
Design and Synthesis of Benzimidazole-Chalcone Derivatives as Potential Anticancer Agents

 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Pharmacology
3. Materials and Methods
3.1. General Procedures
3.2. Synthesis
3.2.1. Synthesis of 1-(1H-benzoimidazol-2-yl)ethan-1-ol (16).
3.2.2. Synthesis of 1-(1H-benzimidazol-2-yl)ethan-1-one (17).
3.2.3. Standard Procedure 1 for the Synthesis of Benzimidazolyl-Chalcone Derivatives 18a–18d.
3.2.4. Standard Procedure 2 for Synthesis of Side Chain Modified Benzimidazolyl-chalcone Derivatives 19–23.
3.3. Compound Data
3.4. MTT Assay
3.5. Flow Cytometry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chimento, A.; Sala, M.; Gomez-Monterrey, I.M.; Musella, S.; Bertamino, A.; Caruso, A.; Sinicropi, M.S.; Sirianni, R.; Puoci, F.; Parisi, O.I.; et al. Biological activity of 3-chloro-azetidin-2-one derivatives having interesting antiproliferative activity on human breast cancer cell lines. Bioorg. Med. Chem. Lett. 2013, 23, 6401–6405. [Google Scholar] [CrossRef]
- Rui, M.; Rossi, D.; Marra, A.; Paolillo, M.; Schinelli, S.; Curti, D.; Tesei, A.; Cortesi, M.; Zamagni, A.; Laurini, E.; et al. Synthesis and biological evaluation of new aryl-alkyl(alkenyl)-4-benzylpiperidines, novel Sigma Receptor (SR) modulators, as potential anticancer-agents. Eur. J. Med. Chem. 2016, 124, 649–665. [Google Scholar] [CrossRef] [Green Version]
- Jukić, M.; Rastija, V.; Opačak-Bernardi, T.; Stolić, I.; Krstulović, L.; Bajić, M.; Glavaš-Obrovac, L. Antitumor activity of 3,4-ethylenedioxythiophene derivatives and quantitative structure-activity relationship analysis. J. Mol. Struct. 2017, 1133, 66–73. [Google Scholar] [CrossRef]
- Townsend, L.B.; Wise, D.S. The synthesis and chemistry of certain anthelmintic benzimidazoles. Parasitol. Today 1990, 6, 107–112. [Google Scholar] [CrossRef]
- Woolley, D.W. Some biological effects produced by benzimidazole and their reversal by purines. J. Biol. Chem. 1944, 152, 225–232. [Google Scholar]
- Fang, Y.; Zhou, H.; Gu, Q.; Xu, J. Synthesis and evaluation of tetrahydroisoquinoline-benzimidazole hybrids as multifunctional agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2019, 167, 133–145. [Google Scholar] [CrossRef]
- Shimomura, I.; Yokoi, A.; Kohama, I.; Kumazaki, M.; Tada, Y.; Tatsumi, K.; Ochiya, T.; Yamamoto, Y. Drug library screen reveals benzimidazole derivatives as selective cytotoxic agents for KRAS-mutant lung cancer. Cancer Lett. 2019, 451, 11–22. [Google Scholar] [CrossRef]
- Tuncbilek, M.; Kiper, T.; Altanlar, N. Synthesis and in vitro antimicrobial activity of some novel substituted benzimidazole derivatives having potent activity against MRSA. Eur. J. Med. Chem. 2009, 44, 1024–1033. [Google Scholar] [CrossRef]
- Shingalapur, R.V.; Hosamani, K.M.; Keri, R.S. Synthesis and evaluation of in vitro anti-microbial and anti-tubercular activity of 2-styryl benzimidazoles. Eur. J. Med. Chem. 2009, 44, 4244–4248. [Google Scholar] [CrossRef]
- Surineni, G.; Gao, Y.; Hussain, M.; Liu, Z.; Lu, Z.; Chhotaray, C.; Islam, M.M.; Hameed, H.M.A.; Zhang, T. Design, synthesis, and in vitro biological evaluation of novel benzimidazole tethered allylidenehydrazinylmethylthiazole derivatives as potent inhibitors of Mycobacterium tuberculosis. Medchemcomm 2019, 10, 49–60. [Google Scholar] [CrossRef]
- Zhang, H.Z.; Damu, G.L.; Cai, G.X.; Zhou, C.H. Design, synthesis and antimicrobial evaluation of novel benzimidazole type of Fluconazole analogues and their synergistic effects with Chloromycin, Norfloxacin and Fluconazole. Eur. J. Med. Chem. 2013, 64, 329–344. [Google Scholar] [CrossRef]
- Hernandez-Luis, F.; Hernandez-Campos, A.; Castillo, R.; Navarrete-Vazquez, G.; Soria-Arteche, O.; Hernandez-Hernandez, M.; Yepez-Mulia, L. Synthesis and biological activity of 2-(trifluoromethyl)-1H-benzimidazole derivatives against some protozoa and Trichinella spiralis. Eur. J. Med. Chem. 2010, 45, 3135–3141. [Google Scholar] [CrossRef]
- Valdez-Padilla, D.; Rodriguez-Morales, S.; Hernandez-Campos, A.; Hernandez-Luis, F.; Yepez-Mulia, L.; Tapia-Contreras, A.; Castillo, R. Synthesis and antiprotozoal activity of novel 1-methylbenzimidazole derivatives. Bioorg. Med. Chem. 2009, 17, 1724–1730. [Google Scholar] [CrossRef]
- Rida, S.M.; El-Hawash, S.A.M.; Fahmy, H.T.Y.; Hazzaa, A.A.; El-Meligy, M.M.M. Synthesis of novel benzofuran and related benzimidazole derivatives for evaluation of in vitro anti-HIV-1, anticancer and antimicrobial activities. Arch. Pharmacal Res. 2006, 29, 826–833. [Google Scholar] [CrossRef]
- Luo, Y.; Yao, J.P.; Yang, L.; Feng, C.L.; Tang, W.; Wang, G.F.; Zuo, J.P.; Lu, W. Design and synthesis of novel benzimidazole derivatives as inhibitors of hepatitis B virus. Bioorg. Med. Chem. 2010, 18, 5048–5055. [Google Scholar] [CrossRef]
- Singla, P.; Luxami, V.; Paul, K. Benzimidazole-biologically attractive scaffold for protein kinase inhibitors. Rsc Adv. 2014, 4, 12422–12440. [Google Scholar] [CrossRef]
- Achar, K.C.; Hosamani, K.M.; Seetharamareddy, H.R. In-vivo analgesic and anti-inflammatory activities of newly synthesized benzimidazole derivatives. Eur. J. Med. Chem. 2010, 45, 2048–2054. [Google Scholar] [CrossRef]
- Refaat, H.M. Synthesis and anticancer activity of some novel 2-substituted benzimidazole derivatives. Eur. J. Med. Chem. 2010, 45, 2949–2956. [Google Scholar] [CrossRef]
- Azam, M.; Khan, A.A.; Al-Resayes, S.I.; Islam, M.S.; Saxena, A.K.; Dwivedi, S.; Musarrat, J.; Trzesowska-Kruszynska, A.; Kruszynski, R. Synthesis and characterization of 2-substituted benzimidazoles and their evaluation as anticancer agent. Spectrochim. Acta Part A 2015, 142, 286–291. [Google Scholar] [CrossRef]
- Pourgholami, M.H.; Woon, L.; Almajd, R.; Akhter, J.; Bowery, P.; Morris, D.L. In vitro and in vivo suppression of growth of hepatocellular carcinoma cells by albendazole. Cancer Lett. 2001, 165, 43–49. [Google Scholar] [CrossRef]
- Di Carlo, G.; Mascolo, N.; Izzo, A.A.; Capasso, F. Flavonoids: Old and new aspects of a class of natural therapeutic drugs. Life Sci. 1999, 65, 337–353. [Google Scholar] [CrossRef]
- Nowakowska, Z. A review of anti-infective and anti-inflammatory chalcones. Eur. J. Med. Chem 2007, 42, 125–137. [Google Scholar] [CrossRef]
- Pettit, G.R.; Rhodes, M.R.; Herald, D.L.; Hamel, E.; Schmidt, J.M.; Pettitt, R.K. Antineoplastic agents. 445. Synthesis and evaluation of structural modifications of (Z)- and (E)-combretastatin A-4. J. Med. Chem. 2005, 48, 4087–4099. [Google Scholar] [CrossRef]
- Sivakumar, P.M.; Ganesan, S.; Veluchamy, P.; Doble, M. Novel chalcones and 1,3,5-triphenyl-2-pyrazoline derivatives as antibacterial agents. Chem. Biol. Drug Des. 2010, 76, 407–411. [Google Scholar] [CrossRef]
- Agarwal, A.; Srivastava, K.; Puri, S.K.; Chauhan, P.M.S. Synthesis of substituted indole derivatives as a new class of antimalarial agents. Bioorg. Med. Chem. Lett. 2005, 15, 3133–3136. [Google Scholar] [CrossRef]
- Suwito, H.; Jumina; Mustofa; Pudjiastuti, P.; Fanani, M.Z.; Kimata-Ariga, Y.; Katahira, R.; Kawakami, T.; Fujiwara, T.; Hase, T.; et al. Design and synthesis of chalcone derivatives as inhibitors of the ferredoxin—ferredoxin-NADP(+) reductase interaction of plasmodium falciparum: Pursuing new antimalarial agents. Molecules 2014, 19, 21473–21488. [Google Scholar]
- Boeck, P.; Leal, P.C.; Yunes, R.A.; Cechinel, V.; Lopez, S.; Sortino, M.; Escalante, A.; Furlan, R.L.E.; Zacchino, S. Antifungal activity and studies on mode of action of novel xanthoxyline-derived chalcones. Arch. Pharm. 2005, 338, 87–95. [Google Scholar] [CrossRef]
- Cheenpracha, S.; Karalai, C.; Ponglimanont, C.; Subhadhirasakul, S.; Tewtrakul, S. Anti-HIV-1 protease activity of compounds from Boesenbergia pandurata. Bioorg. Med. Chem. 2006, 14, 1710–1714. [Google Scholar] [CrossRef]
- Mateeva, N.; Gangapuram, M.; Mazzio, E.; Eyunni, S.; Soliman, K.F.A.; Redda, K.K. Biological evaluation of synthetic chalcone and flavone derivatives as anti-inflammatory agents. Med. Chem. Res. 2015, 24, 1672–1680. [Google Scholar] [CrossRef]
- Kamal, A.; Prabhakar, S.; Ramaiah, M.J.; Reddy, P.V.; Reddy, C.R.; Mallareddy, A.; Shankaraiah, N.; Reddy, T.L.N.; Pushpavalli, S.N.C.V.L.; Pal-Bhadra, M. Synthesis and anticancer activity of chalcone-pyrrolobenzodiazepine conjugates linked via 1,2,3-triazole ring side-armed with alkane spacers. Eur. J. Med. Chem. 2011, 46, 3820–3831. [Google Scholar] [CrossRef]
- Kim, T.H.; Seo, W.D.; Ryu, H.W.; Seo, H.R.; Jin, Y.B.; Lee, M.; Ji, Y.H.; Park, K.H.; Lee, Y.S. Anti-tumor effects by a synthetic chalcone compound is mediated by c-Myc-mediated reactive oxygen species production. Chem.-Biol. Interact. 2010, 188, 111–118. [Google Scholar] [CrossRef]
- Modzelewska, A.; Pettit, C.; Achanta, G.; Davidson, N.E.; Huang, P.; Khan, S.R. Anticancer activities of novel chalcone and bis-chalcone derivatives. Bioorg. Med. Chem. 2006, 14, 3491–3495. [Google Scholar] [CrossRef]
- Quintin, J.; Desrivot, J.; Thoret, S.; le Menez, P.; Cresteil, T.; Lewin, G. Synthesis and biological evaluation of a series of tangeretin-derived chalcones. Bioorg. Med. Chem. Lett. 2009, 19, 167–169. [Google Scholar] [CrossRef]
- He, S.W.; Hong, Q.M.; Lai, Z.; Yang, D.X.; Ting, P.C.; Kuethe, J.T.; Cernak, T.A.; Dykstra, K.D.; Sperbeck, D.M.; Wu, Z.C.; et al. Discovery of a potent and selective DGAT1 inhibitor with a piperidinyl-oxy-cyclohexanecarboxylic acid moiety. ACS Med. Chem. Lett. 2014, 5, 1082–1087. [Google Scholar] [CrossRef]
- Shaharyar, M.; Abdullah, M.M.; Bakht, M.A.; Majeed, J. Pyrazoline bearing benzimidazoles: Search for anticancer agent. Eur. J. Med. Chem. 2010, 45, 114–119. [Google Scholar] [CrossRef]
- Solomon, V.R.; Hu, C.K.; Lee, H. Hybrid pharmacophore design and synthesis of isatin-benzothiazole analogs for their anti-breast cancer activity. Bioorg. Med. Chem. 2009, 17, 7585–7592. [Google Scholar] [CrossRef]
- Wan, M.S.; Xu, L.Y.; Hua, L.; Li, A.L.; Li, S.Q.; Lu, W.J.; Pang, Y.; Cao, C.B.; Liu, X.G.; Jiao, P. Synthesis and evaluation of novel isoxazolyl chalcones as potential anticancer agents. Bioorg. Chem. 2014, 54, 38–43. [Google Scholar] [CrossRef]
- Ouattara, M.; Sissouma, D.; Kone, M.W.; Menan, H.E.; Toure, S.A.; Ouattara, L. Synthesis and anthelmintic activity of some hybrid Benzimidazolyl-chalcone derivatives. Trop. J. Pharm. Res. 2011, 10, 767–775. [Google Scholar] [CrossRef]
- Reddy, V.M.; Reddy, K.R. Synthesis and biological evaluation of some novel-3-(5-substituted benzimidazol-2-yl)-5-arylisoxazolines. Chin. Chem. Lett. 2010, 21, 1145–1148. [Google Scholar] [CrossRef]
- Mathew, B.; Suresh, J.; Vinod, D. Antitumour activity of 5-[(2E)-1-(1H-benzimidazol-2-yl)-3-substituted phenylprop-2-en-1-ylidene] pyrimidine-2,4,6(1H,3H,5H)-triones against Dalton’s ascitic lymphoma in mice. Med. Chem. Res. 2013, 22, 3911–3917. [Google Scholar] [CrossRef]
- Woo, H.B.; Eom, Y.W.; Park, K.S.; Ham, J.; Ahn, C.M.; Lee, S. Synthesis of substituted benzimidazolyl curcumin mimics and their anticancer activity. Bioorg. Med. Chem. Lett. 2012, 22, 933–936. [Google Scholar] [CrossRef]
- Hayes, M.E.; Wallace, G.A.; Grongsaard, P.; Bischoff, A.; George, D.M.; Miao, W.Y.; McPherson, M.J.; Stoffel, R.H.; Green, D.W.; Roth, G.P. Discovery of small molecule benzimidazole antagonists of the chemokine receptor CXCR3. Bioorg. Med. Chem. Lett. 2008, 18, 1573–1576. [Google Scholar] [CrossRef]
- Mehboob, S.; Song, J.H.; Hevener, K.E.; Su, P.C.; Boci, T.; Brubaker, L.; Truong, L.; Mistry, T.; Deng, J.P.; Cook, J.L.; et al. Structural and biological evaluation of a novel series of benzimidazole inhibitors of Francisella tularensis enoyl-ACP reductase (FabI). Bioorg. Med. Chem. Lett. 2015, 25, 1292–1296. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Luxami, V.; Paul, K. Synthesis, single crystal and antitumor activities of benzimidazole-quinazoline hybrids. Bioorg. Med. Chem. Lett. 2013, 23, 3288–3294. [Google Scholar] [CrossRef]
- Hwu, J.R.; Hsu, M.H.; Huang, R.C.C. New nordihydroguaiaretic acid derivatives as anti-HIV agents. Bioorg. Med. Chem. Lett. 2008, 18, 1884–1888. [Google Scholar] [CrossRef]
- Orjales, A.; Alonso-Cires, L.; Lopez-Tudanca, P.; Tapia, I.; Mosquera, R.; Labeaga, L. Benzimidazole-2-carboxylic acid amides and esters: A new structural class of 5-HT(3) ligands. Eur. J. Med. Chem. 1999, 34, 415–422. [Google Scholar] [CrossRef]
- Ganot, N.; Meker, S.; Reytman, L.; Tzubery, A.; Tshuva, E.Y. Anticancer metal complexes: Synthesis and cytotoxicity evaluation by the MTT assay. J. Vis. Exp. 2013, 81, 507–567. [Google Scholar] [CrossRef]
- Grigalius, I.; Petrikaite, V. Relationship between antioxidant and anticancer activity of trihydroxyflavones. Molecules 2017, 22, 2169. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Li, F.Y.; Wang, X.; Duan, W.G.; Lin, G.S. Synthesis and in vitro anticancer activity of novel dehydroabietic acid-based acylhydrazones. Molecules 2017, 22, 1087. [Google Scholar] [CrossRef]
- Sliwka, L.; Wiktorska, K.; Suchocki, P.; Milczarek, M.; Mielczarek, S.; Lubelska, K.; Cierpial, T.; Lyzwa, P.; Kielbasinski, P.; Jaromin, A.; et al. The comparison of MTT and CVS assays for the assessment of anticancer agent interactions. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Murakami, T. Absorption sites of orally administered drugs in the small intestine. Expert Opin. Drug Discov. 2017, 12, 1219–1232. [Google Scholar] [CrossRef]
- Tetko, I.V.; Bruneau, P. Application of ALOGPS to predict 1-octanol/water distribution coefficients, logP, and logD, of AstraZeneca in-house database. J. Pharm. Sci. 2004, 93, 3103–3110. [Google Scholar] [CrossRef]
- Tetko, I.V.; Tanchuk, V.Y. Application of associative neural networks for prediction of lipophilicity in ALOGPS 2.1 program. J. Chem. Inf. Comput. Sci. 2002, 42, 1136–1145. [Google Scholar] [CrossRef]
- Tetko, I.V.; Tanchuk, V.Y.; Kasheva, T.N.; Villa, A.E. Internet software for the calculation of the lipophilicity and aqueous solubility of chemical compounds. J. Chem. Inf. Comput. Sci. 2001, 41, 246–252. [Google Scholar] [CrossRef]
- Desai, N.C.; Pandya, D.D.; Joshi, V.V.; Rajpara, K.M.; Vaghani, H.V.; Satodiya, H.M. Synthesis, characterization and antimicrobial screening of hybrid molecules containing benzimidazole-pyrazole and pyridine nucleus. Med. Chem. Res. 2012, 21, 4463–4472. [Google Scholar] [CrossRef]
- Schwertheim, S.; Wein, F.; Lennartz, K.; Worm, K.; Schmid, K.W.; Sheu-Grabellus, S.Y. Curcumin induces G2/M arrest, apoptosis, NF-kappaB inhibition, and expression of differentiation genes in thyroid carcinoma cells. J. Cancer Res. Clin. Oncol. 2017, 143, 1143–1154. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Lipophilicity and Water Solubility | Cancer Cells (IC50 μM) | ||||||
|---|---|---|---|---|---|---|---|---|
| M.W. | clogP | clogS | S(cal) (mmol/L) | A549 | MCF-7 | HEP-G2 | OVCAR-3 | |
| 18a | 248.29 | 3.51 | −4.38 | 10.35 | 119.3 ± 29.9 | 13.49 ± 0.16 | 24.2 ± 0.32 | 16.91 ± 0.37 |
| 18b | 262.31 | 3.80 | −4.67 | 5.61 | 19.17 ± 0.43 | 18.09 ± 0.28 | 59.13 ± 0.92 | 24.7 ± 1.69 |
| 18c | 278.31 | 3.41 | −4.56 | 7.67 | 17.41 ± 0.16 | 16.04 ± 0.24 | 140.85 ± 0.88 | 34.44 ± 1.55 |
| 18d | 282.73 | 4.01 | −5.07 | 2.41 | 35.89 ± 0.84 | 32.55 ± 3.26 | 36.54 ± 1.35 | 36.48 ± 1.36 |
| 19a | 288.35 | 4.19 | −4.58 | 7.58 | 12.47 ± 0.18 | 12.12 ± 0.10 | 15.44 ± 0.25 | 16.09 ± 0.39 |
| 19b | 302.38 | 4.30 | −4.90 | 3.81 | 41.05 ± 1.61 | 53.54 ± 1.12 | 117.28 ± 2.42 | 59.01 ± 8.91 |
| 19c | 318.38 | 4.20 | −4.81 | 4.93 | >314 | 254.9 ± 13.6 | >314 | 299.52 ± 9.27 |
| 19d | 322.79 | 4.57 | −5.04 | 2.94 | 15.79 ± 0.49 | 13.42 ± 0.24 | 17.6 ± 0.25 | 16.13 ± 0.32 |
| 20a | 345.45 | 4.20 | −4.61 | 8.48 | 10.3 ± 0.13 | 9.65 ± 0.06 | 10.16 ± 0.08 | 10.5 ± 0.10 |
| 20b | 359.47 | 4.55 | −4.90 | 4.53 | 54.12 ± 1.20 | 53.19 ± 0.77 | 64.91 ± 0.24 | 28.71 ± 1.44 |
| 20c | 375.47 | 4.28 | −4.80 | 5.95 | 56.21 ± 0.96 | 56.09 ± 0.14 | 36.61 ± 1.89 | 11.4 ± 0.24 |
| 20d | 379.89 | 4.71 | −5.03 | 3.55 | 19.53 ± 0.71 | 14.73±0.09 | 15.49±0.16 | 14.04 ± 0.29 |
| 21a | 359.47 | 4.59 | −4.87 | 4.85 | 10.73 ± 0.58 | 9.73 ± 0.16 | 10.33 ± 0.06 | 10.34 ± 0.19 |
| 21b | 373.5 | 4.90 | −5.10 | 2.97 | 11.64 ± 0.25 | 11.14 ± 0.07 | 32.16 ± 1.83 | 12.55 ± 0.12 |
| 21c | 389.50 | 4.70 | −5.07 | 3.32 | 22.36 ± 0.54 | 21.12 ± 0.53 | 58.74 ± 0.75 | 13.29 ± 0.47 |
| 21d | 393.92 | 5.09 | −5.27 | 2.12 | 50.45 ± 0.82 | 54.41 ± 0.72 | 56.45 ± 0.86 | 33.13 ± 0.14 |
| 22a | 361.45 | 3.37 | −4.33 | 16.91 | 14.59 ± 0.40 | 10.38 ± 0.08 | 36.13 ± 0.75 | 22.44±0.47 |
| 22b | 375.47 | 3.74 | −4.65 | 8.41 | 10.76 ± 0.29 | 10.15 ± 0.06 | 42.05 ± 0.91 | 16.32 ± 0.45 |
| 22c | 391.47 | 3.35 | −4.52 | 11.82 | 10.27 ± 0.15 | 11.12 ± 0.20 | 50.24 ± 0.88 | 14.88 ± 0.67 |
| 22d | 395.89 | 3.92 | −4.83 | 5.86 | 24.06 ± 0.08 | 22.93 ± 0.49 | 21.38 ± 0.68 | 14.22 ± 0.33 |
| 23a | 375.47 | 3.82 | −4.67 | 8.03 | 9.73 ± 0.07 | 8.91 ± 0.07 | 10.93 ± 0.10 | 10.76 ± 0.12 |
| 23b | 389.5 | 4.12 | −4.93 | 4.58 | 11.79 ± 0.27 | 11.34 ± 0.17 | 47.88 ± 0.76 | 13.76 ± 0.27 |
| 23c | 405.5 | 3.84 | −4.83 | 6.00 | 16.92 ± 0.61 | 11.93 ± 0.14 | 32.92 ± 0.38 | 13.4 ± 0.33 |
| 23d | 409.91 | 4.32 | −5.10 | 3.26 | 81.48 ± 1.40 | 35.69 ± 0.47 | 95.7 ± 2.44 | 42.24 ± 2.43 |
| DOX | 0.46 ± 0.01 | 0.42 ± 0.01 | 0.72 ± 0.01 | 3.95 ± 0.09 | ||||
| Cisplatin | 7.31 ± 0.44 | 11.7 ± 0.12 | 3.97 ± 0.04 | 16.04 ± 0.74 | ||||
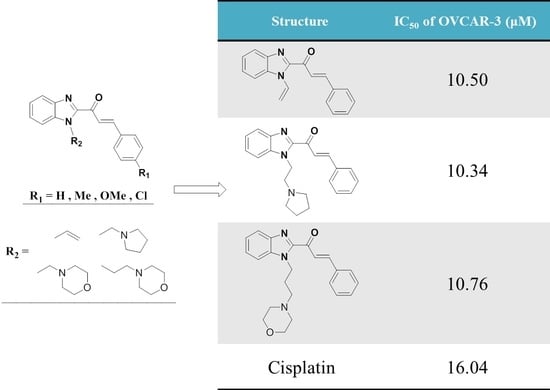
| Treatment | G1 (%) | S (%) | G2/M (%) | IC50 (μM) |
|---|---|---|---|---|
| Control (PBS) | 55.26 | 8.00 | 36.74 | − |
| 20a | 37.03 | 8.00 | 54.96 | 10.50 |
| 21a | 29.37 | 8.00 | 62.63 | 10.34 |
| 22a | 62.58 | 8.00 | 29.42 | 22.44 |
| 23a | 60.24 | 8.00 | 31.76 | 10.76 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsieh, C.-Y.; Ko, P.-W.; Chang, Y.-J.; Kapoor, M.; Liang, Y.-C.; Chu, H.-L.; Lin, H.-H.; Horng, J.-C.; Hsu, M.-H. Design and Synthesis of Benzimidazole-Chalcone Derivatives as Potential Anticancer Agents. Molecules 2019, 24, 3259. https://doi.org/10.3390/molecules24183259
Hsieh C-Y, Ko P-W, Chang Y-J, Kapoor M, Liang Y-C, Chu H-L, Lin H-H, Horng J-C, Hsu M-H. Design and Synthesis of Benzimidazole-Chalcone Derivatives as Potential Anticancer Agents. Molecules. 2019; 24(18):3259. https://doi.org/10.3390/molecules24183259
Chicago/Turabian StyleHsieh, Cheng-Ying, Pi-Wen Ko, Yu-Jui Chang, Mohit Kapoor, Yu-Chuan Liang, Hsueh-Liang Chu, Hui-Hsien Lin, Jia-Cherng Horng, and Ming-Hua Hsu. 2019. "Design and Synthesis of Benzimidazole-Chalcone Derivatives as Potential Anticancer Agents" Molecules 24, no. 18: 3259. https://doi.org/10.3390/molecules24183259