
Comparison of Data Fusion Methods as Consensus Scores for Ensemble Docking



Abstract
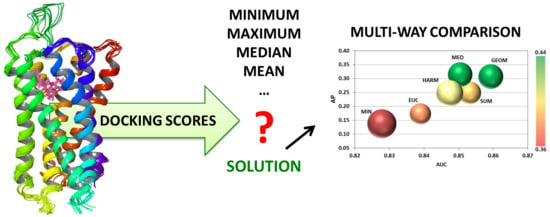
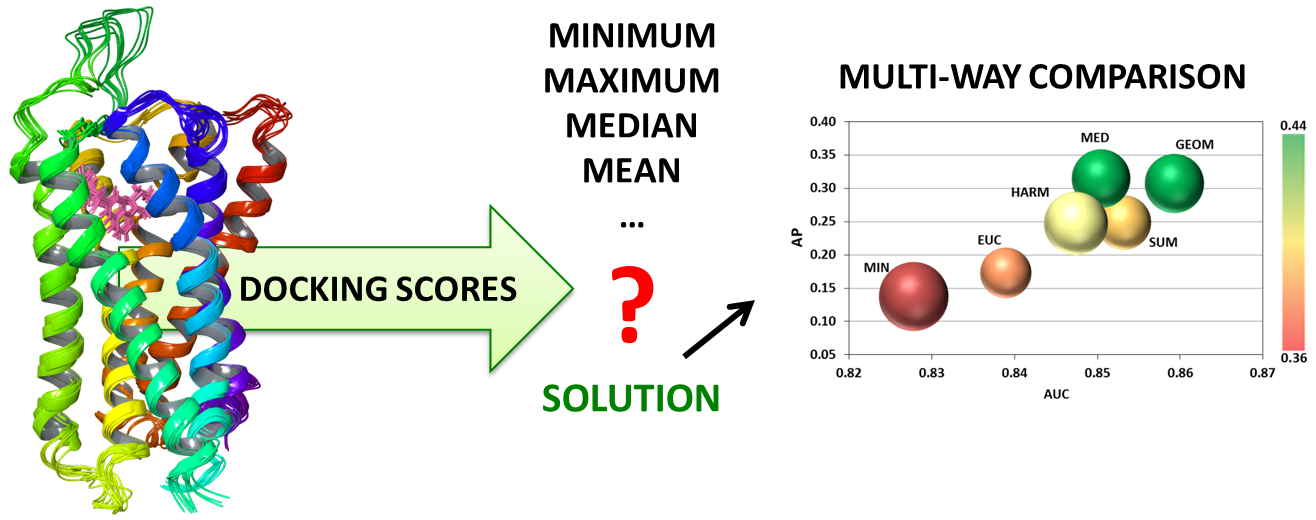
1. Introduction
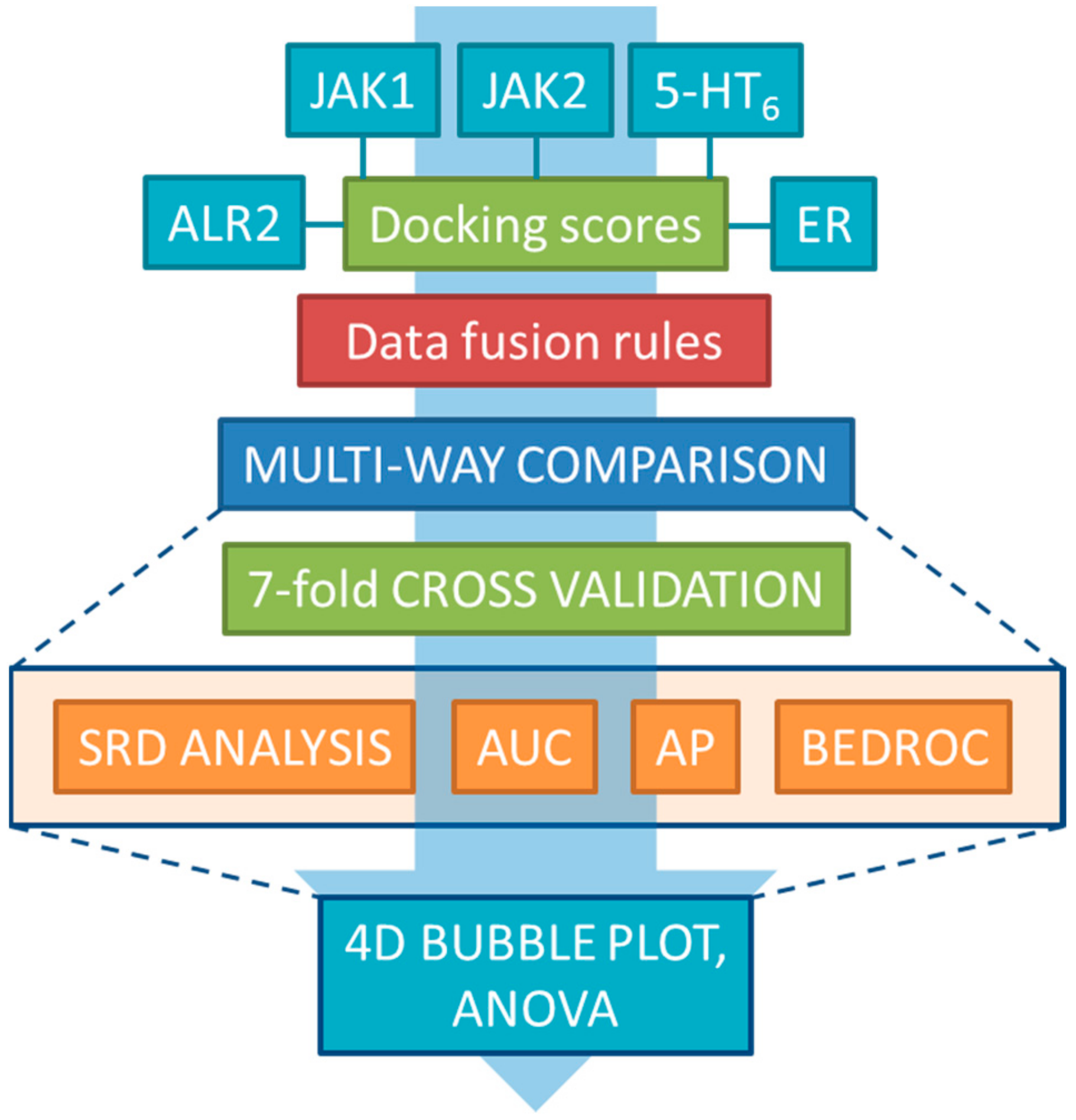
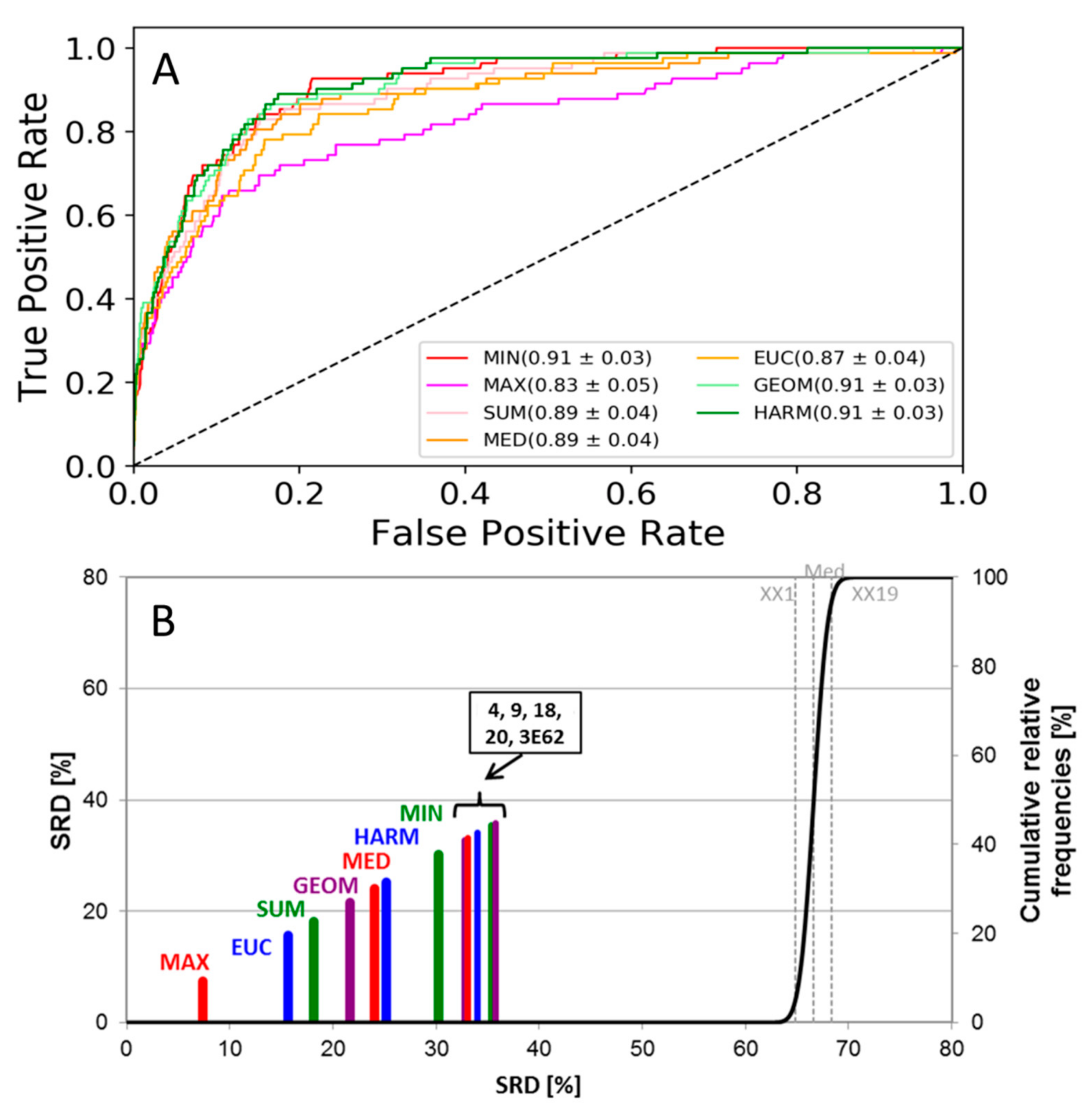
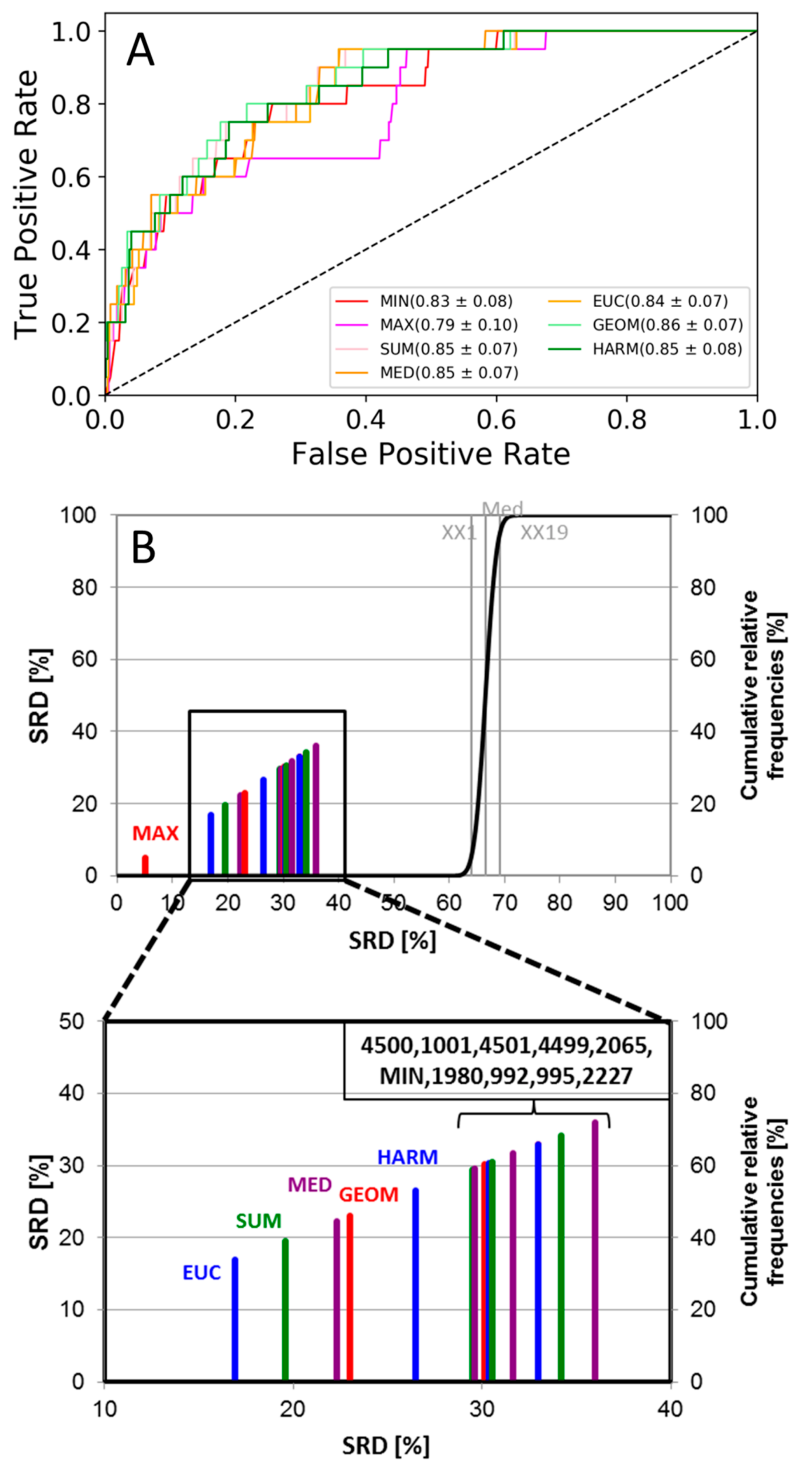
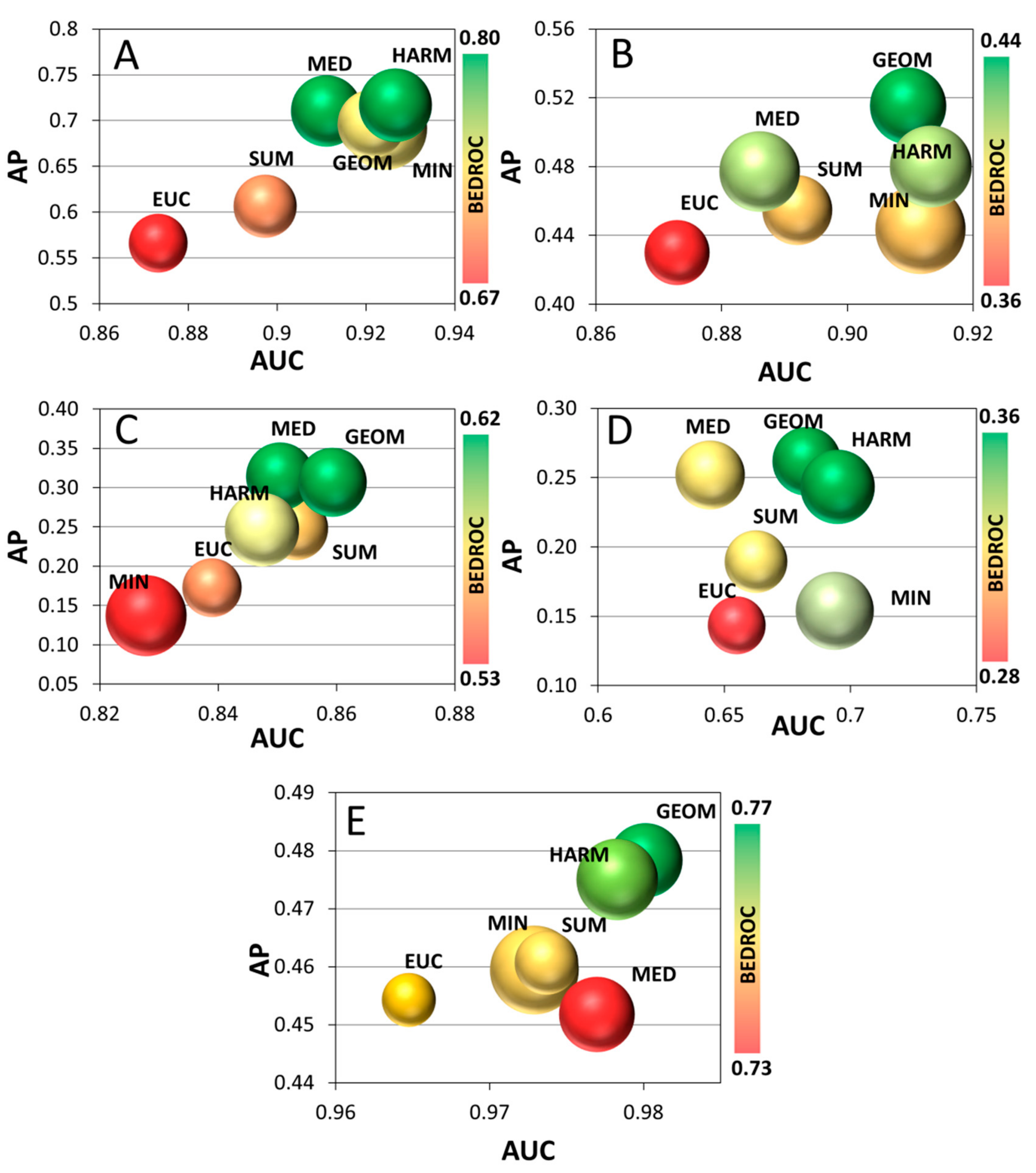
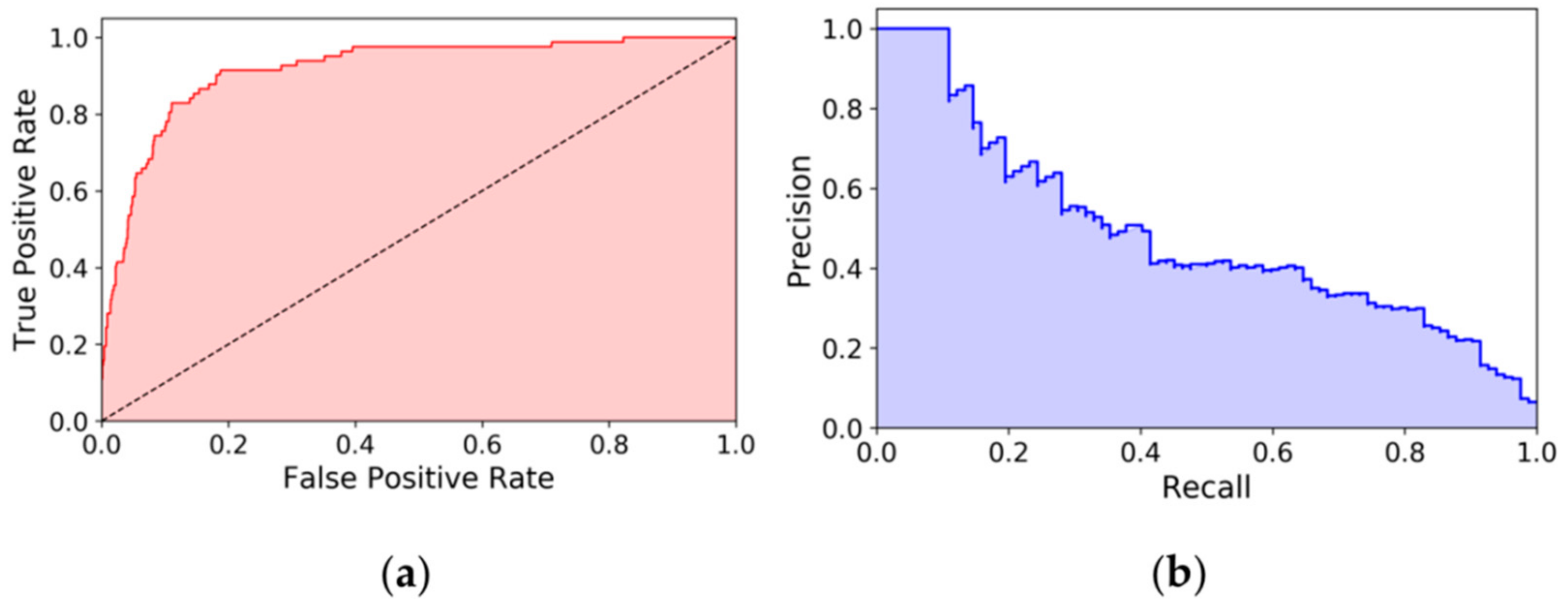
2. Results and Discussion
3. Methods
3.1. Dataset Generation and Docking
3.2. Data Fusion Methods
3.3. Performance Metrics and Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sotriffer, C. Virtual Screening: Principles, Challenges, and Practical Guidelines; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011. [Google Scholar]
- Bajusz, D.; Ferenczy, G.; Keserű, G. Structure-Based Virtual Screening Approaches in Kinase-Directed Drug Discovery. Curr. Top. Med. Chem. 2017, 17, 2235–2259. [Google Scholar] [CrossRef] [PubMed]
- Cross, J.B. Methods for Virtual Screening of GPCR Targets: Approaches and Challenges. In Computational Methods for GPCR Drug Discovery; Heifetz, A., Ed.; Humana Press: New York, NY, USA, 2018; pp. 233–264. [Google Scholar]
- Amaro, R.E.; Baudry, J.; Chodera, J.; Demir, Ö.; McCammon, J.A.; Miao, Y.; Smith, J.C. Ensemble Docking in Drug Discovery. Biophys. J. 2018, 114, 2271–2278. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-Y.; Zou, X. Ensemble docking of multiple protein structures: Considering protein structural variations in molecular docking. Proteins Struct. Funct. Bioinform. 2007, 66, 399–421. [Google Scholar] [CrossRef] [PubMed]
- Swift, R.V.; Jusoh, S.A.; Offutt, T.L.; Li, E.S.; Amaro, R.E. Knowledge-Based Methods To Train and Optimize Virtual Screening Ensembles. J. Chem. Inf. Model. 2016, 56, 830–842. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Wang, L.; Xie, X.-Q. ProSelection: A Novel Algorithm to Select Proper Protein Structure Subsets for in Silico Target Identification and Drug Discovery Research. J. Chem. Inf. Model. 2017, 57, 2686–2698. [Google Scholar] [CrossRef]
- Craig, I.R.; Essex, J.W.; Spiegel, K. Ensemble Docking into Multiple Crystallographically Derived Protein Structures: An Evaluation Based on the Statistical Analysis of Enrichments. J. Chem. Inf. Model. 2010, 50, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Novoa, E.M.; de Pouplana, L.R.; Barril, X.; Orozco, M. Ensemble Docking from Homology Models. J. Chem. Theory Comput. 2010, 6, 2547–2557. [Google Scholar] [CrossRef]
- Korb, O.; Olsson, T.S.G.; Bowden, S.J.; Hall, R.J.; Verdonk, M.L.; Liebeschuetz, J.W.; Cole, J.C. Potential and limitations of ensemble docking. J. Chem. Inf. Model. 2012, 52, 1262–1274. [Google Scholar] [CrossRef]
- Tarcsay, Á.; Paragi, G.; Vass, M.; Jójárt, B.; Bogár, F.; Keserű, G.M. The Impact of Molecular Dynamics Sampling on the Performance of Virtual Screening against GPCRs. J. Chem. Inf. Model. 2013, 53, 2990–2999. [Google Scholar] [CrossRef]
- Cavasotto, C.N. Normal mode-based approaches in receptor ensemble docking. Methods Mol. Biol. 2012, 819, 157–168. [Google Scholar]
- Kurczab, R.; Smusz, S.; Bojarski, A.J. The influence of negative training set size on machine learning-based virtual screening. J. Cheminform. 2014, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Smusz, S.; Kurczab, R.; Bojarski, A.J. The influence of the inactives subset generation on the performance of machine learning methods. J. Cheminform. 2013, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Réau, M.; Langenfeld, F.; Zagury, J.-F.; Lagarde, N.; Montes, M. Decoys Selection in Benchmarking Datasets: Overview and Perspectives. Front. Pharmacol. 2018, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Willett, P. Combination of similarity rankings using data fusion. J. Chem. Inf. Model. 2013, 53, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Willett, P. Fusing similarity rankings in ligand-based virtual screening. Comput. Struct. Biotechnol. J. 2013, 5, e201302002. [Google Scholar] [CrossRef] [PubMed]
- Bajusz, D.; Ferenczy, G.G.; Keserű, G.M. Ensemble docking-based virtual screening yields novel spirocyclic JAK1 inhibitors. J. Mol. Graph. Model. 2016, 70, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Bajusz, D.; Ferenczy, G.G.; Keserű, G.M. Discovery of subtype selective Janus kinase (JAK) inhibitors by structure-based virtual screening. J. Chem. Inf. Model. 2016, 56, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Vass, M.; Jójárt, B.; Bogár, F.; Paragi, G.; Keserű, G.M.; Tarcsay, Á. Dynamics and structural determinants of ligand recognition of the 5-HT6 receptor. J. Comput. Aided Mol. Des. 2015, 29, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Shoichet, B.; Irwin, J. Benchmarking sets for molecular docking. J. Med. Chem. 2006, 49, 6789–6801. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E.J.; Kerr, I.M.; Stark, G.R. Jak-STAT Pathways and Transcriptional Activation to IFNs Response and Other Transcriptional Activation in Extracellular Signaling Proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2018-4: Glide, LigPrep, Schrödinger; LLC: New York, NY, USA, 2018.
- Marazziti, D.; Baroni, S.; Borsini, F.; Picchetti, M.; Vatteroni, E.; Falaschi, V.; Catena-Dell’Osso, M. Serotonin Receptors of Type 6 (5-HT6): From Neuroscience to Clinical Pharmacology. Curr. Med. Chem. 2013, 20, 371–377. [Google Scholar] [PubMed]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Bioinform. 2004, 55, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- ChEMBL Database (Release 21). Available online: https://www.ebi.ac.uk/chembl/ (accessed on 15 July 2016).
- Tang, W.H.; Martin, K.A.; Hwa, J. Aldose Reductase, Oxidative Stress, and Diabetic Mellitus. Front. Pharmacol. 2012, 3, 87. [Google Scholar] [CrossRef] [PubMed]
- Deroo, B.J.; Korach, K.S. Estrogen receptors and human disease. J. Clin. Invest. 2006, 116, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Williams, N.K.; Bamert, R.S.; Patel, O.; Wang, C.; Walden, P.M.; Wilks, A.F.; Fantino, E.; Rossjohn, J.; Lucet, I.S. Dissecting specificity in the Janus kinases: The structures of JAK-specific inhibitors complexed to the JAK1 and JAK2 protein tyrosine kinase domains. J. Mol. Biol. 2009, 387, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Zak, M.; Hurley, C.; Ward, S. Identification of C-2 hydroxyethyl imidazopyrrolopyridines as potent JAK1 inhibitors with favorable physicochemical properties and high selectivity over JAK2. J. Med. Chem. 2013, 56, 4764–4785. [Google Scholar] [CrossRef]
- Antonysamy, S.; Hirst, G.; Park, F.; Sprengeler, P.; Stappenbeck, F.; Steensma, R.; Wilson, M.; Wong, M. Fragment-based discovery of JAK-2 inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 279–282. [Google Scholar] [CrossRef]
- Schenkel, L.B.; Huang, X.; Cheng, A.; Deak, H.L.; Doherty, E.; Emkey, R.; Gu, Y.; Gunaydin, H.; Kim, J.L.; Lee, J.; et al. Discovery of potent and highly selective thienopyridine Janus kinase 2 inhibitors. J. Med. Chem. 2011, 54, 8440–8450. [Google Scholar] [CrossRef]
- Wacker, D.; Wang, C.; Katritch, V.; Han, G.W.; Huang, X.-P.; Vardy, E.; McCorvy, J.D.; Jiang, Y.; Chu, M.; Siu, F.Y.; et al. Structural Features for Functional Selectivity at Serotonin Receptors. Science 2013, 340, 615–619. [Google Scholar] [CrossRef]
- Kinoshita, T.; Miyake, H.; Fujii, T.; Takakura, S.; Goto, T. The structure of human recombinant aldose reductase complexed with the potent inhibitor zenarestat. Acta Crystallogr. Sect. D 2002, 58, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Steuber, H.; Zentgraf, M.; Gerlach, C.; Sotriffer, C.A.; Heine, A.; Klebe, G. Expect the Unexpected or Caveat for Drug Designers: Multiple Structure Determinations Using Aldose Reductase Crystals Treated under Varying Soaking and Co-crystallisation Conditions. J. Mol. Biol. 2006, 363, 174–187. [Google Scholar] [CrossRef] [PubMed]
- Cousido-Siah, A.; Petrova, T.; Hazemann, I.; Mitschler, A.; Ruiz, F.X.; Howard, E.; Ginell, S.; Atmanene, C.; Van Dorsselaer, A.; Sanglier-Cianferani, S.; et al. Crystal packing modifies ligand binding affinity: The case of aldose reductase. Proteins 2012, 80, 2552–2561. [Google Scholar] [CrossRef] [PubMed]
- Koch, C.; Heine, A.; Klebe, G. Tracing the detail: How mutations affect binding modes and thermodynamic signatures of closely related aldose reductase inhibitors. J. Mol. Biol. 2011, 406, 700–712. [Google Scholar] [CrossRef] [PubMed]
- Koch, C.; Heine, A.; Klebe, G. Ligand-induced fit affects binding modes and provokes changes in crystal packing of aldose reductase. Biochim. Biophys. Acta 2011, 1810, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Kong, E.; Heldring, N.; Gustafsson, J.A.; Treuter, E.; Hubbard, R.E.; Pike, A.C.W. Delineation of a Unique Protein-Protein Interaction Site on the Surface of the Estrogen Receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 3593. [Google Scholar] [CrossRef] [PubMed]
- Nwachukwu, J.C.; Srinivasan, S.; Bruno, N.E.; Nowak, J.; Wright, N.J.; Minutolo, F.; Rangarajan, E.S.; Izard, T.; Yao, X.Q.; Grant, B.J.; et al. Systems Structural Biology Analysis of Ligand Effects on ER alpha Predicts Cellular Response to Environmental Estrogens and Anti-hormone Therapies. Cell Chem. Biol. 2017, 24, 35–45. [Google Scholar] [CrossRef]
- Puyang, X.; Furman, C.; Zheng, G.Z.; Wu, Z.J.; Banka, D.; Aithal, K.; Agoulnik, S.; Bolduc, D.M.; Buonamici, S.; Caleb, B.; et al. Discovery of Selective Estrogen Receptor Covalent Antagonists for the Treatment of ER alphaWTand ER alphaMUTBreast Cancer. Cancer Discov. 2018, 8, 1176–1193. [Google Scholar] [CrossRef]
- Truchon, J.-F.; Bayly, C.I. Evaluating Virtual Screening Methods: Good and Bad Metrics for the “Early Recognition” Problem. J. Chem. Inf. Model. 2007, 47, 488–508. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Héberger, K. Sum of ranking differences compares methods or models fairly. TrAC Trends Anal. Chem. 2010, 29, 101–109. [Google Scholar] [CrossRef]
- Kollár-Hunek, K.; Héberger, K. Method and model comparison by sum of ranking differences in cases of repeated observations (ties). Chemom. Intell. Lab. Syst. 2013, 127, 139–146. [Google Scholar] [CrossRef]
- Bajusz, D.; Rácz, A.; Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 2015, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Rácz, A.; Andrić, F.; Bajusz, D.; Héberger, K. Binary similarity measures for fingerprint analysis of qualitative metabolomic profiles. Metabolomics 2018, 14, 29. [Google Scholar] [CrossRef] [PubMed]
- Rácz, A.; Bajusz, D.; Héberger, K. Life beyond the Tanimoto coefficient: Similarity measures for interaction fingerprints. J. Cheminform. 2018, 10, 48. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Dataset Name | Protein Structures | Actives | Inactives | ||
|---|---|---|---|---|---|---|
| X-ray | MD Frames | MD Starting Structure | ||||
| 1 | JAK1 | 3EYG [30], 4IVC [31] | 3 | 4IVC [31] | 115 | 1644 |
| 2 | JAK2 | 3E62 [32] | 4 | 3TJD [33] | 82 | 1437 |
| 3 | 5-HT6 | - | 9 | homology model based on 4IB4 [34] | 20 | 689 |
| 4 | ALR2 | 1IEI [35], 2FZD [36], 2PFH [37], 3LZ5 [38], 3M0I [39], 4GCA | - | - | 26 | 917 |
| 5 | ER | 2BJ4 [40], 5TM9 [41], 6CHZ [42] | - | - | 38 | 1344 |
| Rule | Full Name | Formula1 |
|---|---|---|
| MIN | Minimum | min(DS1, DS2 … DSn) |
| MAX | Maximum | max(DS1, DS2 … DSn) |
| SUM | Arithmetic mean | |
| GEOM | Geometric mean | |
| HARM | Harmonic mean | |
| MED | Median | median(DS1, DS2 … DSn) |
| EUC | Euclidean norm |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bajusz, D.; Rácz, A.; Héberger, K. Comparison of Data Fusion Methods as Consensus Scores for Ensemble Docking. Molecules 2019, 24, 2690. https://doi.org/10.3390/molecules24152690
Bajusz D, Rácz A, Héberger K. Comparison of Data Fusion Methods as Consensus Scores for Ensemble Docking. Molecules. 2019; 24(15):2690. https://doi.org/10.3390/molecules24152690
Chicago/Turabian StyleBajusz, Dávid, Anita Rácz, and Károly Héberger. 2019. "Comparison of Data Fusion Methods as Consensus Scores for Ensemble Docking" Molecules 24, no. 15: 2690. https://doi.org/10.3390/molecules24152690
APA StyleBajusz, D., Rácz, A., & Héberger, K. (2019). Comparison of Data Fusion Methods as Consensus Scores for Ensemble Docking. Molecules, 24(15), 2690. https://doi.org/10.3390/molecules24152690