
Antiproliferative S-Trityl-l-Cysteine -Derived Compounds as SIRT2 Inhibitors: Repurposing and Solubility Enhancement

 ,
,  ,
,  ,
, 
Abstract
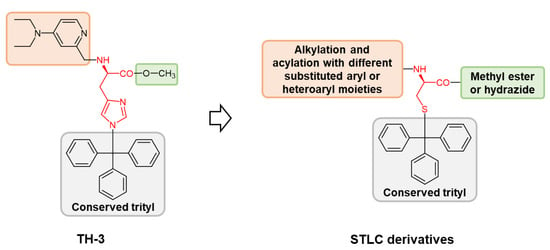
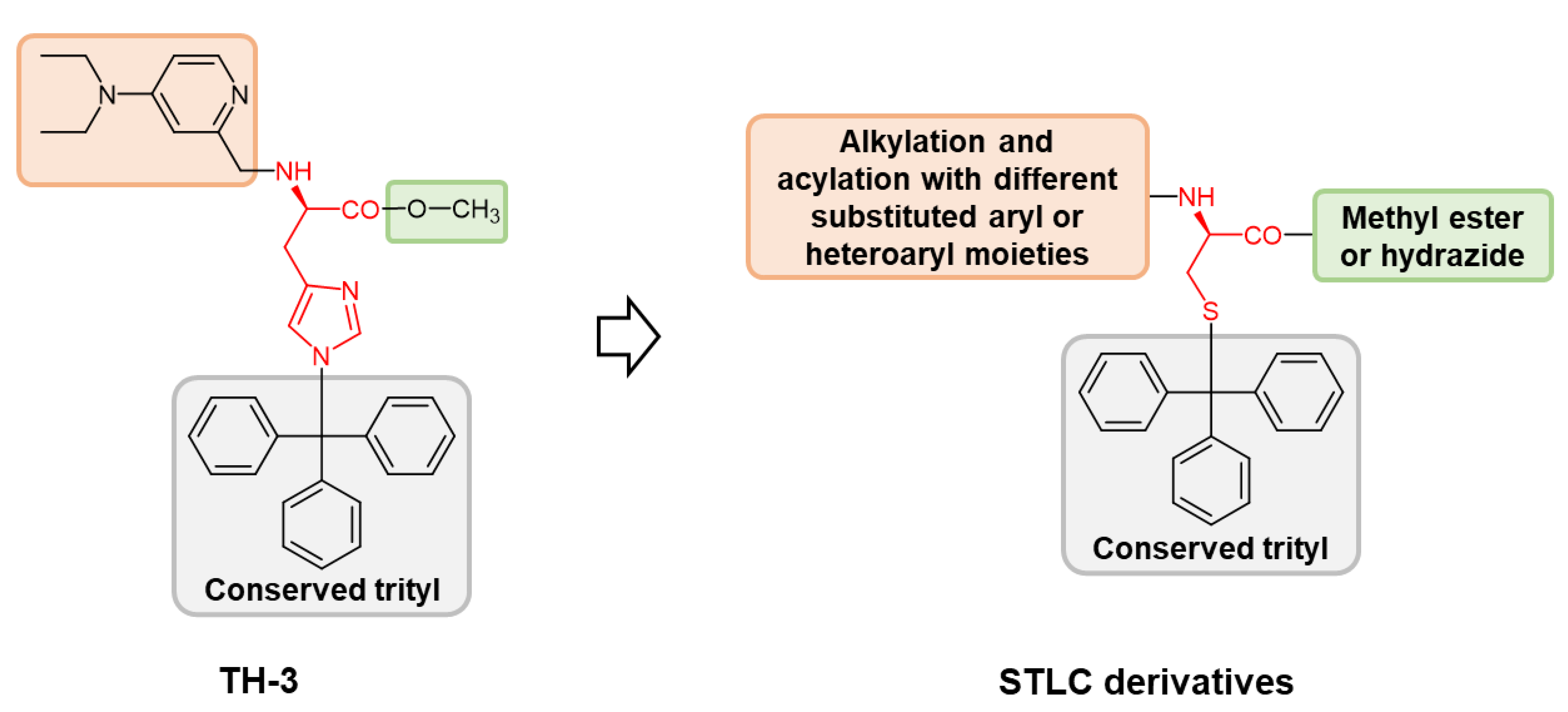
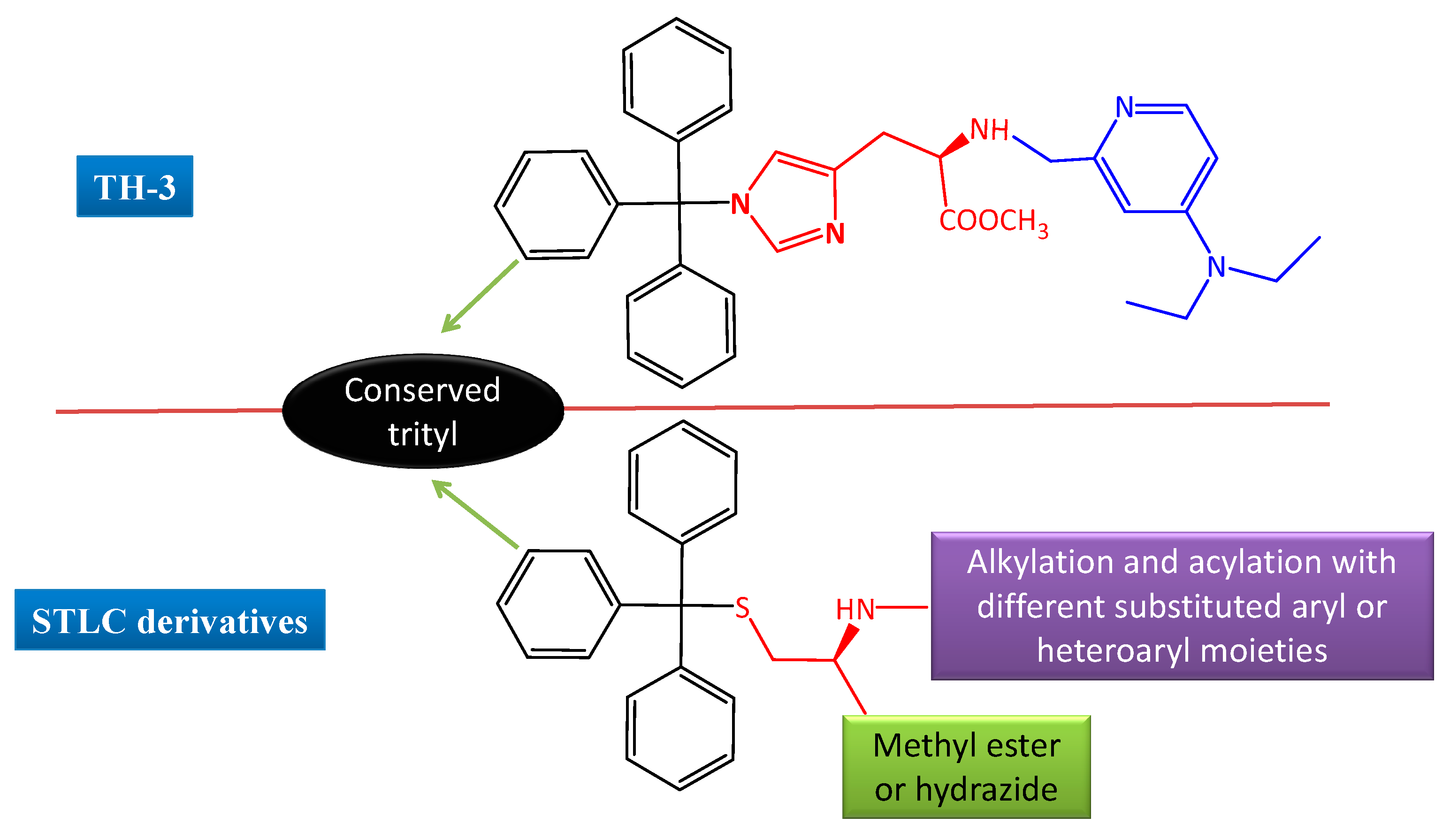
:
1. Introduction
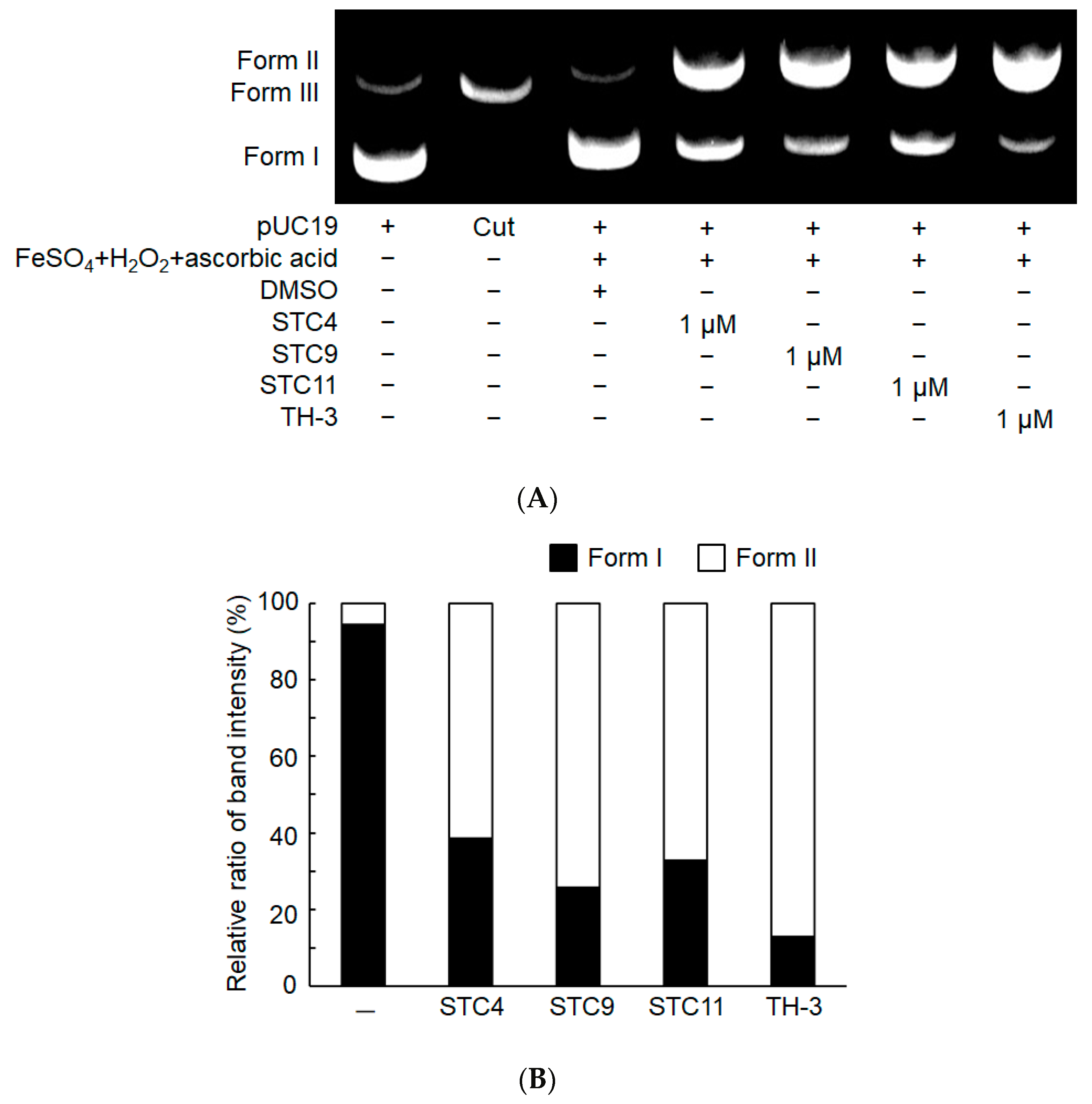
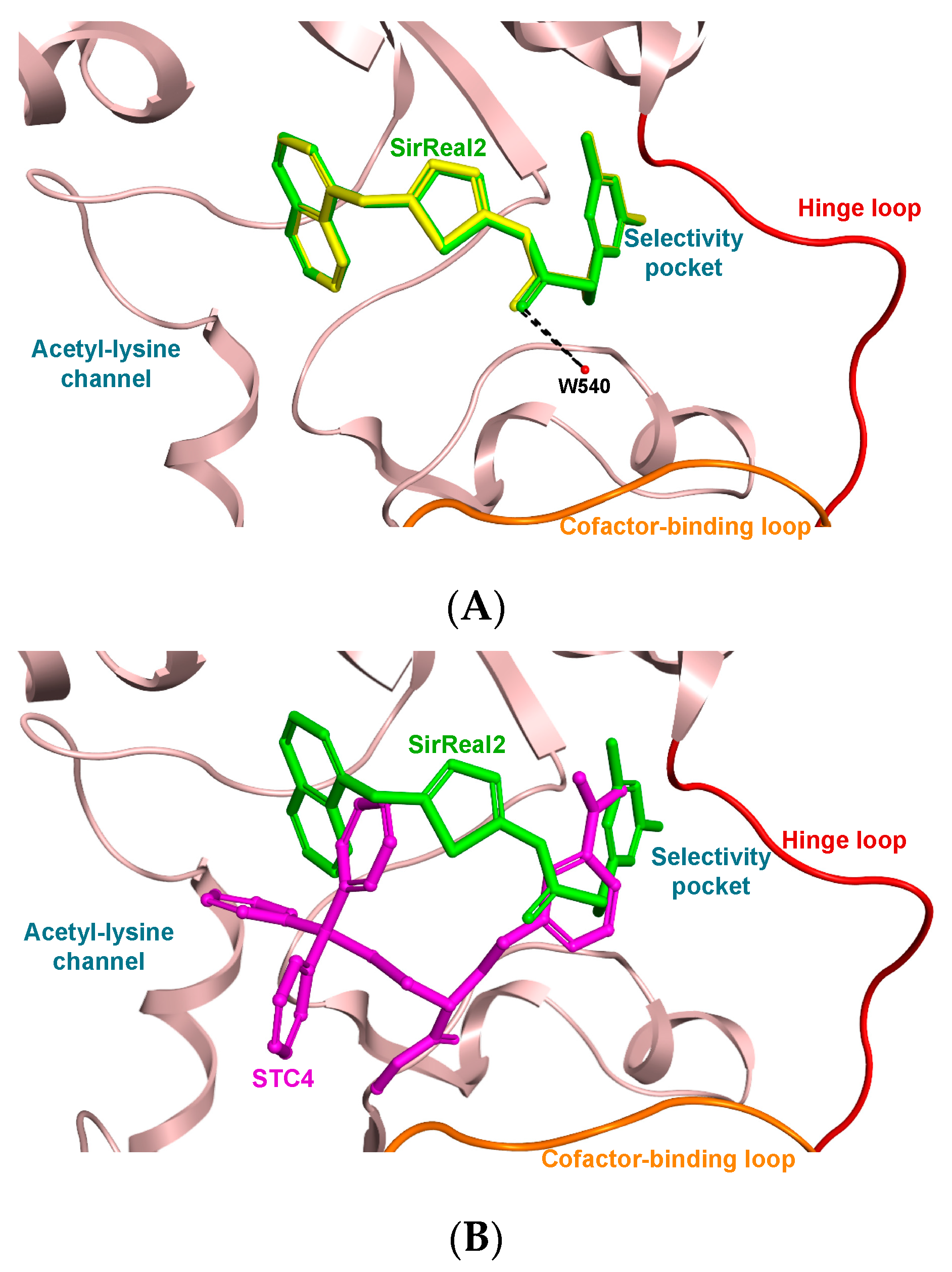
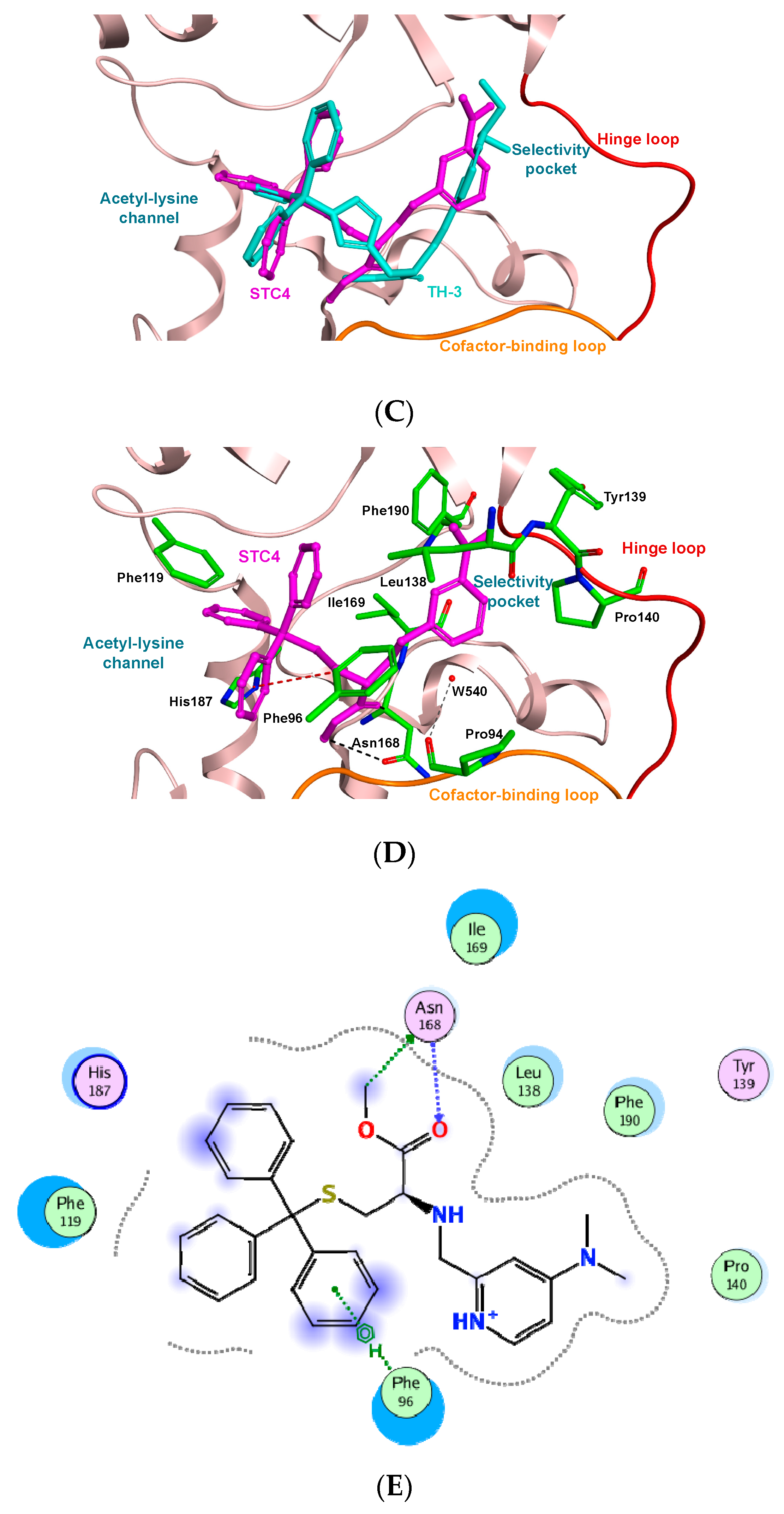
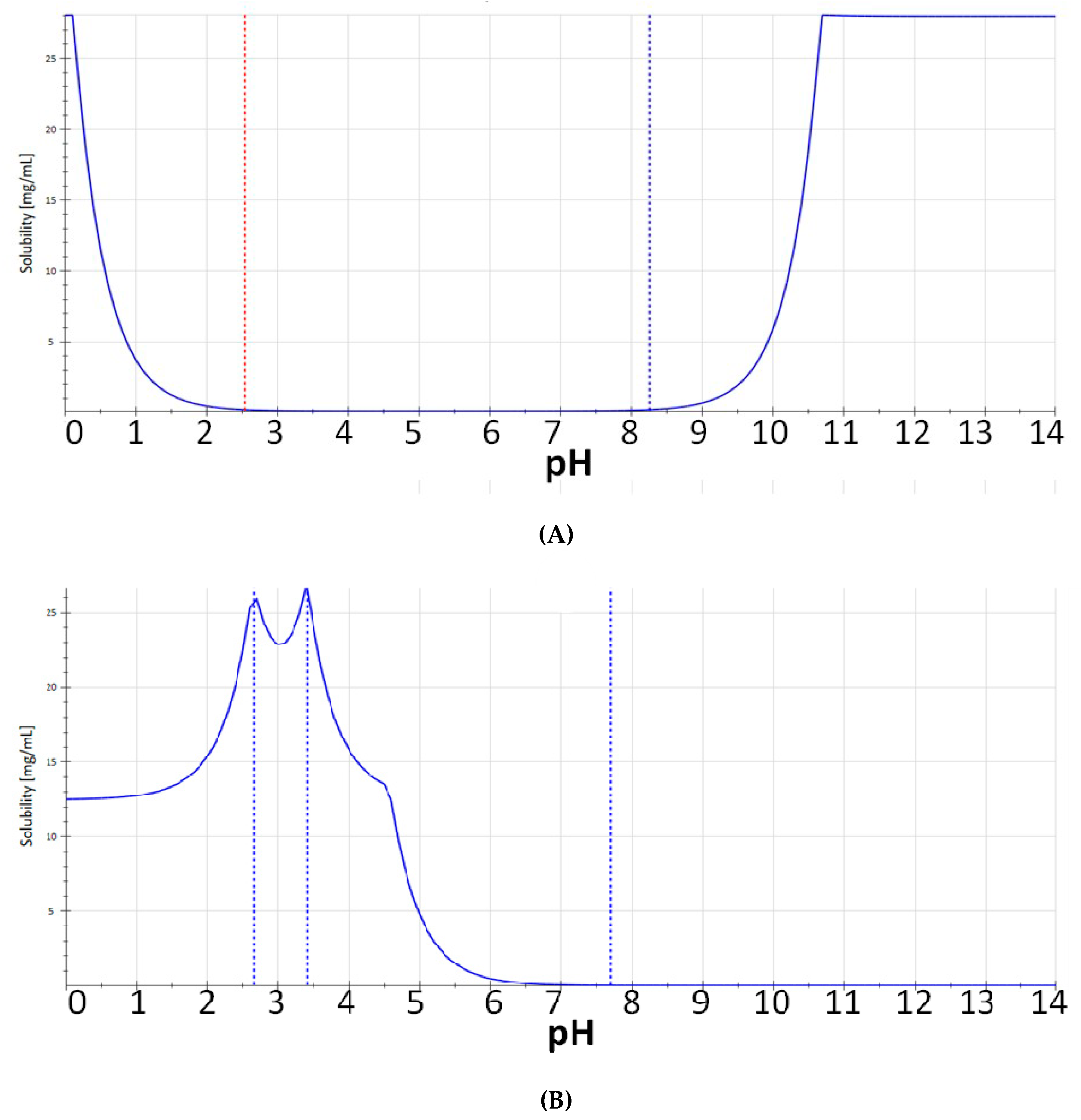
2. Results and Discussion
3. Materials and Methods
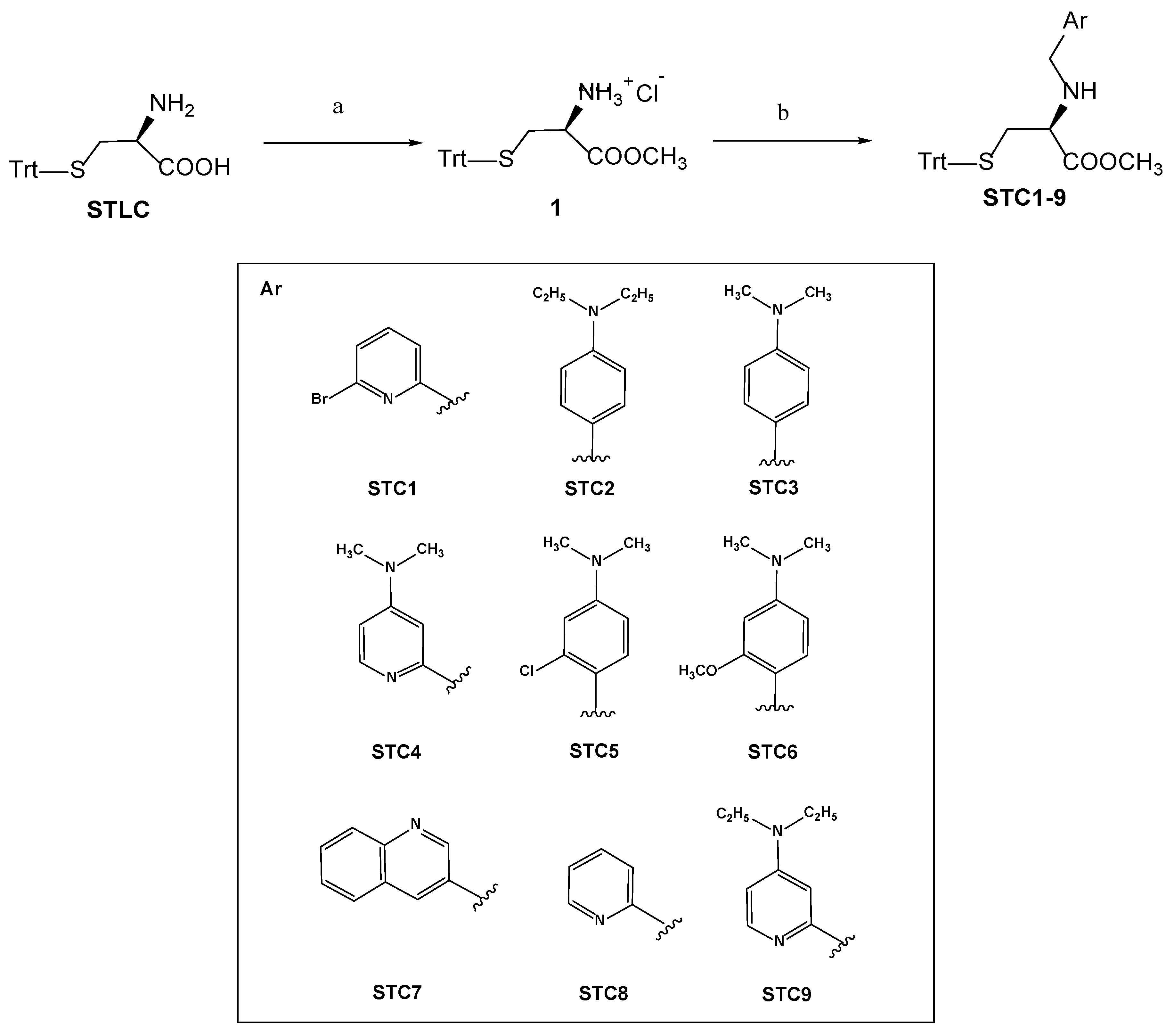
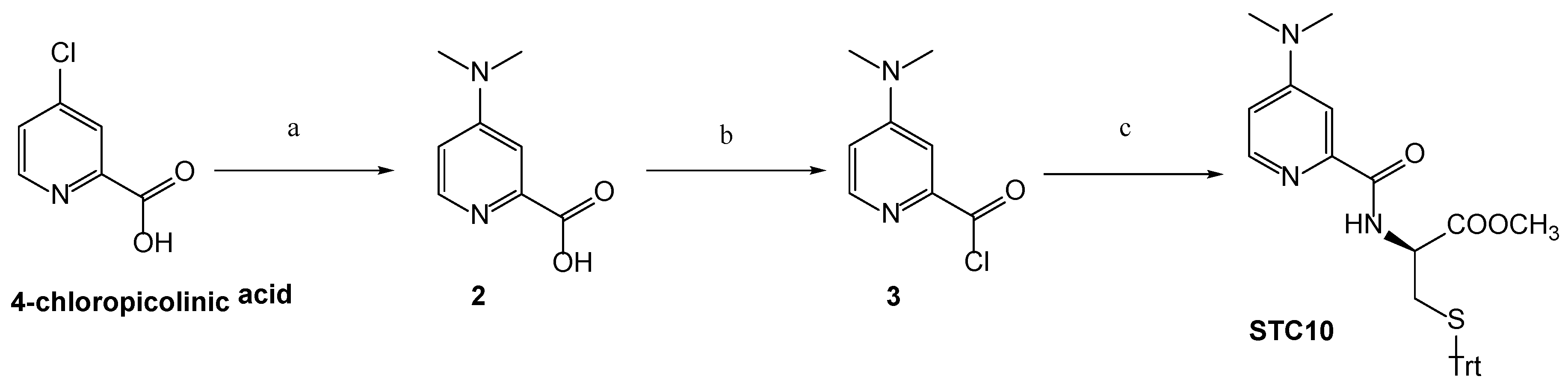
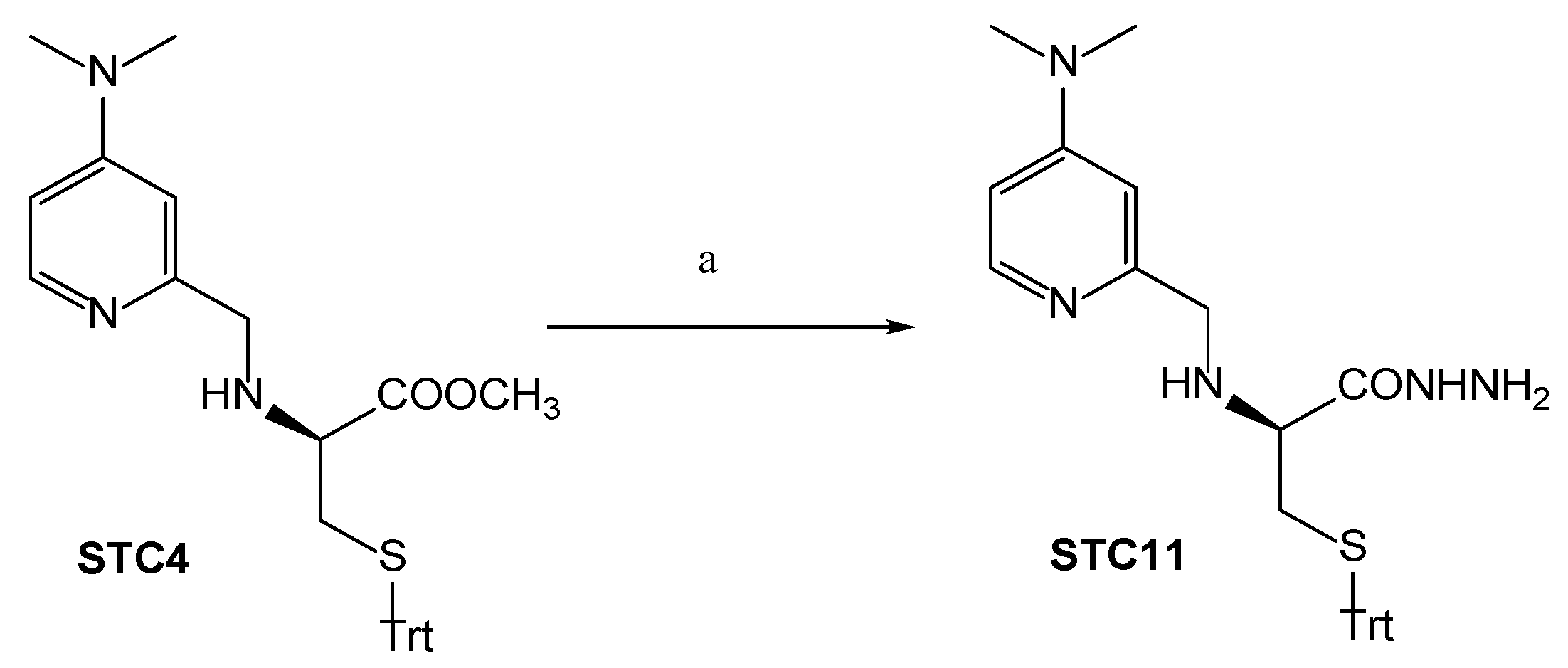
3.1. Chemistry
Synthetic Procedures
3.2. Biological Assays
3.2.1. In Vitro Inhibitory Activities against SIRT2
3.2.2. Cell Culture and Drug Treatment
3.2.3. MTT Assay
3.2.4. DNA Cleavage
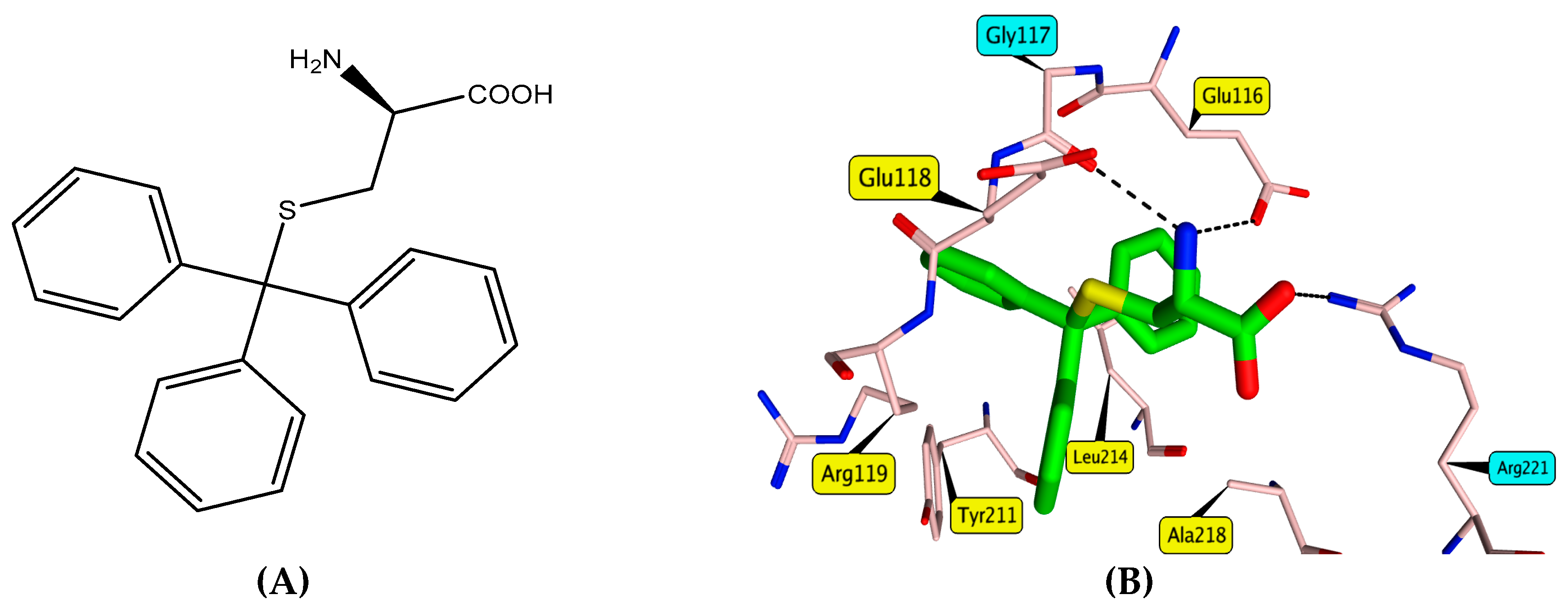
3.3. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- DeBonis, S.; Skoufias, D.A.; Lebeau, L.; Lopez, R.; Robin, G.; Margolis, R.L.; Wade, R.H.; Kozielski, F. In Vitro Screening for Inhibitors of the Human Mitotic Kinesin Eg5 with Antimitotic and Antitumor Activities. Mol. Cancer Ther. 2004, 3, 1079–1090. [Google Scholar] [PubMed]
- Skoufias, D.A.; DeBonis, S.; Saoudi, Y.; Lebeau, L.; Crevel, I.; Cross, R.; Wade, R.H.; Hackney, D.; Kozielski, F. S -Trityl-l-Cysteine Is a Reversible, Tight Binding Inhibitor of the Human Kinesin Eg5 That Specifically Blocks Mitotic Progression. J. Biol. Chem. 2006, 281, 17559–17569. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Ishii, H.; Ogo, N.; Unno, Y.; Matsuno, K.; Sawada, J.; Akiyama, Y.; Asai, A. S-trityl- l -cysteine derivative induces caspase-independent cell death in K562 human chronic myeloid leukemia cell line. Cancer Lett. 2010, 298, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Ogo, N.; Oishi, S.; Matsuno, K.; Sawada, J.; Fujii, N.; Asai, A. Synthesis and Biological Evaluation of l-Cysteine Derivatives as Mitotic Kinesin Eg5 Inhibitors. Bioorganic Med. Chem. Lett. 2007, 17, 3921–3924. [Google Scholar] [CrossRef] [PubMed]
- Boyd, M.R.; Paull, K.D. Some Practical Considerations and Applications of the National Cancer Institute in Vitro Anticancer Drug Discovery Screen. Drug Dev. Res. 1995, 34, 91–109. [Google Scholar] [CrossRef]
- Purcell, J.W.; Davis, J.; Reddy, M.; Martin, S.; Samayoa, K.; Vo, H.; Thomsen, K.; Bean, P.; Kuo, W.L.; Ziyad, S.; et al. Activity of the Kinesin Spindle Protein Inhibitor Ispinesib (SB-715992) in Models of Breast Cancer. Clin. Cancer Res. 2010, 16, 566–576. [Google Scholar] [CrossRef]
- El-Nassan, H.B. Advances in the Discovery of Kinesin Spindle Protein (Eg5) Inhibitors as Antitumor Agents. Eur. J. Med. Chem. 2013, 62, 614–631. [Google Scholar] [CrossRef]
- Rath, O.; Kozielski, F. Kinesins and Cancer. Nat. Rev. Cancer 2012, 12, 527–539. [Google Scholar] [CrossRef]
- Perez-Melero, C. KSP Inhibitors as Antimitotic Agents. Curr. Top. Med. Chem. 2014, 14, 2286–2311. [Google Scholar] [CrossRef]
- Rodriguez, D.; Ramesh, C.; Henson, L.H.; Wilmeth, L.; Bryant, B.K.; Kadavakollu, S.; Hirsch, R.; Montoya, J.; Howell, P.R.; George, J.M.; et al. Synthesis and Characterization of Tritylthioethanamine Derivatives with Potent KSP Inhibitory Activity. Bioorganic Med. Chem. 2011, 19, 5446–5453. [Google Scholar] [CrossRef]
- DeBonis, S.; Skoufias, D.A.; Indorato, R.L.; Liger, F.; Marquet, B.; Laggner, C.; Joseph, B.; Kozielski, F. Structure-Activity Relationship of S-Trityl-l-Cysteine Analogues as Inhibitors of the Human Mitotic Kinesin Eg5. J. Med. Chem. 2008, 51, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Kaan, H.Y.K.; Weiss, J.; Menger, D.; Ulaganathan, V.; Tkocz, K.; Laggner, C.; Popowycz, F.; Joseph, B.; Kozielski, F. Structure−Activity Relationship and Multidrug Resistance Study of New S -Trityl- l -Cysteine Derivatives As Inhibitors of Eg5. J. Med. Chem. 2011, 54, 1576–1586. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.M.; Collins, I. Recent Findings and Future Directions for Interpolar Mitotic Kinesin Inhibitors in Cancer Therapy. Future Med. Chem. 2016, 8, 463–489. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.D.; Buckley, R.; Learman, S.; Richard, J.; Parke, C.; Worthylake, D.K.; Wojcik, E.J.; Walker, R.A.; Kim, S. Allosteric Drug Discrimination Is Coupled to Mechanochemical Changes in the Kinesin-5 Motor Core. J. Biol. Chem. 2010, 285, 18650–18661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abualhasan, M.N.; Good, J.A.D.; Wittayanarakul, K.; Anthony, N.G.; Berretta, G.; Rath, O.; Kozielski, F.; Sutcliffe, O.B.; Mackay, S.P. Doing the Methylene Shuffle – Further Insights into the Inhibition of Mitotic Kinesin Eg5 with S-Trityl- l-Cysteine. Eur. J. Med. Chem. 2012, 54, 483–498. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Ma, T.; Zhu, Q.; Xu, Y.; Zha, X. Recent Advances in Inhibitors of Sirtuin1/2: An Update and Perspective. Future Med. Chem. 2018, 10, 907–934. [Google Scholar] [CrossRef] [PubMed]
- Schiedel, M.; Robaa, D.; Rumpf, T.; Sippl, W.; Jung, M. The Current State of NAD + -Dependent Histone Deacetylases (Sirtuins) as Novel Therapeutic Targets. Med. Res. Rev. 2018, 38, 147–200. [Google Scholar] [CrossRef]
- H Beumer, J.; Tawbi, H. Role of Histone Deacetylases and Their Inhibitors in Cancer Biology and Treatment. Curr. Clin. Pharmacol. 2010, 5, 196–208. [Google Scholar] [CrossRef]
- North, B.J.; Marshall, B.L.; Borra, M.T.; Denu, J.M.; Verdin, E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 2003, 11, 437–444, PMID: 12620231. [Google Scholar] [CrossRef]
- Ito, A.; Shimazu, T.; Maeda, S.; Shah, A.A.; Tsunoda, T.; Iemura, S.; Natsume, T.; Suzuki, T.; Motohashi, H.; Yamamoto, M.; et al. The subcellular localization and activity of cortactin is regulated by acetylation and interaction with Keap1. Sci. Signal. 2015, 24, PMID: 26602019. [Google Scholar] [CrossRef]
- Deng, A.; Ning, Q.; Zhou, L.; Liang, Y. SIRT2 Is an Unfavorable Prognostic Biomarker in Patients with Acute Myeloid Leukemia. Sci. Rep. 2016, 6, 27694. [Google Scholar] [CrossRef] [PubMed]
- Jing, H.; Hu, J.; He, B.; Negrón Abril, Y.L.; Stupinski, J.; Weiser, K.; Carbonaro, M.; Chiang, Y.-L.; Southard, T.; Giannakakou, P.; et al. A SIRT2-Selective Inhibitor Promotes c-Myc Oncoprotein Degradation and Exhibits Broad Anticancer Activity. Cancer Cell 2016, 29, 297–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carafa, V.; Rotili, D.; Forgione, M.; Cuomo, F.; Serretiello, E.; Hailu, G.S.; Jarho, E.; Lahtela-Kakkonen, M.; Mai, A.; Altucci, L. Sirtuin Functions and Modulation: From Chemistry to the Clinic. Clin. Epigenetics 2016, 8, 61. [Google Scholar] [CrossRef]
- Shah, A.A.; Ito, A.; Nakata, A.; Yoshida, M. Identification of a Selective SIRT2 Inhibitor and Its Anti-Breast Cancer Activity. Biol. Pharm. Bull. 2016, 39, 1739–1742. [Google Scholar] [CrossRef] [PubMed]
- Sunami, Y.; Araki, M.; Hironaka, Y.; Morishita, S.; Kobayashi, M.; Liew, E.L.; Edahiro, Y.; Tsutsui, M.; Ohsaka, A.; Komatsu, N. Inhibition of the NAD-Dependent Protein Deacetylase SIRT2 Induces Granulocytic Differentiation in Human Leukemia Cells. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Kozako, T.; Mellini, P.; Ohsugi, T.; Aikawa, A.; Uchida, Y.; Honda, S.; Suzuki, T. Novel Small Molecule SIRT2 Inhibitors Induce Cell Death in Leukemic Cell Lines. Bmc Cancer 2018, 18, 791. [Google Scholar] [CrossRef] [PubMed]
- Moniot, S.; Forgione, M.; Lucidi, A.; Hailu, G.S.; Nebbioso, A.; Carafa, V.; Baratta, F.; Altucci, L.; Giacché, N.; Passeri, D.; et al. Development of 1,2,4-Oxadiazoles as Potent and Selective Inhibitors of the Human Deacetylase Sirtuin 2: Structure–Activity Relationship, X-Ray Crystal Structure, and Anticancer Activity. J. Med. Chem. 2017, 60, 2344–2360. [Google Scholar] [CrossRef]
- Li, Y.; Matsumori, H.; Nakayama, Y.; Osaki, M.; Kojima, H.; Kurimasa, A.; Ito, H.; Mori, S.; Katoh, M.; Oshimura, M.; et al. SIRT2 Down-Regulation in HeLa Can Induce P53 Accumulation via P38 MAPK Activation-Dependent P300 Decrease, Eventually Leading to Apoptosis. Genes Cells 2011, 16, 34–45. [Google Scholar] [CrossRef]
- Ali, T.F.S.; Ciftci, H.I.; Radwan, M.O.; Koga, R.; Ohsugi, T.; Okiyama, Y.; Honma, T.; Nakata, A.; Ito, A.; Yoshida, M.; et al. New SIRT2 Inhibitors: Histidine-Based Bleomycin Spin-Off. Bioorganic Med. Chem. 2019, 27, 1767–1775. [Google Scholar] [CrossRef]
- Rumpf, T.; Schiedel, M.; Karaman, B.; Roessler, C.; North, B.J.; Lehotzky, A.; Oláh, J.; Ladwein, K.I.; Schmidtkunz, K.; Gajer, M.; et al. Selective Sirt2 Inhibition by Ligand-Induced Rearrangement of the Active Site. Nat. Commun. 2015, 6, 6263. [Google Scholar] [CrossRef]
- Radwan, M.O.; Sonoda, S.; Ejima, T.; Tanaka, A.; Koga, R.; Okamoto, Y.; Fujita, M.; Otsuka, M. Zinc-Mediated Binding of a Low-Molecular-Weight Stabilizer of the Host Anti-Viral Factor Apolipoprotein B MRNA-Editing Enzyme, Catalytic Polypeptide-like 3G. Bioorganic Med. Chem. 2016, 24, 4398–4405. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.F.S.; Iwamaru, K.; Ciftci, H.I.; Koga, R.; Matsumoto, M.; Oba, Y.; Kurosaki, H.; Fujita, M.; Okamoto, Y.; Umezawa, K.; et al. Novel Metal Chelating Molecules with Anticancer Activity. Striking Effect of the Imidazole Substitution of the Histidine–Pyridine–Histidine System. Bioorganic Med. Chem. 2015, 23, 5476–5482. [Google Scholar] [CrossRef] [PubMed]
- Radwan, M.O.; Koga, R.; Hida, T.; Ejima, T.; Kanemaru, Y.; Tateishi, H.; Okamoto, Y.; Inoue, J.; Fujita, M.; Otsuka, M. Minimum Structural Requirements for Inhibitors of the Zinc Finger Protein TRAF6. Bioorganic Med. Chem. Lett. 2019, 29, 2162–2167. [Google Scholar] [CrossRef] [PubMed]
- Yeoh, K.K.; Chan, M.C.; Thalhammer, A.; Demetriades, M.; Chowdhury, R.; Tian, Y.-M.; Stolze, I.; McNeill, L.A.; Lee, M.K.; Woon, E.C.Y.; et al. Dual-Action Inhibitors of HIF Prolyl Hydroxylases That Induce Binding of a Second Iron Ion. Org. Biomol. Chem. 2013, 11, 732–745. [Google Scholar] [CrossRef] [PubMed]
- Radwan, M.O.; Ismail, M.A.H.; El-Mekkawy, S.; Ismail, N.S.M.; Hanna, A.G. Synthesis and Biological Activity of New 18 β-Glycyrrhetinic Acid Derivatives. Arab. J. Chem. 2016, 9, 390–399. [Google Scholar] [CrossRef]
- Schiedel, M.; Rumpf, T.; Karaman, B.; Lehotzky, A.; Oláh, J.; Gerhardt, S.; Ovádi, J.; Sippl, W.; Einsle, O.; Jung, M. Aminothiazoles as Potent and Selective Sirt2 Inhibitors: A Structure− Activity Relationship Study. J. Med. Chem. 2016, 59, 1599–1612. [Google Scholar] [CrossRef] [PubMed]
- Swarbrick, J.D.; Ung, P.; Chhabra, S.; Graham, B. An Iminodiacetic Acid Based Lanthanide Binding Tag for Paramagnetic Exchange NMR Spectroscopy. Angew. Chem. Int. Ed. 2011, 50, 4403–4406. [Google Scholar] [CrossRef]
- Mundinger, S.; Jakob, U.; Bichovski, P.; Bannwarth, W. Modification and Optimization of the Bis-Picolylamide-Based Relay Protection for Carboxylic Acids to Be Cleaved by Unusual Complexation with Cu 2+ Salts. J. Org. Chem. 2012, 77, 8968–8979. [Google Scholar] [CrossRef]
- Khan, K.M.; Rasheed, M.; Ullah, Z.; Hayat, S.; Kaukab, F.; Choudhary, M.I.; Ur-Rahman, A.; Perveen, S. Synthesis and in Vitro Leishmanicidal Activity of Some Hydrazides and Their Analogues. Bioorganic Med. Chem. 2003, 11, 1381–1387. [Google Scholar] [CrossRef]
- Kudo, N.; Ito, A.; Arata, M.; Nakata, A.; Yoshida, M. Identification of a Novel Small Molecule That Inhibits Deacetylase but Not Defatty-Acylase Reaction Catalysed by SIRT2. Philos. Trans. R. Soc. B: Biol. Sci. 2018, 373, 20170070. [Google Scholar] [CrossRef]
- Altıntop, M.D.; Ciftci, H.I.; Radwan, M.O.; Sever, B.; Kaplancıklı, Z.A.; Ali, T.F.S.; Koga, R.; Fujita, M.; Otsuka, M.; Zdemir, A. Design, Synthesis, and Biological Evaluation of Novel 1,3,4-Thiadiazole Derivatives as Potential Antitumor Agents against Chronic Myelogenous Leukemia: Striking Effect of Nitrothiazole Moiety. Molecules 2018, 23, 59. [Google Scholar] [CrossRef]
- Bayrak, N.; Yıldırım, H.; Tuyun, A.F.; Kara, E.M.; Celik, B.O.; Gupta, G.K.; Ciftci, H.I.; Fujita, M.; Otsuka, M.; Nasiri, H.R. Synthesis, Computational Study, and Evaluation of In Vitro Antimicrobial, Antibiofilm, and Anticancer Activities of New Sulfanyl Aminonaphthoquinone Derivatives. Lett. Drug Des. Discov. 2017, 14. [Google Scholar] [CrossRef]
- Ciftci, H.I.; Ozturk, S.E.; Ali, T.F.S.; Radwan, M.O.; Tateishi, H.; Koga, R.; Ocak, Z.; Can, M.; Otsuka, M.; Fujita, M. The First Pentacyclic Triterpenoid Gypsogenin Derivative Exhibiting Anti-ABL1 Kinase and Anti-Chronic Myelogenous Leukemia Activities. Biol. Pharm. Bull. 2018, 41, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Karabacak, M.; Altintop, M.D.; Çiftçi, H.I.; Koga, R.; Otsuka, M.; Fujita, M.; Özdemir, A. Synthesis and Evaluation of New Pyrazoline Derivatives as Potential Anticancer Agents. Molecules 2015, 20, 19066–19084. [Google Scholar] [CrossRef] [PubMed]
- Ida, S.; Iwamaru, K.; Fujita, M.; Okamoto, Y.; Kudo, Y.; Kurosaki, H.; Otsuka, M. l-Histidyl-glycyl-glycyl-l-histidine. Amino-acid structuring of the bleomycin-type pentadentate metal-binding environment capable of efficient double-strand cleavage of plasmid DNA. Bioorganic Chem. 2015, 62, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Radwan, M.O.; Hamasaki, A.; Ejima, T.; Obata, E.; Koga, R.; Tateishi, H.; Okamoto, Y.; Fujita, M.; Nakao, M.; et al. A Novel Inhibitor of Farnesyltransferase with a Zinc Site Recognition Moiety and a Farnesyl Group. Bioorganic Med. Chem. Lett. 2017, 27, 3862–3866. [Google Scholar] [CrossRef] [PubMed]
- Koga, R.; Radwan, M.O.; Ejima, T.; Kanemaru, Y.; Tateishi, H.; Ali, T.F.S.; Ciftci, H.I.; Shibata, Y.; Taguchi, Y.; Inoue, J.-I.; et al. A Dithiol Compound Binds to the Zinc Finger Protein TRAF6 and Suppresses Its Ubiquitination. ChemMedChem 2017, 12, 1935–1941. [Google Scholar] [CrossRef]
- El-Shaheny, R.; Fuchigami, T.; Yoshida, S.; Radwan, M.O.; Nakayama, M. Complementary HPLC, in Silico Toxicity, and Molecular Docking Studies for Investigation of the Potential Influences of Gastric Acidity and Nitrite Content on Paracetamol Safety. Microchem. J. 2019, 150, 104–107. [Google Scholar] [CrossRef]
- Nishimura, N.; Radwan, M.O.; Amano, M.; Endo, S.; Fujii, E.; Hayashi, H.; Ueno, S.; Ueno, N.; Tatetsu, H.; Hata, H.; et al. Novel p97/VCP inhibitor induces endoplasmic reticulum stress and apoptosis in both bortezomib-sensitive and-resistant multiple myeloma cells. Cancer Sci. 2019, 00, 1–13. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 against SIRT2 (μM) | Compound | IC50 against SIRT2 (μM) |
|---|---|---|---|
| 1 | > 100 | STC7 | > 100 |
| STC1 | > 100 | STC8 | > 100 |
| STC2 | > 100 | STC9 | 17.2 ± 1.2 |
| STC3 | > 100 | STC10 | > 100 |
| STC4 | 10.8 ± 1.9 | STC11 | 9.5 ± 1.2 |
| STC5 | > 100 | TH-3 | 1.3 ± 0.2 |
| STC6 | > 100 |
| IC50 µM | |||||
|---|---|---|---|---|---|
| MCF-7 | HeLa | K562 | MT-2 | HL-60 | |
| SirReal2 | 17.08 ± 2.15 | 10.37 ± 0.94 | 13.65 ± 0.44 | 17.86 ± 1.52 | 90.6 ± 8.77 |
| STC4 | 3.16 ± 0.26 | 1.56 ± 0.17 | 2.17 ± 0.25 | 3.15 ± 0.13 | 0.45 ± 0.05 |
| STC9 | 3.32 ± 0.41 | 2.72 ± 0.19 | 2.53 ± 0.31 | 2.55 ± 0.28 | 1.19 ± 0.09 |
| STC11 | 10.03 ± 1.12 | 7.95 ± 0.81 | 14.99 ± 1.17 | 16.82 ± 1.04 | 12.78 ± 0.95 |
| TH-3 | 0.71 ± 0.08 | 0.37 ± 0.04 | 0.30 ± 0.02 | 0.17 ± 0.02 | 0.28 ± 0.04 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radwan, M.O.; Ciftci, H.I.; Ali, T.F.S.; Ellakwa, D.E.; Koga, R.; Tateishi, H.; Nakata, A.; Ito, A.; Yoshida, M.; Okamoto, Y.; et al. Antiproliferative S-Trityl-l-Cysteine -Derived Compounds as SIRT2 Inhibitors: Repurposing and Solubility Enhancement. Molecules 2019, 24, 3295. https://doi.org/10.3390/molecules24183295
Radwan MO, Ciftci HI, Ali TFS, Ellakwa DE, Koga R, Tateishi H, Nakata A, Ito A, Yoshida M, Okamoto Y, et al. Antiproliferative S-Trityl-l-Cysteine -Derived Compounds as SIRT2 Inhibitors: Repurposing and Solubility Enhancement. Molecules. 2019; 24(18):3295. https://doi.org/10.3390/molecules24183295
Chicago/Turabian StyleRadwan, Mohamed O., Halil I. Ciftci, Taha F. S. Ali, Doha E. Ellakwa, Ryoko Koga, Hiroshi Tateishi, Akiko Nakata, Akihiro Ito, Minoru Yoshida, Yoshinari Okamoto, and et al. 2019. "Antiproliferative S-Trityl-l-Cysteine -Derived Compounds as SIRT2 Inhibitors: Repurposing and Solubility Enhancement" Molecules 24, no. 18: 3295. https://doi.org/10.3390/molecules24183295
APA StyleRadwan, M. O., Ciftci, H. I., Ali, T. F. S., Ellakwa, D. E., Koga, R., Tateishi, H., Nakata, A., Ito, A., Yoshida, M., Okamoto, Y., Fujita, M., & Otsuka, M. (2019). Antiproliferative S-Trityl-l-Cysteine -Derived Compounds as SIRT2 Inhibitors: Repurposing and Solubility Enhancement. Molecules, 24(18), 3295. https://doi.org/10.3390/molecules24183295