
Synthesis, Anti-Cancer and Anti-Migratory Evaluation of 3,6-Dibromocarbazole and 5-Bromoindole Derivatives

 and
and 
Abstract
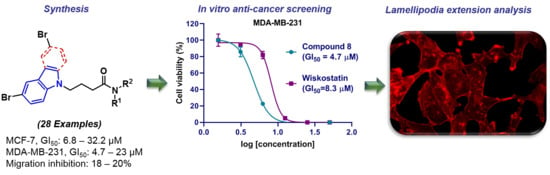
:
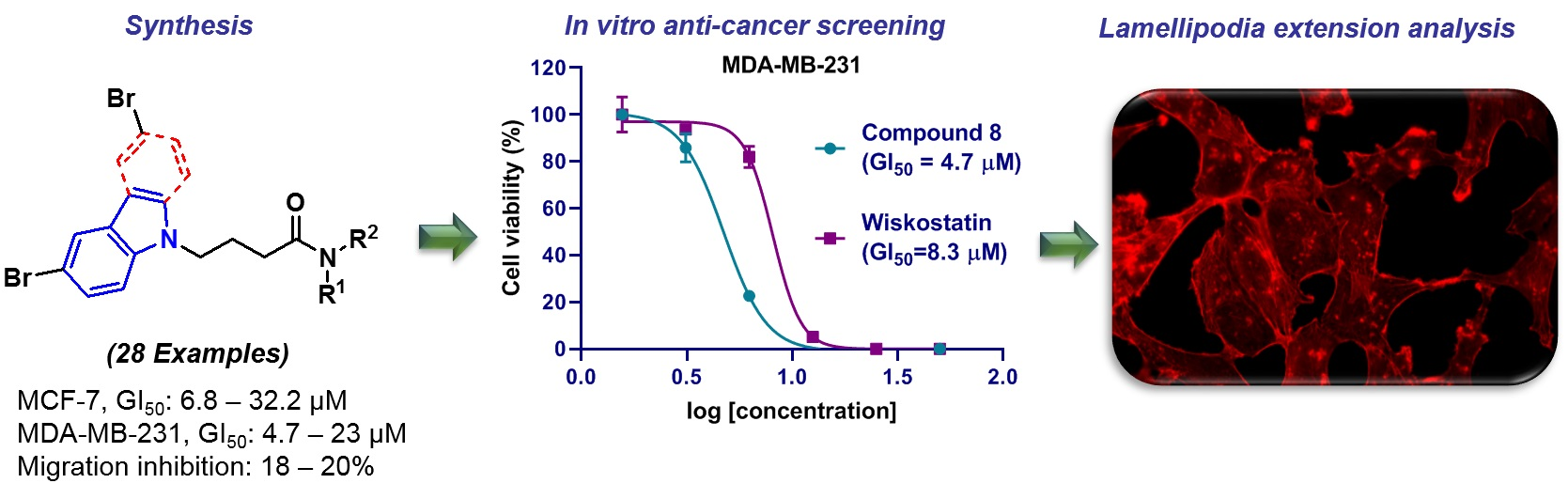
1. Introduction
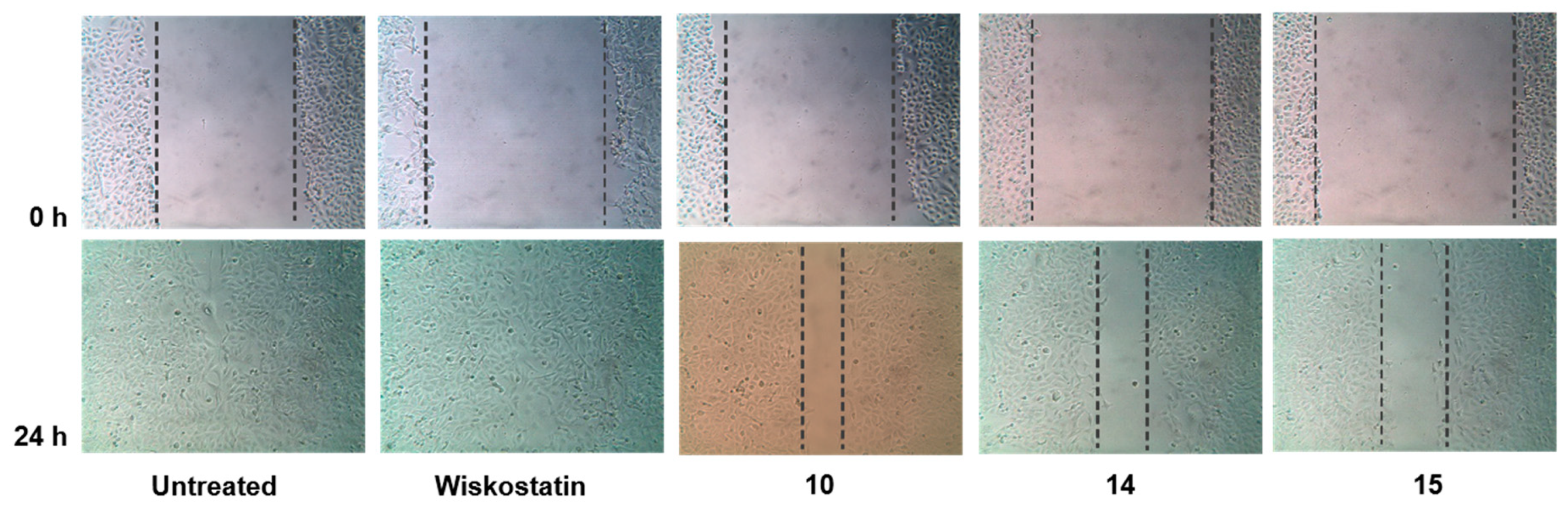
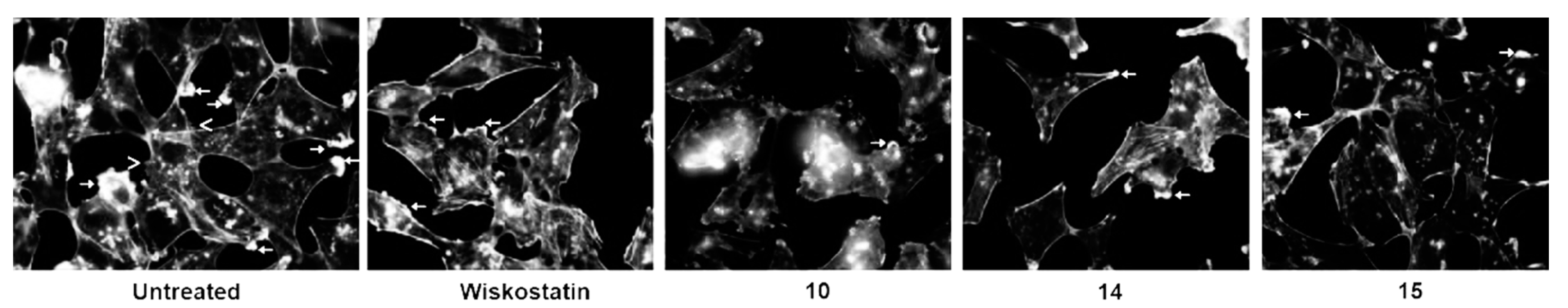
2. Results and Discussion
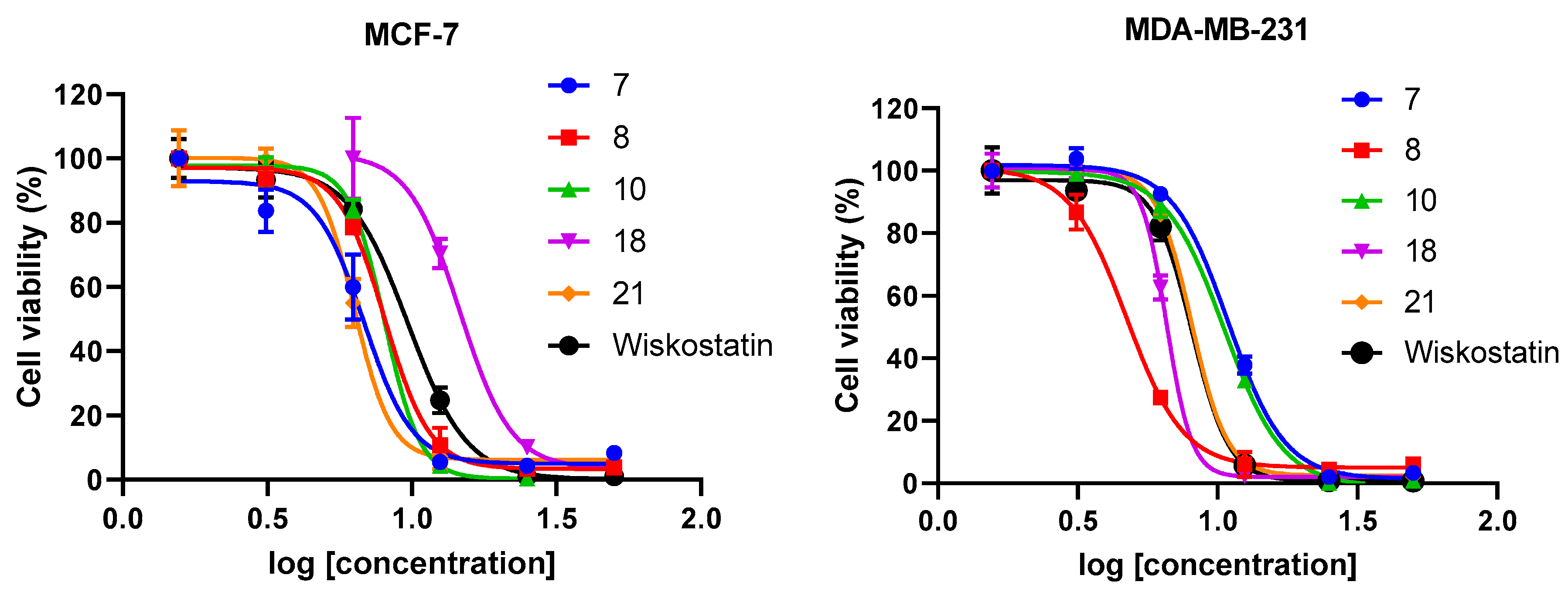
2.1. N-Alkyl-3,6-Dibromocarbazole Derivatives
2.2. N-alkyl-5-Bromoindole Derivatives
3. Materials and Methods
3.1. General Methods
3.2. Synthesis Methods
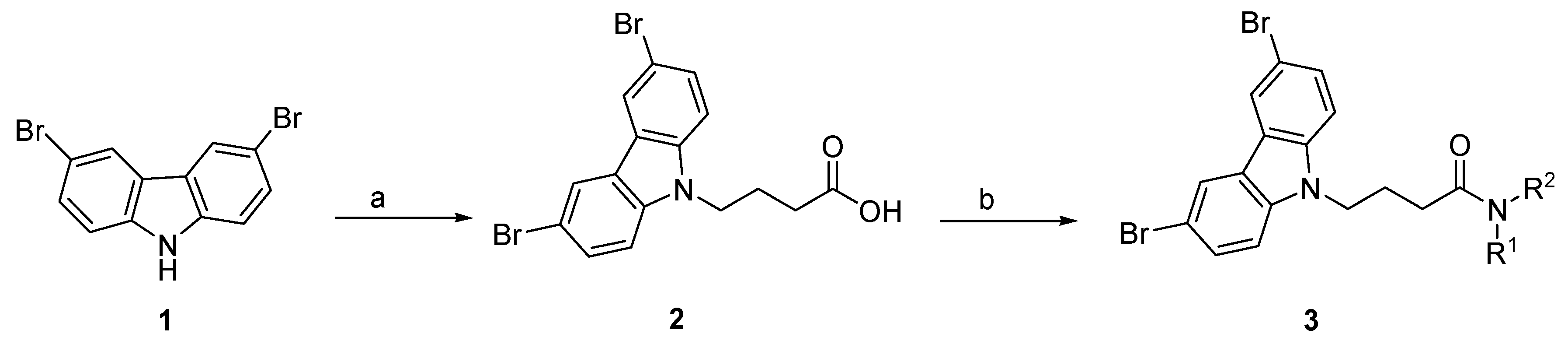
3.2.1. General Procedure for the Synthesis of 4-(3,6-Dibromo-Carbazol-9-yl)-Butyric Acid (2)
3.2.2. Synthesis of 3,6-Dibromocarbazole-4-butyramide Derivatives (4–27)
3.2.3. General Procedure for the Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-N-(2-Morpholin-4-Ylethyl)Butyramide (4), and for Compounds 5–27
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-N-(2-methoxyethyl)butyramide (5)
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-N-(3-imidazol-1-yl-propyl)butyramide (6)
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-N-(2-piperidin-1-ylethyl)butyramide (7)
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-N-(2-piperazin-1-ylethyl)butyramide (8)
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-N-(3-morpholin-4-ylpropyl)butyramide (9)
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-N-(4-diethylamino-1-methylbutyl)butyramide (10)
Synthesis of 4-[4-(3,6-Dibromocarbazol-9-yl)-butyrylamino]piperidine-1-carboxylic acid ethyl ester (11)
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-1-morpholin-4-ylbutan-1-one (12)
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-1-thiomorpholin-4-ylbutan-1-one (13)
Synthesis of 1-[4-(3,6-Dibromocarbazol-9-yl)butyryl]piperidine-4-carbonitrile (14)
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-1-(4-phenylpiperazin-1-yl)butan-1-one (15)
Synthesis of 1-(4-Acetylpiperazin-1-yl)-4-(3,6-dibromocarbazol-9-yl)butan-1-one (16)
Synthesis of 4-[4-(3,6-Dibromocarbazol-9-yl)butyryl]piperazine-1-carboxylic acid tert-butyl ester (17)
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-1-piperazin-1-ylbutan-1-one (18)
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-1-[4-(2-morpholin-4-ylethyl)piperazin-1-yl]butan-1-one (19)
Synthesis of 2-{4-[4-(3,6-Dibromocarbazol-9-yl)-butyryl]piperazin-1-yl}-N-isopropylacetamide (20)
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-1-[4-(2-dimethylaminoethyl)piperazin-1-yl]butan-1-one (21)
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-N-(4-morpholin-4-ylphenyl)butyramide (22)
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-N-(3,4,5-trimethoxyphenyl)butyramide (23)
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-N-quinolin-3-ylbutyramide (24)
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-N-(1H-indol-6-yl)butyramide (25)
Synthesis of N-(3H-Benzoimidazol-5-yl)-4-(3,6-dibromocarbazol-9-yl)butyramide (26)
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-N-(4-methoxyphenyl)butyramide (27)
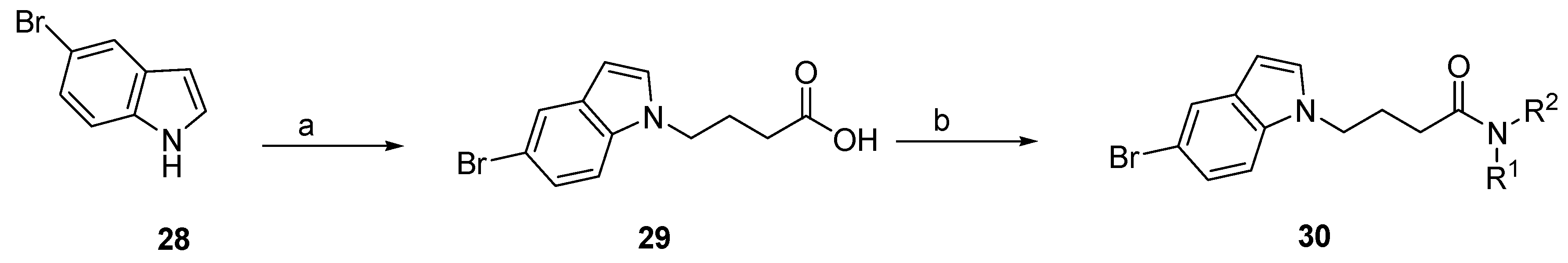
3.2.4. Synthesis of N-Alkyl-5-Bromoindole Derivatives (31–34)
3.2.5. General Procedure for the Synthesis of 4-(5-Bromoindol-1-yl)Butyric Acid (29)
3.2.6. General Procedure for the Synthesis of 4-(5-Bromoindol-1-yl)-N-(3-Morpholin-4-ylpropyl)Butyramide (31), and for Compounds 32–34
Synthesis of 1-[4-(5-Bromoindol-1-yl)butyryl]piperidine-4-carbonitrile (32)
Synthesis of 4-(5-Bromoindol-1-yl)-N-(3-imidazol-1-yl-propyl)butyramide (33)
Synthesis of 4-(5-Bromoindol-1-yl)-N-(2-piperidin-1-ylethyl)butyramide (34)
3.3. Biological Evaluation
3.3.1. Cell Culture
3.3.2. Sulforhodamine B (SRB) Assay
3.3.3. Wound Healing Assay (Scratch Method) Using MDA-MB-231 Cancer Cells
3.3.4. Actin Polymerization Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gupta, G.P.; Massagué, J. Cancer Metastasis: Building a Framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, C.; Mojares, E.; Hernández, A.D.R. Role of Extracellular Matrix in Development and Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3028. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.T. Proteolytic activity of specialized surface protrusions formed at rosette contact sites of transformed cells. J. Exp. Zool. 1989, 251, 167–185. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D.; Borisy, G.G. Cellular Motility Driven by Assembly and Disassembly of Actin Filaments. Cell 2003, 113, 549. [Google Scholar] [CrossRef] [Green Version]
- Wertheimer, E.; Gutierrez-Uzquiza, A.; Rosemblit, C.; Lopez-Haber, C.; Sosa, M.S.; Kazanietz, M.G. Rac signaling in breast cancer: A tale of GEFs and GAPs. Cell. Signal. 2012, 24, 353–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muise, A.M.; Walters, T.; Xu, W.; Shen-Tu, G.; Guo, C.H.; Fattouh, R.; Lam, G.Y.; Wolters, V.M.; Bennitz, J.; Van Limbergen, J.; et al. Single nucleotide polymorphisms that increase expression of the guanosine triphosphatase RAC1 are associated with ulcerative colitis. Gastroenterology 2011, 141, 633–641. [Google Scholar]
- (a) Worthylake, R.A.; Lemoine, S.; Watson, J.M.; Burridge, K. RhoA is required for monocyte tail retraction during transendothelial migration. J. Cell Biol. 2001, 154, 147–160, (b) Takenawa, T.; Miki, H. WASP and WAVE family proteins: key molecules for rapid rearrangement of cortical actin filaments and cell movement. J. Cell Sci. 2001, 114, 1801–1809. [Google Scholar] [CrossRef] [Green Version]
- Rohatgi, R.; Ma, L.; Miki, H.; Lopez, M.; Kirchhausen, T.; Takenawa, T.; Kirschner, M.W. The Interaction between N-WASP and the Arp2/3 Complex Links Cdc42-Dependent Signals to Actin Assembly. Cell 1999, 97, 221–231. [Google Scholar] [CrossRef] [Green Version]
- Vlaar, C.P.; Castillo-Pichardo, L.; Medina, J.I.; Marrero-Serra, C.M.; Velez, E.; Ramos, Z.; Hernández, E. Design, synthesis and biological evaluation of new carbazole derivatives as anti-cancer and anti-migratory agents. Biorganic. Med. Chem. 2018, 26, 884–890. [Google Scholar] [CrossRef]
- Montalvo-Ortiz, B.L.; Castillo-Pichardo, L.; Hernández, E.; Humphries-Bickley, T.; De La Mota-Peynado, A.; Cubano, L.A.; Vlaar, C.P.; Dharmawardhane, S. Characterization of EHop-016, novel small molecule inhibitor of Rac GTPase. J. Biol. Chem. 2012, 287, 13228–13238. [Google Scholar] [CrossRef]
- Castillo-Pichardo, L.; Humphries-Bickley, T.; De La Parra, C.; Forestier-Roman, I.; Martinez-Ferrer, M.; Hernandez, E.; Vlaar, C.; Ferrer-Acosta, Y.; Washington, A.V.; Cubano, L.A.; et al. The Rac inhibitor EHop-016 inhibits mammary tumor growth and metastasis in a nude mouse model. Transl. Oncol. 2014, 7, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Dharmawardhane, S.; Hernandez, E.; Vlaar, C. Development of EHop-016: a small molecule inhibitor of Rac. Enzymes 2013, 33, 117–146. [Google Scholar] [PubMed]
- Humphries-Bickley, T.; Castillo-Pichardo, L.; Corujo-Carro, F.; Duconge, J.; Hernandez-O’Farrill, E.; Vlaar, C.; Rodriguez-Orengo, J.F.; Cubano, L.; Dharmawardhane, S. Pharmacokinetics of Rac inhibitor EHop-016 in mice by ultra-performance liquid chromatography tandem mass spectrometry. J. Chromatogr. B 2015, 981, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Głuszyńska, A. Biological potential of carbazole derivatives. Eur. J. Med. Chem. 2015, 94, 405–426. [Google Scholar] [CrossRef] [PubMed]
- Guillonneau, C.; Pierré, A.; Charton, Y.; Guilbaud, N.; Kraus-Berthier, L.; Léonce, S.; Michel, A.; Bisagni, E.; Atassi, G. Synthesis of 9-O-substituted derivatives of 9-hydroxy-5,6-dimethyl-6H-pyrido[4,3-b]carbazole-1-carboxylic acid (2-(Dimethylamino)-ethyl) amide and their 10-and 11-methyl analogues with improved antitumor activity. J. Med. Chem. 1999, 42, 2191–2203. [Google Scholar] [CrossRef] [PubMed]
- Saturnino, C.; Iacopetta, D.; Sinicropi, M.; Rosano, C.; Caruso, A.; Caporale, A.; Marra, N.; Marengo, B.; Pronzato, M.; Parisi, O.; et al. N-alkyl carbazole derivatives as new tools for Alzheimer’s disease: preliminary studies. Molecules 2014, 19, 9307–9317. [Google Scholar] [CrossRef] [PubMed]
- Bandgar, B.P.; Adsul, L.K.; Chavan, H.V.; Jalde, S.S.; Shringare, S.N.; Shaikh, R.; Meshram, R.J.; Gacche, R.N.; Masand, V. Synthesis, biological evaluation, and docking studies of 3-(substituted)-aryl-5-(9-methyl-3-carbazole)-1H-2-pyrazolines as potent anti-inflammatory and antioxidant agents. Bioorganic Med. Chem. Lett. 2012, 22, 5839–5844. [Google Scholar] [CrossRef] [PubMed]
- Biamonte, M.A.; Wanner, J.; Le Roch, K.G. Recent advances in malaria drug discovery. Bioorganic Med. Chem. Lett. 2013, 23, 2829–2843. [Google Scholar] [CrossRef] [Green Version]
- Caruso, A.; Voisin-Chiret, A.S.; Lancelot, J.-C.; Sinicropi, M.S.; Garofalo, A.; Rault, S. Efficient and Simple Synthesis of 6-Aryl-1,4-dimethyl-9H-carbazoles. Molecules 2008, 13, 1312–1320. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, M.; Ghosh, N.; Harigaya, Y. A clay-mediated, regioselective synthesis of 2-(aryl/alkyl) amino-thiazolo [4, 5-c] carbazoles. Tetrahedron Lett. 2004, 45, 4955–4957. [Google Scholar] [CrossRef]
- Caruso, A.; Sinicropi, M.S.; Lancelot, J.C.; El-Kashef, H.; Saturnino, C.; Aubert, G.; Ballandonne, C.; Lesnard, A.; Cresteil, T.; Dallemagne, P.; et al. Synthesis and evaluation of cytotoxic activities of new guanidines derived from carbazoles. Bioorganic Med. Chem. Lett. 2014, 24, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Iacopetta, D.; Rosano, C.; Puoci, F.; Parisi, O.I.; Saturnino, C.; Caruso, A.; Longo, P.; Ceramella, J.; Malzert-Fréon, A.; Dallemagne, P.; et al. Multifaceted properties of 1, 4-dimethylcarbazoles: Focus on trimethoxybenzamide and trimethoxyphenylurea derivatives as novel human topoisomerase II inhibitors. Eur. J. Pharm. Sci. 2017, 96, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Saturnino, C.; Palladino, C.; Napoli, M.; Sinicropi, M.S.; Botta, A.; Sala, M.; Carcereri de Prati, A.; Novellino, E.; Suzuki, H. Synthesis and biological evaluation of new N-alkylcarbazole derivatives as STAT3 inhibitors: Preliminary study. Eur. J. Pharm. Sci. 2013, 60, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Rizza, P.; Pellegrino, M.; Caruso, A.; Iacopetta, D.; Sinicropi, M.S.; Rault, S.; Lancelot, J.C.; El-Kashef, H.; Lesnard, A.; Rochais, C.; et al. 3-(Dipropylamino)-5-hydroxybenzofuro [2, 3-f] quinazolin-1 (2H)-one (DPA-HBFQ-1) plays an inhibitory role on breast cancer cell growth and progression. Eur. J. Pharm. Sci. 2016, 107, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Issa, S.; Walchshofer, N.; Kassab, I.; Termoss, H.; Chamat, S.; Geahchan, A.; Bouaziz, Z. Synthesis and antiproliferative activity of oxazinocarbazole and N,N-bis(carbazolylmethyl)amine derivatives. Eur. J. Med. Chem. 2010, 45, 2567–2577. [Google Scholar] [CrossRef] [PubMed]
- Danish, I.A.; Prasad, K.J.R. A one-pot synthesis of 1, 2, 4, 5-tetraazaspiro [5.5]-6, 7, 8, 9-tetrahydrocarbazol-3-thiones and their antibacterial activities. Indian J. Heterocycl. Chemstry 2004, 14, 19–22. [Google Scholar]
- Indumathi, T.; Fronczek, F.R.; Prasad, K.R. Synthesis of 2-amino-8-chloro-4-phenyl-5, 11-dihydro-6H-pyrido[2,3-a]carbazole-3-carbonitrile: Structural and biological evaluation. J. Mol. Struct. 2012, 1016, 134–139. [Google Scholar] [CrossRef]
- Kantevari, S.; Yempala, T.; Surineni, G.; Sridhar, B.; Yogeeswari, P.; Sriram, D. Synthesis and antitubercular evaluation of novel dibenzo[b,d]furan and 9-methyl-9H-carbazole derived hexahydro-2H-pyrano[3,2-c]quinolines via Povarov reaction. Eur. J. Med. Chem. 2011, 46, 4827–4833. [Google Scholar] [CrossRef] [PubMed]
- Woon, K.L.; Ariffin, A.; Ho, K.W.; Chen, S.-A. Effect of conjugation and aromaticity of 3,6 di-substituted carbazoles on triplet energy and the implication of triplet energy in multiple-cyclic aromatic compounds. RSC Adv. 2018, 8, 9850–9857. [Google Scholar] [CrossRef] [Green Version]
- Bashir, M.; Bano, A.; Ijaz, A.S.; Chaudhary, B.A. Recent Developments and Biological Activities of N-Substituted Carbazole Derivatives: A Review. Molecules 2015, 20, 13496–13517. [Google Scholar] [CrossRef] [Green Version]
- Mac Millan, K.S.; Naidoo, J.; Liang, J.; Melito, L.; Williams, L.S.; Morlock, L.; Huntington, P.J.; Estill, S.J.; Longood, J.; Becker, G.; et al. Development of Proneurogenic, Neuroprotective Small Molecules. J. Am. Chem. Soc. 2011, 133, 1428–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molette, J.; Routier, J.; Abla, N.; Besson, D.; Bombrun, A.; Brun, R.; Burt, H.; Georgi, K.; Kaiser, M.; Nwaka, S.; et al. Identification and Optimization of an Aminoalcohol-Carbazole Series with Antimalarial Properties. ACS Med. Chem. Lett. 2013, 4, 1037–1041. [Google Scholar]
- Saturnino, C.; Caruso, A.; Iacopetta, D.; Rosano, C.; Caramella, J.; Muia, N.; Mariconda, A.; Grazia Bonomo, M.; Ponassi, M.; Rosace, G.; et al. Inhibition of Human Topoisomerase II by N,N,N-Trimethylethanammonium Iodide Alkylcarbazole Derivatives. Chem. Med. Chem. 2018, 13, 2635–2643. [Google Scholar] [CrossRef] [PubMed]
- Caruso, A.; Iacopetta, D.; Pouci, F.; Cappello, A.R.; Saturnino, C.; Sinicropi, M.S. Carbazole derivatives: A promising scenario for breast cancer treatment. Mini Rev. Med. Chem. 2016, 16, 630. [Google Scholar] [CrossRef] [PubMed]
- Bombrun, A.; Gerber, P.; Casi, G.; Terradillos, O.; Antonsson, B.; Halazy, S. 3,6-dibromocarbazole piperazine derivatives of 2-propanol as first inhibitors of cytochrome c release via Bax channel modulation. J. Med. Chem. 2003, 46, 4365–4368. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tangadanchu, V.K.R.; Cheng, Y.; Yang, R.G.; Lin, J.M.; Zhou, C.H. Potential Antimicrobial Isopropanol-Conjugated Carbazole Azoles as Dual Targeting Inhibitors of Enterococcus faecalis. ACS Med. Chem. Lett. 2018, 9, 244–249. [Google Scholar] [CrossRef]
- Pieper, A.A.; Xie, S.; Capota, E.; Estill, S.J.; Zhong, J.; Long, J.M.; Becker, G.L.; Huntington, P.; Goldman, S.E.; Shen, C.-H.; et al. Discovery of a proneurogenic, neuroprotective chemical. Cell 2010, 142, 39. [Google Scholar] [CrossRef]
- Peterson, J.R.; Bickford, L.C.; Morgan, D.; Kim, A.S.; Ouerfelli, O.; Kirschner, M.W.; Rosen, M.K. Chemical inhibition of N-WASP by stabilization of a native autoinhibited conformation. Nat. Struct. Mol. Boil. 2004, 11, 747–755. [Google Scholar] [CrossRef]
- Guerriero, C.J.; Weisz, O.A. N-WASP inhibitor wiskostatin non-selectively perturbs membrane transport by decreasing cellular ATP levels. Am. J. Physiol. Cell Physiol. 2007, 292, C1562–C1566. [Google Scholar] [CrossRef]
- Orellana, E.A.; Kasinski, A.L. Sulforhodamine B (SRB) Assay in Cell Culture to Investigate Cell Proliferation. Bio-protocol 2016, 6, e1984. [Google Scholar] [CrossRef]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.F.; Sahai, E. The actin cytoskeleton in cancer cell motility. Clin. Exp. Metastasis 2009, 26, 273. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Boil. 2006, 16, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Takenawa, T.; Suetsugu, S. The WASP–WAVE protein network: Connecting the membrane to the cytoskeleton. Nat. Rev. Mol. Cell Boil. 2007, 8, 37–48. [Google Scholar] [CrossRef]
- Frugtniet, B.; Jiang, W.G.; Martin, T.A.; Martin, T. Role of the WASP and WAVE family proteins in breast cancer invasion and metastasis. Breast Cancer: Targets Ther. 2015, 7, 99–109. [Google Scholar] [Green Version]
Sample Availability: Not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. |  R = | GI50 (µM)a | Migration (%)b,c | |
|---|---|---|---|---|
| MCF-7 | MDA-MB-231 | |||
| 4 |  | >50 | >50 | 99 ± 6.03 |
| 5 |  | >50 | >50 | 99 ± 5.98 |
| 6 |  | 16.8 | 16 | 94 ± 3.93 |
| 7 |  | 6.8 | 10 | 99 ± 0.03 (at 2 μM) |
| 8 |  | 8 | 4.7 | 97 ± 4.90 (at 1 μM) |
| 9 |  | 13.4 | 15.4 | 99 ± 1.58 (at 3.1 μM) |
| 10 |  | 8.1 | 10.5 | 87 ± 4.65 (at 2.1 μM) |
| 11 |  | >50 | >50 | 99 ± 0.02 |
| 12 |  | >50 | >50 | 97 ± 6.41 |
| 13 |  | >50 | 25 | 99 ± 0.03 |
| 14 |  | 11.8 | 16.7 | 81 ± 8.96 |
| 15 |  | >50 | >50 | 80 ± 5.87 |
| 16 |  | 18.2 | 23 | 82 ± 7.52 |
| 17 |  | 17.5 | >50 | 99 ± 2.09 |
| 18 |  | 7.5 | 6.7 | 99 ± 0.02 |
| 19 |  | 12.4 | >50 | 99 ± 0.97 |
| 20 |  | 9.1 | 13.4 | 99 ± 0.05 (at 2.7 μM) |
| 21 |  | 6.5 | 8 | 99 ± 0.48 (at 1.6 μM) |
| 22 |  | >50 | >50 | 97 ± 4.19 |
| 23 |  | >50 | 19 | 90 ± 6.60 |
| 24 |  | 32.2 | >50 | 82 ± 5.19 |
| 25 |  | >50 | >50 | 96 ± 4.69 |
| 26 |  | >50 | >50 | 99 ± 6.52 |
| 27 |  | >50 | >50 | 99 ± 0.05 |
| Wiskostatin | 9.7 | 8.3 | 95 ± 6.79 (at 2 μM) | |
| Comp. |  R = | GI50 (µM)a | Migration (%)b,c | |
|---|---|---|---|---|
| MCF-7 | MDA-MB-231 | |||
| 31 |  | >50 | >50 | 99 ± 0.05 |
| 32 |  | >50 | >50 | 99 ± 7.78 |
| 33 |  | >50 | >50 | 99 ± 0.02 |
| 34 |  | 18.4 | >50 | 99 ± 6.60 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butler-Fernández, K.M.; Ramos, Z.; Francis-Malavé, A.M.; Bloom, J.; Dharmawardhane, S.; Hernández, E. Synthesis, Anti-Cancer and Anti-Migratory Evaluation of 3,6-Dibromocarbazole and 5-Bromoindole Derivatives. Molecules 2019, 24, 2686. https://doi.org/10.3390/molecules24152686
Butler-Fernández KM, Ramos Z, Francis-Malavé AM, Bloom J, Dharmawardhane S, Hernández E. Synthesis, Anti-Cancer and Anti-Migratory Evaluation of 3,6-Dibromocarbazole and 5-Bromoindole Derivatives. Molecules. 2019; 24(15):2686. https://doi.org/10.3390/molecules24152686
Chicago/Turabian StyleButler-Fernández, Krystal M., Zulma Ramos, Adela M. Francis-Malavé, Joseph Bloom, Suranganie Dharmawardhane, and Eliud Hernández. 2019. "Synthesis, Anti-Cancer and Anti-Migratory Evaluation of 3,6-Dibromocarbazole and 5-Bromoindole Derivatives" Molecules 24, no. 15: 2686. https://doi.org/10.3390/molecules24152686
APA StyleButler-Fernández, K. M., Ramos, Z., Francis-Malavé, A. M., Bloom, J., Dharmawardhane, S., & Hernández, E. (2019). Synthesis, Anti-Cancer and Anti-Migratory Evaluation of 3,6-Dibromocarbazole and 5-Bromoindole Derivatives. Molecules, 24(15), 2686. https://doi.org/10.3390/molecules24152686