Evaluation of In-Batch and In-Flow Synthetic Strategies towards the Stereoselective Synthesis of a Fluorinated Analogue of Retro-Thiorphan

 , and
, and
Abstract
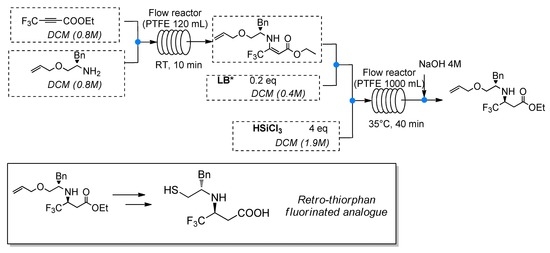
:
1. Introduction
2. Results and Discussion
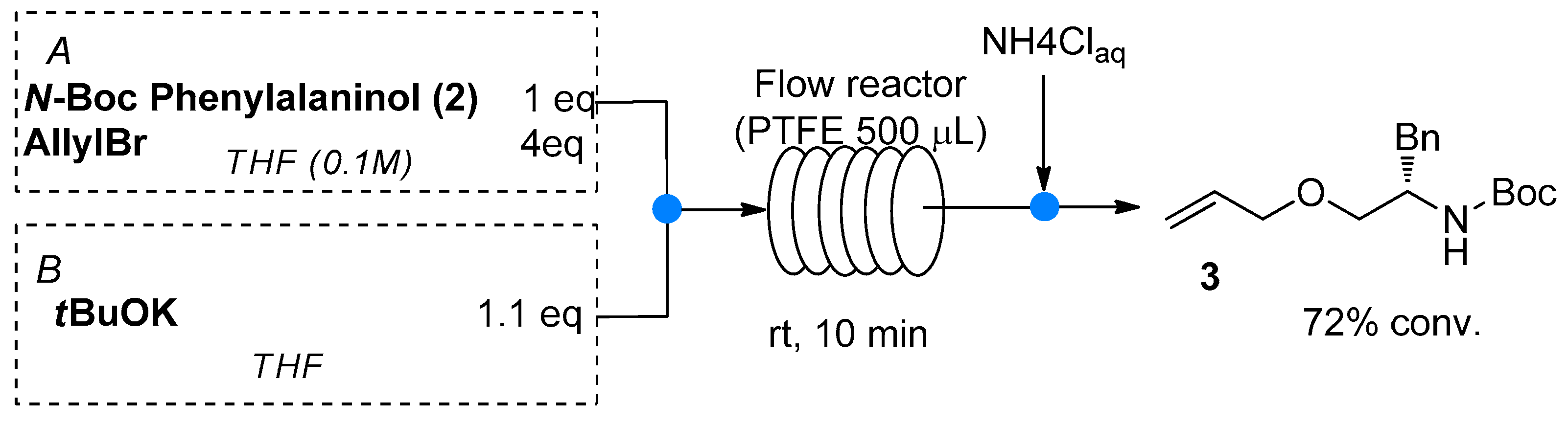
2.1. Synthetic Strategy: In Batch Optimization
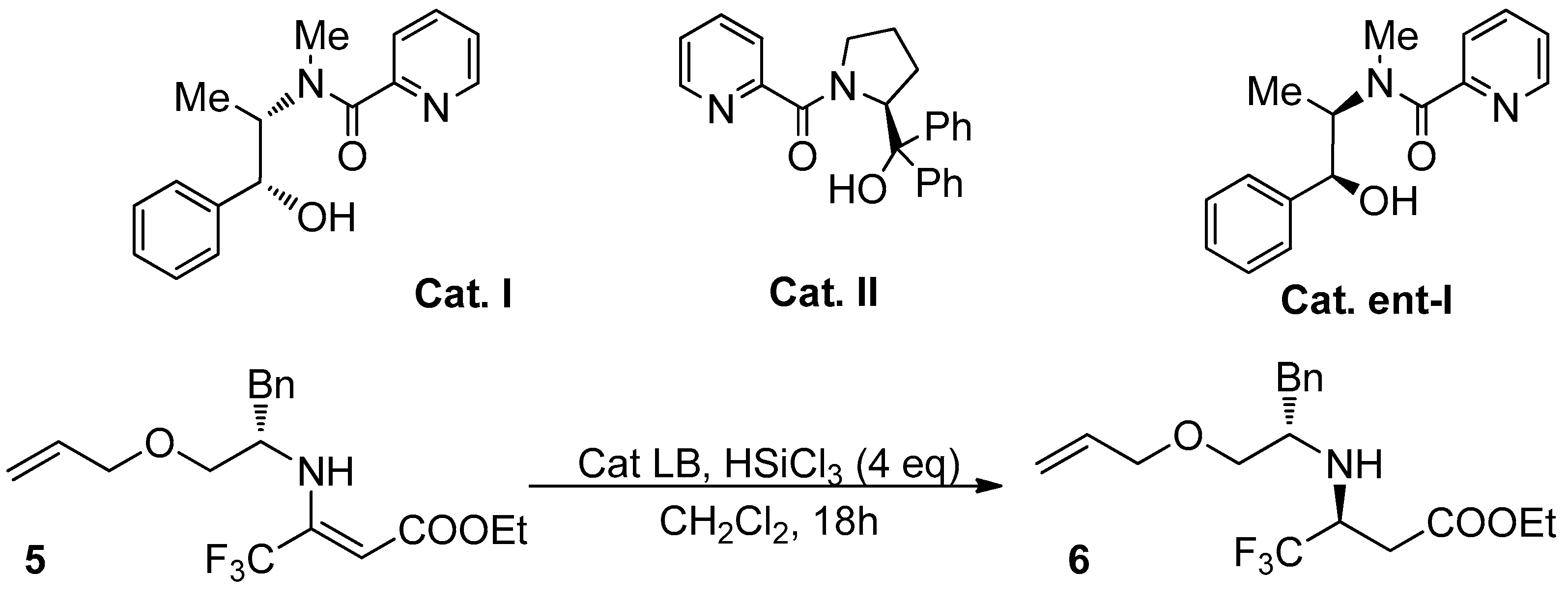
2.2. Catalytic Tests
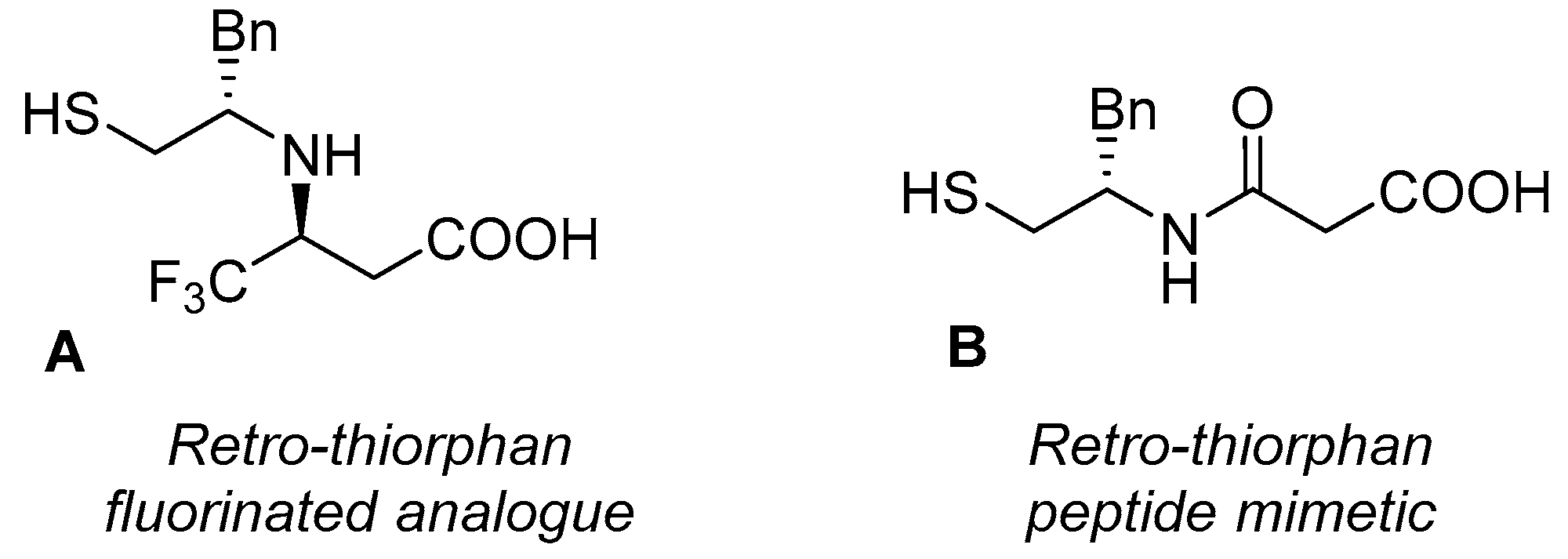
2.3. Derivatization
2.4. Continuous Flow Strategy
3. Conclusions
4. Materials and Methods
4.1. General Procedure for the Stereoselective Catalytic Reduction of Enamine 5 under Batch Conditions
4.2. General Procedure for the Multistep Formation and Stereoselective Catalytic Reduction of Enamine 5 under Flow Conditions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell: Chichester, UK, 2009. [Google Scholar]
- Cahard, D.; Bizet, V. The influence of fluorine in asymmetric catalysis. Chem. Soc. Rev. 2014, 43, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Zanda, M. Trifluoromethyl group: An effective xenobiotic function for peptide backbone modification. New J. Chem. 2004, 28, 1401–1411. [Google Scholar] [CrossRef]
- Begue, J.-P.; Bonnet-Delpon, D.; Crousse, B.; Legros, J. The chemistry of trifluoromethyl imines and related acetals derived from fluoral. Chem. Soc. Rev. 2005, 34, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Huguenot, F.; Brigaud, T. Convenient Asymmetric Synthesis of β-Trifluoromethyl-β-amino Acid, β-Amino Ketones, and γ-Amino Alcohols via Reformatsky and Mannich-Type Reactions from 2-Trifluoromethyl-1,3-oxazolidines. J. Org. Chem. 2006, 71, 2159–2162. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, T.V.; Venugopalan, P. Microwave assisted fluorofunctionalization of phenyl substituted alkenes using selectfluor™. J. Fluor. Chem. 2013, 150, 72–77. [Google Scholar] [CrossRef]
- Molteni, M.; Volonterio, A.; Fossati, G.; Lazzari, P.; Zanda, M. Conjugated additions of amines and β-amino alcohols to trifluorocrotonic acid derivatives: Synthesis of ψ[NHCH(CF3)]-retro-thiorphan. Tetrahedron Lett. 2007, 48, 589–593. [Google Scholar] [CrossRef]
- Genoni, A.; Benaglia, M.; Massolo, E.; Rossi, S. Stereoselective metal-free catalytic synthesis of chiral trifluoromethyl and alkyl amines. Chem. Commun. 2013, 49, 8365–8367. [Google Scholar] [CrossRef] [PubMed]
- Massolo, E.; Benaglia, M.; Orlandi, M.; Rossi, S.; Celentano, G. Enantioselective organocatalytic reduction of β-trifluoromethyl nitroalkenes: An efficient strategy for the synthesis of chiral β-trifluoromethyl amines. Chem. Eur. J. 2015, 21, 3589–3595. [Google Scholar] [CrossRef]
- Abubakar, S.; Benaglia, M.; Rossi, S.; Annunziata, R. Organocatalytic α-trifluoromethylthiolation of silylenol ethers: Batch vs continuous flow reactions. Catal. Today 2018, 308, 94–101. [Google Scholar] [CrossRef]
- Rossi, S.; Puglisi, A.; Raimondi, L.; Benaglia, M. Synthesis of alpha-trifluoromethylthio carbonyl compounds: A survey of the methods for the direct introduction of the SCF3 group on to organic molecules. ChemCatChem 2018, 10, 2717–2733. [Google Scholar] [CrossRef]
- Brenna, D.; Porta, R.; Massolo, E.; Raimondi, L.; Benaglia, M. A new class of low-loading catalysts for a highly enantioselective, metal-free imine reduction of wide general applicability. ChemCatChem 2017, 9, 941–945. [Google Scholar] [CrossRef]
- Review: Rossi, S.; Benaglia, M.; Massolo, E.; Raimondi, L. Organocatalytic strategies for enantioselective metal-free reduction. Catal. Sci. Technol. 2014, 9, 2708–2723. [Google Scholar] [CrossRef]
- Jogula, S.; Dasari, B.; Khatravath, M.; Chandrasekar, G.; Kitambi, S.S.; Arya, P. Building a Macrocyclic Toolbox from C-Linked Carbohydrates Identifies Antiangiogenesis Agents from Zebrafish Assay. Eur. J. Org. Chem. 2013, 23, 5036–5040. [Google Scholar] [CrossRef]
- Guizzetti, S.; Benaglia, M.; Rossi, S. Highly stereoselective metal-free catalytic reduction of imines: An easy entry to enantiomerically pure amines and natural and unnatural α-amino esters. Org. Lett. 2009, 11, 2928–2931. [Google Scholar] [CrossRef] [PubMed]
- Porta, R.; Benaglia, M.; Puglisi, A. Flow chemistry: Recent developments in the synthesis of pharmaceutical products. Org. Process Res. Dev. 2016, 20, 2–25. [Google Scholar] [CrossRef]
- Gutmann, B.; Cantillo, D.; Kappe, C.O. Continuous-flow technology—A tool for the safe manufacturing of active pharmaceutical ingredients. Angew. Chem. Int. Ed. 2015, 54, 6688–6728. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.; Baxendale, I.R.; Ley, S.V. The flow synthesis of heterocycles for natural product and medicinal chemistry applications. Mol. Divers. 2011, 15, 613–630. [Google Scholar] [CrossRef] [PubMed]
- Richard, S.; Prié, G.; Guignard, A.; Thibonnet, J.; Parrain, J.L.; Duchêne, A.; Abarbri, M. A new synthesis of optically active 3-substituted (3S)-3,4-dihydro-5-(perfluoroalkyl)-2H-[1,4]oxazepin-7-ones. Helv. Chim. Acta 2003, 86, 726–732. [Google Scholar] [CrossRef]
- Brenna, D.; Pirola, M.; Raimondi, L.; Burke, A.J.; Benaglia, M. A stereoselective, catalytic strategy for the in-flow synthesis of advanced precursors of Rasagiline and Tamsulosin. Bioorg. Med. Chem. 2017, 25, 6242–6247. [Google Scholar] [CrossRef]
- Review: Jones, S.; Warner, C.J.A. Trichlorosilane mediated asymmetric reductions of the C=N bond. Org. Biomol. Chem. 2012, 10, 2189–2200. [Google Scholar] [CrossRef]
- Zheng, H.; Deng, J.; Lin, W.; Zhang, X. Enantioselective hydrosilylation of ketimines with trichlorosilane promoted by chiral N-picolinoylaminoalcohols. Tetrahedron Lett. 2007, 48, 7934–7937. [Google Scholar] [CrossRef]
- Malkov, A.V.; Mariani, A.; MacDougal, K.N.; Kočovský, P. Role of noncovalent interactions in enantioselective reduction of aromatic ketimines with trichlorosilane. Org. Lett. 2004, 6, 2253–2256. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Wang, C.; Chen, L.; Wu, X.; Zhou, L.; Sun, J. Chiral Lewis Base-catalyzed, enantioselective reduction of unprotected β-enamino esters with trichlorosilane. Adv. Synth. Catal. 2016, 358, 1042–1048. [Google Scholar] [CrossRef]
- Chelouan, A.; Recio, R.; Borrego, L.; Álvarez, E.; Khiar, N.; Fernández, I. Sulfinamide Phosphinates as chiral catalysts for the enantioselective organocatalytic reduction of imines. Org. Lett. 2016, 18, 3258–3261. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Chen, X.; Zheng, Y.; Xue, Z.; Shu, C.; Yuan, W.; Zhang, X. Asymmetric Synthesis of anti-β-Amino-α-Hydroxy Esters via Dynamic Kinetic Resolution of β-Amino-α-Keto Esters. Angew. Chem. Int. Ed. 2011, 50, 7304–7307. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of all of the prepared compounds are available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Lewis Base | Cat. eq | T (°C) | d.r.1 | Yield (%) 2 |
|---|---|---|---|---|---|
| 1 3 | DMF | 5 | 0 | 67:34 | 22 |
| 2 | Cat. I | 0.2 | 0 to rt | 34:66 | 40 |
| 3 | Cat. II | 0.2 | 0 to rt | 81:19 | 38 |
| 4 | Cat. ent-I | 0.2 | 0 to rt | 83:17 | 70 |
| 5 | Cat. ent-I | 0.2 | -10 | 95:5 | 60 |
| 6 | Cat. ent-I | 0.1 | -10 | 95:5 | 53 |
| 7 | Cat. ent-I | 0.1 | -20 | 96:4 | 37 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pirola, M.; Puglisi, A.; Raimondi, L.; Forni, A.; Benaglia, M. Evaluation of In-Batch and In-Flow Synthetic Strategies towards the Stereoselective Synthesis of a Fluorinated Analogue of Retro-Thiorphan. Molecules 2019, 24, 2260. https://doi.org/10.3390/molecules24122260
Pirola M, Puglisi A, Raimondi L, Forni A, Benaglia M. Evaluation of In-Batch and In-Flow Synthetic Strategies towards the Stereoselective Synthesis of a Fluorinated Analogue of Retro-Thiorphan. Molecules. 2019; 24(12):2260. https://doi.org/10.3390/molecules24122260
Chicago/Turabian StylePirola, Margherita, Alessandra Puglisi, Laura Raimondi, Alessandra Forni, and Maurizio Benaglia. 2019. "Evaluation of In-Batch and In-Flow Synthetic Strategies towards the Stereoselective Synthesis of a Fluorinated Analogue of Retro-Thiorphan" Molecules 24, no. 12: 2260. https://doi.org/10.3390/molecules24122260