.; Meszaros, B.; Simon, I. ANCHOR: Web server for predicting protein binding regions in disordered proteins. Bioinformatics 2009, 25, 2745–2746. [Google Scholar] [CrossRef]
- Meszaros, B.; Erdos, G.; Dosztanyi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Arzola, K.; Diaz-Moreno, I.; Cano-Gonzalez, A.; Diaz-Quintana, A.; Velazquez-Campoy, A.; Moreno-Beltran, B.; Lopez-Rivas, A.; De la Rosa, M.A. Structural basis for inhibition of the histone chaperone activity of SET/TAF-Ibeta by cytochrome c. Proc. Natl. Acad. Sci. USA 2015, 112, 9908–9913. [Google Scholar] [CrossRef] [PubMed]
- Ladenson, R.C.; Crimmins, D.L.; Landt, Y.; Ladenson, J.H. Isolation and characterization of a thermally stable recombinant anti-caffeine heavy-chain antibody fragment. Anal. Chem. 2006, 78, 4501–4508. [Google Scholar] [CrossRef]
- Olichon, A.; Schweizer, D.; Muyldermans, S.; de Marco, A. Heating as a rapid purification method for recovering correctly-folded thermotolerant VH and VHH domains. BMC Biotechnol. 2007, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, A.; Sauer-Eriksson, A.E. Heating of proteins as a means of improving crystallization: A successful case study on a highly amyloidogenic triple mutant of human transthyretin. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2007, 63 Pt 8, 695–700. [Google Scholar] [CrossRef]
- Hammond, C.M.; Sundaramoorthy, R.; Larance, M.; Lamond, A.; Stevens, M.A.; El-Mkami, H.; Norman, D.G.; Owen-Hughes, T. The histone chaperone Vps75 forms multiple oligomeric assemblies capable of mediating exchange between histone H3-H4 tetramers and Asf1-H3-H4 complexes. Nucleic Acids Res. 2016, 44, 6157–6172. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E. Crystal contacts as nature’s docking solutions. J. Comput. Chem. 2010, 31, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Meeth, K.; Jiang, E.; Luo, C.; Marmorstein, R. Structure of Vps75 and implications for histone chaperone function. Proc. Natl. Acad. Sci. USA 2008, 105, 12206–12211. [Google Scholar] [CrossRef] [Green Version]
- Franke, D.; Petoukhov, M.V.; Konarev, P.V.; Panjkovich, A.; Tuukkanen, A.; Mertens, H.D.T.; Kikhney, A.G.; Hajizadeh, N.R.; Franklin, J.M.; Jeffries, C.M.; et al. ATSAS 2.8: A comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 2017, 50 Pt 4, 1212–1225. [Google Scholar] [CrossRef]
- Franke, D.; Svergun, D.I. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2009, 42 Pt 2, 342–346. [Google Scholar] [CrossRef]
- Volkov, V.V.; Svergun, D.I. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2003, 36, 860–864. [Google Scholar] [CrossRef]
- Svergun, D.; Barberato, C.; Koch, M.H.J. CRYSOL—A program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 1995, 28, 768–773. [Google Scholar] [CrossRef]
- Panjkovich, A.; Svergun, D.I. Deciphering conformational transitions of proteins by small angle X-ray scattering and normal mode analysis. Phys. Chem. Chem. Phys. 2016, 18, 5707–5719. [Google Scholar] [CrossRef]
- Laskey, R.A.; Honda, B.M.; Mills, A.D.; Finch, J.T. Nucleosomes are assembled by an acidic protein which binds histones and transfers them to DNA. Nature 1978, 275, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Zhu, Y.; Dong, A.; Shen, W.H. Histone H2A/H2B chaperones: From molecules to chromatin-based functions in plant growth and development. Plant J. 2015, 83, 78–95. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Gurrieri, C.; Larabi, A.; Vinayachandran, V.; Patel, N.A.; Yen, K.; Reja, R.; Ebong, I.O.; Schoehn, G.; Robinson, C.V.; Pugh, B.F.; et al. Structural evidence for Nap1-dependent H2A-H2B deposition and nucleosome assembly. EMBO J. 2016, 35, 1465–1482. [Google Scholar] [CrossRef] [PubMed]
- Elsasser, S.J.; D’Arcy, S. Towards a mechanism for histone chaperones. Biochim. Biophys. Acta 2013, 1819, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Are, V.N.; Ghosh, B.; Kumar, A.; Gadre, R.; Makde, R.D. Crystal structure and dynamics of Spt16N-domain of FACT complex from Cicer arietinum. Int. J. Biol. Macromol. 2016, 88, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Muto, S.; Miyamoto, S.; Aizawa, K.; Horikoshi, M.; Nagai, R. Functional interaction of the DNA-binding transcription factor Sp1 through its DNA-binding domain with the histone chaperone TAF-I. J. Biol. Chem. 2003, 278, 28758–28764. [Google Scholar] [CrossRef] [PubMed]
- Magnusdottir, A.; Johansson, I.; Dahlgren, L.G.; Nordlund, P.; Berglund, H. Enabling IMAC purification of low abundance recombinant proteins from E. coli lysates. Nat. Methods 2009, 6, 477–478. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 2, 125–132. [Google Scholar] [CrossRef]
- Vagin, A.; Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 1, 22–25. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67 Pt 4, 235–242. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997, 53 Pt 3, 240–255. [Google Scholar] [CrossRef]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 2012, 68 Pt 4, 352–367. [Google Scholar] [CrossRef]
- Headd, J.J.; Echols, N.; Afonine, P.V.; Grosse-Kunstleve, R.W.; Chen, V.B.; Moriarty, N.W.; Richardson, D.C.; Richardson, J.S.; Adams, P.D. Use of knowledge-based restraints in phenix.refine to improve macromolecular refinement at low resolution. Acta Crystallogr. D Biol. Crystallogr. 2012, 68 Pt 4, 381–390. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 1, 12–21. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Schuck, P. Size-distribution analysis of macromolecules by sedimentation velocity ultra-centrifugation and lamm equation modeling. Biophys. J. 2000, 78, 1606–1619. [Google Scholar] [CrossRef]
- Brown, P.H.; Schuck, P. Macromolecular size-and-shape distributions by sedimentation velocity analytical ultracentrifugation. Biophys. J. 2006, 90, 4651–4661. [Google Scholar] [CrossRef] [PubMed]
- Konarev, P.V.; Volkov, V.V.; Sokolova, A.V.; Koch, M.H.J.; Svergun, D.I. PRIMUS—A Windows-PC based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003, 36, 1277–1282. [Google Scholar] [CrossRef]
- Svergun, D.I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 1992, 25, 495–503. [Google Scholar] [CrossRef]
- Fujii-Nakata, T.; Ishimi, Y.; Okuda, A.; Kikuchi, A. Functional analysis of nucleosome assembly protein, NAP-1. The negatively charged COOH-terminal region is not necessary for the intrinsic assembly activity. J. Biol. Chem. 1992, 267, 20980–20986. [Google Scholar] [PubMed]
- Terakura, S.; Ueno, Y.; Tagami, H.; Kitakura, S.; Machida, C.; Wabiko, H.; Aiba, H.; Otten, L.; Tsukagoshi, H.; Nakamura, K.; et al. An oncoprotein from the plant pathogen agrobacterium has histone chaperone-like activity. Plant Cell 2007, 19, 2855–2865. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | AtNRP2 |
|---|---|
| Data collection and Processing | |
| Beam line | ID30A-3, ESRF, Grenoble, France |
| Detector type | Dectris pixel (Eiger_4M) |
| Wavelength (Å) | 0.9794 |
| Data Collection temperature (K) | 100 K |
| Space Group | H32 |
| α,β,γ a,b,c | 90.00°, 90.00°, 120.00° 123.45 Å, 123.45 Å, 228.71 Å |
| Resolution (Å) | 42.04–3.42 (3.50–3.42) |
| Rmerge (%) | 13% (51%) |
| I/σI | 1.93 |
| CC (1/2) | 0.999 (0.722) |
| Total number of reflections | 105,656 (10,947) |
| Completeness (%) | 99.69 (100.00) |
| Multiplicity | 11.4 |
| Wilson B factor (Å2) | 116.5 |
| Average B, all atoms (Å2) | 90.0 |
| Anisotropy | 0.027 |
| Number of molecules in asymmetric unit | 2 |
| Refinement | |
| Number of unique reflections | 9304 (914) |
| R work/R free (%) | 0.272/0.320 (0.277/0.327) |
| Total number of non-H atoms | 2346 |
| r.m.s. deviations: Bond lengths (Å) Bond angles (o) | 0.004 0.96 |
| Ramachandran plot values (%) Favored/Outliers | 99.04/0.96 |
| Protein Complex | Kd1(nM) | Kd2(µM) | ∆H1 (Kcal/mol) | ∆H2 (Kcal/mol) | −T∆S1 (Kcal/mol) | −T∆S2 (Kcal/mol) | Reduced Chi Square Value |
|---|---|---|---|---|---|---|---|
| NRP1:H2A-H2B | 130 ± 0.0005 | 2.4 ± 0.12 | 8.51 ± 0.107 | 6.00 ± 0.110 | −17.9 | −13.7 | 2.2 × 10−2 |
| NRP2:H2A-H2B | 39.6 ± 0.455 | 1.0 ± 0.0004 | 7.28 ± 0.04 | 1.33 ± 0.06 | −17.4 | −9.51 | 9.8 × 10−3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, A.; Kumar Singh, A.; Chandrakant Bobde, R.; Vasudevan, D. Structural Characterization of Arabidopsis thaliana NAP1-Related Protein 2 (AtNRP2) and Comparison with Its Homolog AtNRP1. Molecules 2019, 24, 2258. https://doi.org/10.3390/molecules24122258
Kumar A, Kumar Singh A, Chandrakant Bobde R, Vasudevan D. Structural Characterization of Arabidopsis thaliana NAP1-Related Protein 2 (AtNRP2) and Comparison with Its Homolog AtNRP1. Molecules. 2019; 24(12):2258. https://doi.org/10.3390/molecules24122258
Chicago/Turabian StyleKumar, Ashish, Ajit Kumar Singh, Ruchir Chandrakant Bobde, and Dileep Vasudevan. 2019. "Structural Characterization of Arabidopsis thaliana NAP1-Related Protein 2 (AtNRP2) and Comparison with Its Homolog AtNRP1" Molecules 24, no. 12: 2258. https://doi.org/10.3390/molecules24122258