
Yeast Models for Amyloids and Prions: Environmental Modulation and Drug Discovery




Abstract
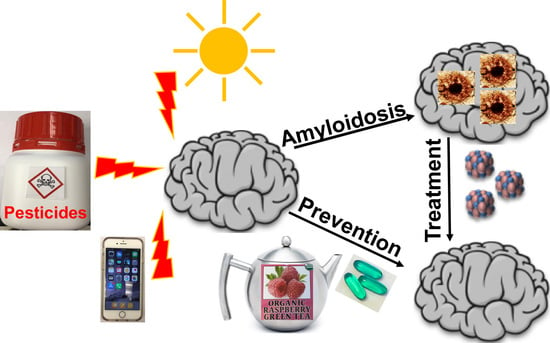
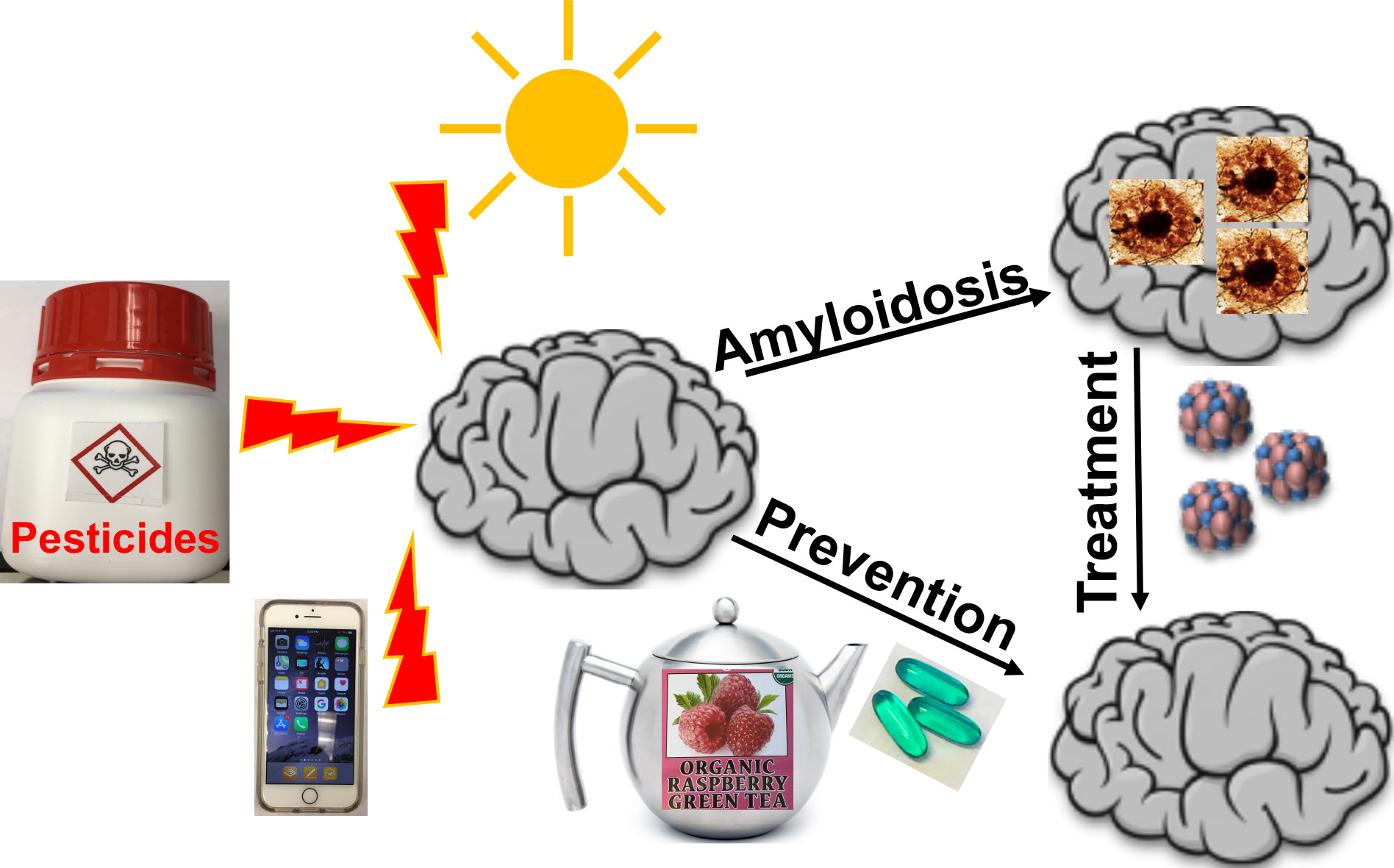
:
{kind=link}
{kind=link}
{kind=link}
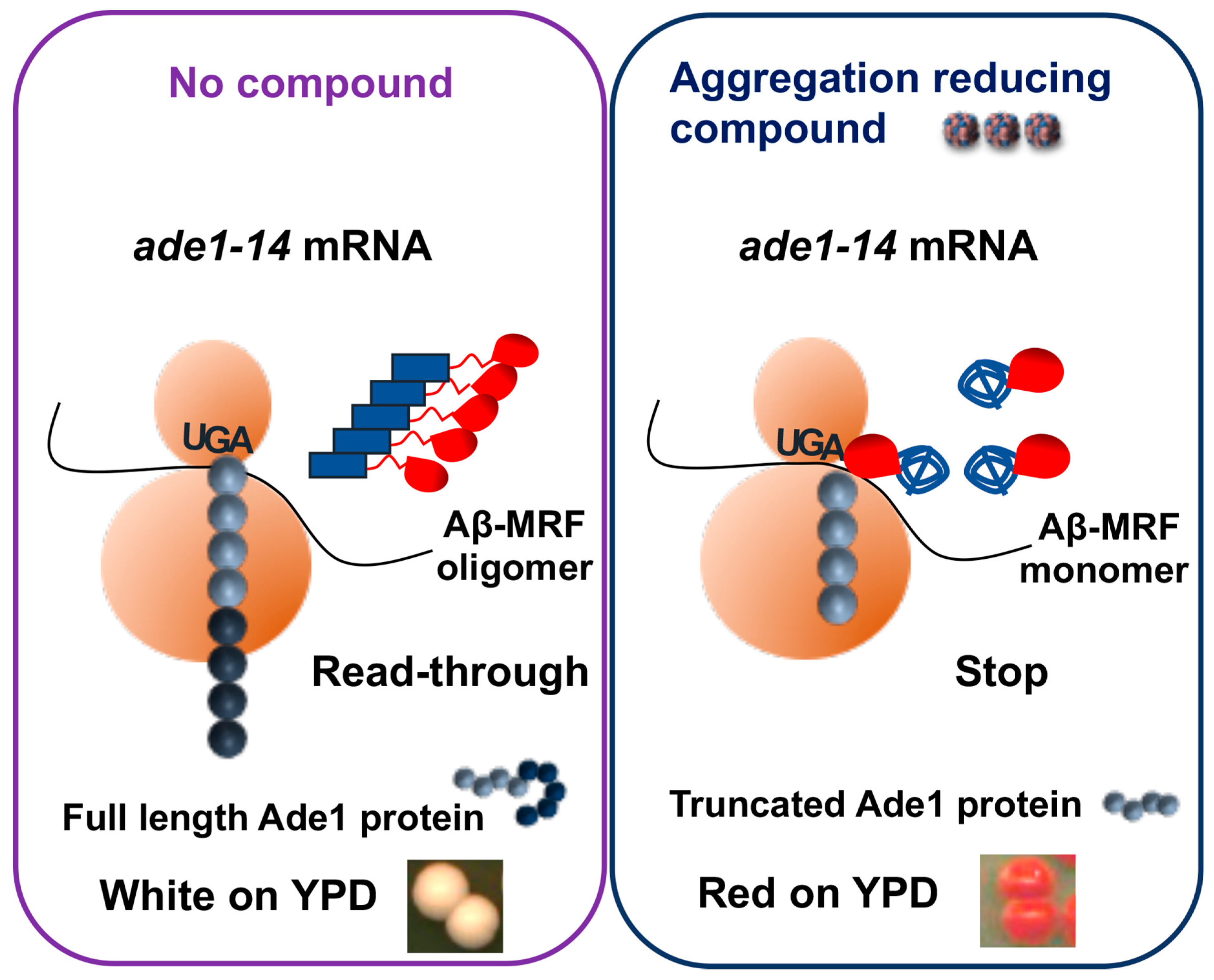{kind=link}
1. Protein Misfolding Diseases
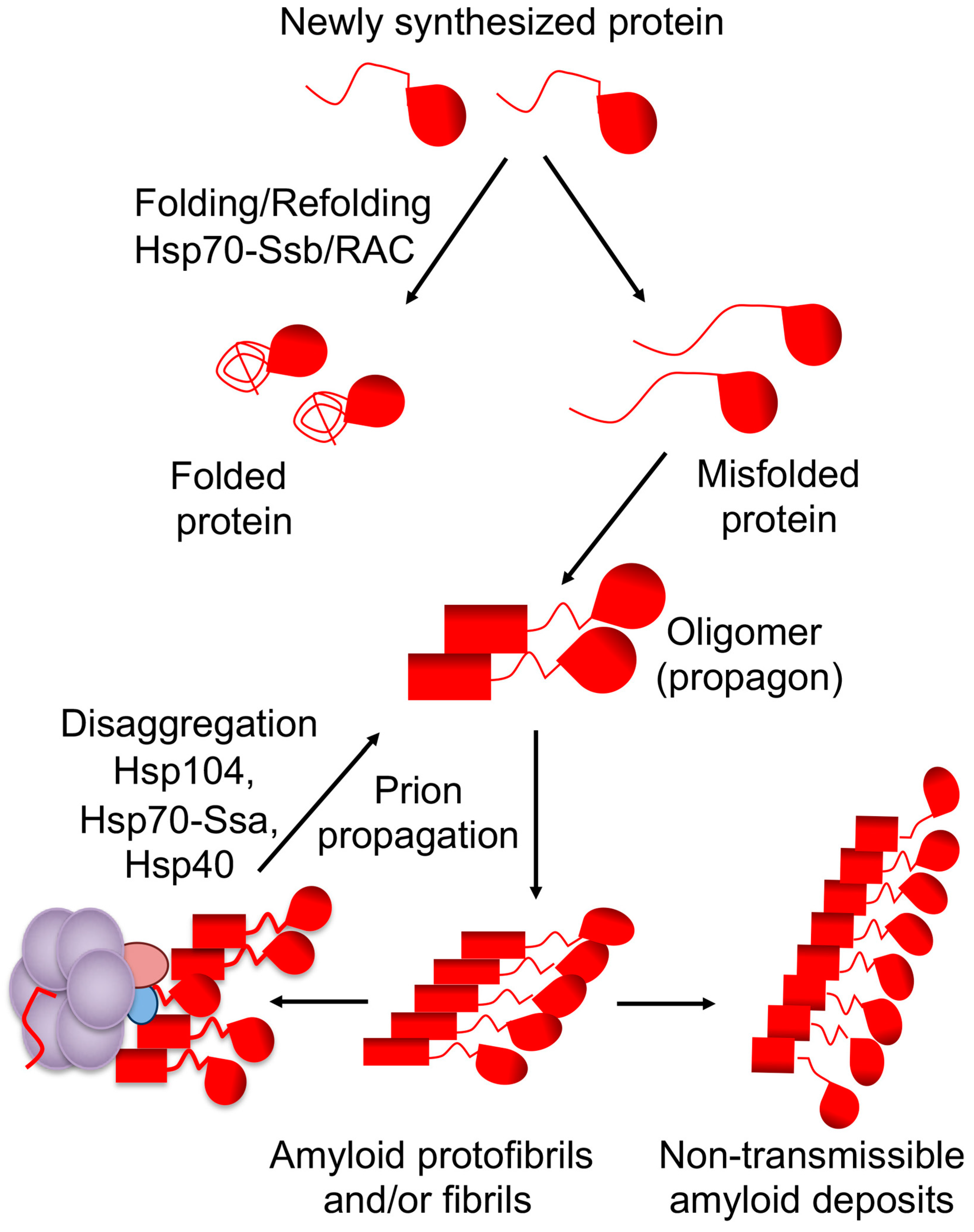
2. Yeast Prions and Protein Quality Control
3. Contribution of Environmental Factors to Amyloid Disease
4. Effects of Chemical Agents and Environmental Factors on the Formation of Yeast Prions
5. Clearance of Yeast Prions by Chemical Agents and Environmental Factors
6. Yeast Models for Discovery of Anti-Prion Drugs
7. Yeast Models for Identifying Candidate Drugs Against Alzheimer’s Disease
8. A Yeast Model for Discovery of Drugs against Huntington’s Disease
9. Drug Discovery in Yeast Model of Parkinson’s Disease
10. General and Specific Patterns of the Yeast Models for Anti-Amyloid Drug Discovery
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wilson, C.J.; Bommarius, A.S.; Champion, J.A.; Chernoff, Y.O.; Lynn, D.G.; Paravastu, A.K.; Liang, C.; Hsieh, M.C.; Heemstra, J.M. Biomolecular Assemblies: Moving from Observation to Predictive Design. Chem. Rev. 2018, 118, 11519–11574. [Google Scholar] [CrossRef] [PubMed]
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prusiner, S.B. Cell biology. A unifying role for prions in neurodegenerative diseases. Science 2012, 336, 1511–1513. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.C.; Jucker, M. Neurodegenerative diseases: Expanding the prion concept. Annu. Rev. Neurosci. 2015, 38, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Masnata, M.; Cicchetti, F. The Evidence for the Spread and Seeding Capacities of the Mutant Huntingtin Protein in in Vitro Systems and Their Therapeutic Implications. Front. Neurosci. 2017, 11, 647. [Google Scholar] [CrossRef]
- Walker, L.C. Prion-like mechanisms in Alzheimer disease. Handb. Clin. Neurol. 2018, 153, 303–319. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Walker, L.C. Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1341–1349. [Google Scholar] [CrossRef]
- Giasson, B.I.; Lee, V.M.; Trojanowski, J.Q. Interactions of amyloidogenic proteins. Neuromolecular Med. 2003, 4, 49–58. [Google Scholar] [CrossRef]
- Stohr, J.; Condello, C.; Watts, J.C.; Bloch, L.; Oehler, A.; Nick, M.; DeArmond, S.J.; Giles, K.; DeGrado, W.F.; Prusiner, S.B. Distinct synthetic Aβ prion strains producing different amyloid deposits in bigenic mice. Proc. Natl. Acad. Sci. USA 2014, 111, 10329–10334. [Google Scholar] [CrossRef]
- Watts, J.C.; Condello, C.; Stohr, J.; Oehler, A.; Lee, J.; DeArmond, S.J.; Lannfelt, L.; Ingelsson, M.; Giles, K.; Prusiner, S.B. Serial propagation of distinct strains of Abeta prions from Alzheimer’s disease patients. Proc. Natl. Acad. Sci. USA 2014, 111, 10323–10328. [Google Scholar] [CrossRef]
- Giles, K.; Olson, S.H.; Prusiner, S.B. Developing Therapeutics for PrP Prion Diseases. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B. Yeast and Fungal Prions. Cold Spring Harb. Perspect. Biol. 2016, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Liebman, S.W.; Chernoff, Y.O. Prions in yeast. Genetics 2012, 191, 1041–1072. [Google Scholar] [CrossRef] [PubMed]
- Kryndushkin, D.; Edskes, H.K.; Shewmaker, F.P.; Wickner, R.B. Prions. Cold Spring Harb. Protoc. 2017, 3, a006833. [Google Scholar] [CrossRef] [PubMed]
- Tenreiro, S.; Munder, M.C.; Alberti, S.; Outeiro, T.F. Harnessing the power of yeast to unravel the molecular basis of neurodegeneration. J. Neurochem. 2013, 127, 438–452. [Google Scholar] [CrossRef] [PubMed]
- Chernoff, Y.O. Stress and prions: Lessons from the yeast model. FEBS Lett. 2007, 581, 3695–3701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernova, T.A.; Wilkinson, K.D.; Chernoff, Y.O. Physiological and environmental control of yeast prions. FEMS Microbiol. Rev. 2014, 38, 326–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernova, T.A.; Wilkinson, K.D.; Chernoff, Y.O. Prions, Chaperones, and Proteostasis in Yeast. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.D.; Wegrzyn, R.D.; Chernova, T.A.; Muller, S.; Newnam, G.P.; Winslett, P.A.; Wittich, K.B.; Wilkinson, K.D.; Chernoff, Y.O. Hsp70 chaperones as modulators of prion life cycle: Novel effects of Ssa and Ssb on the Saccharomyces cerevisiae prion [PSI+]. Genetics 2005, 169, 1227–1242. [Google Scholar] [CrossRef] [PubMed]
- Chernova, T.A.; Allen, K.D.; Wesoloski, L.M.; Shanks, J.R.; Chernoff, Y.O.; Wilkinson, K.D. Pleiotropic effects of Ubp6 loss on drug sensitivities and yeast prion are due to depletion of the free ubiquitin pool. J. Biol. Chem. 2003, 278, 52102–52115. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.D.; Chernova, T.A.; Tennant, E.P.; Wilkinson, K.D.; Chernoff, Y.O. Effects of ubiquitin system alterations on the formation and loss of a yeast prion. J. Biol. Chem. 2007, 282, 3004–3013. [Google Scholar] [CrossRef] [PubMed]
- Chernoff, Y.O.; Lindquist, S.L.; Ono, B.; Inge-Vechtomov, S.G.; Liebman, S.W. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science 1995, 268, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Matveenko, A.G.; Barbitoff, Y.A.; Jay-Garcia, L.M.; Chernoff, Y.O.; Zhouravleva, G.A. Differential effects of chaperones on yeast prions: CURrent view. Curr. Genet. 2018, 64, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Newnam, G.P.; Wegrzyn, R.D.; Lindquist, S.L.; Chernoff, Y.O. Antagonistic interactions between yeast chaperones Hsp104 and Hsp70 in prion curing. Mol. Cell. Biol. 1999, 19, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Jung, G.; Jones, G.; Wegrzyn, R.D.; Masison, D.C. A role for cytosolic hsp70 in yeast [PSI+] prion propagation and [PSI+] as a cellular stress. Genetics 2000, 156, 559–570. [Google Scholar] [PubMed]
- Jones, G.; Song, Y.; Chung, S.; Masison, D.C. Propagation of Saccharomyces cerevisiae [PSI+] prion is impaired by factors that regulate Hsp70 substrate binding. Mol. Cell. Biol. 2004, 24, 3928–3937. [Google Scholar] [CrossRef] [PubMed]
- Hines, J.K.; Li, X.; Du, Z.; Higurashi, T.; Li, L.; Craig, E.A. [SWI], the prion formed by the chromatin remodeling factor Swi1, is highly sensitive to alterations in Hsp70 chaperone system activity. PLoS Genet. 2011, 7, e1001309. [Google Scholar] [CrossRef]
- Sporn, Z.A.; Hines, J.K. Hsp40 function in yeast prion propagation: Amyloid diversity necessitates chaperone functional complexity. Prion 2015, 9, 80–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astor, M.T.; Kamiya, E.; Sporn, Z.A.; Berger, S.E.; Hines, J.K. Variant-specific and reciprocal Hsp40 functions in Hsp104-mediated prion elimination. Mol. Microbiol. 2018, 109, 41–62. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.E.; Nolte, A.M.; Kamiya, E.; Hines, J.K. Three J-proteins impact Hsp104-mediated variant-specific prion elimination: A new critical role for a low-complexity domain. Curr. Genet. 2019, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.; Tyedmers, J.; Bukau, B.; Mogk, A. Hsp70 targets Hsp100 chaperones to substrates for protein disaggregation and prion fragmentation. J. Cell Biol. 2012, 198, 387–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kryndushkin, D.S.; Engel, A.; Edskes, H.; Wickner, R.B. Molecular chaperone Hsp104 can promote yeast prion generation. Genetics 2011, 188, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.N.; Zhao, X.; Yim, Y.I.; Todor, H.; Ellerbrock, R.; Reidy, M.; Eisenberg, E.; Masison, D.C.; Greene, L.E. Hsp104 overexpression cures Saccharomyces cerevisiae [PSI+] by causing dissolution of the prion seeds. Eukaryot. Cell 2014, 13, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Newnam, G.P.; Birchmore, J.L.; Chernoff, Y.O. Destabilization and recovery of a yeast prion after mild heat shock. J. Mol. Biol. 2011, 408, 432–448. [Google Scholar] [CrossRef] [PubMed]
- Ness, F.; Cox, B.S.; Wongwigkarn, J.; Naeimi, W.R.; Tuite, M.F. Over-expression of the molecular chaperone Hsp104 in Saccharomyces cerevisiae results in the malpartition of [PSI+] propagons. Molec. Microbiol. 2017, 104, 125–143. [Google Scholar] [CrossRef] [PubMed]
- Bukau, B.; Weissman, J.; Horwich, A. Molecular chaperones and protein quality control. Cell 2006, 125, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.; Tyedmers, J.; Bukau, B.; Mogk, A. Chaperone networks in protein disaggregation and prion propagation. J. Struct. Biol. 2012, 179, 152–160. [Google Scholar] [CrossRef]
- Rampelt, H.; Kirstein-Miles, J.; Nillegoda, N.B.; Chi, K.; Scholz, S.R.; Morimoto, R.I.; Bukau, B. Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. EMBO J. 2012, 31, 4221–4235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattoo, R.U.H.; Goloubinoff, P. Molecular chaperones are nanomachines that catalytically unfold misfolded and alternatively folded proteins. Cell Mol. Life Sci. 2014, 71, 3311–3325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrente, M.P.; Shorter, J. The metazoan protein disaggregase and amyloid depolymerase system: Hsp110, Hsp70, Hsp40, and small heat shock proteins. Prion 2013, 7, 457–463. [Google Scholar] [CrossRef]
- Zaarur, N.; Xu, X.; Lestienne, P.; Meriin, A.B.; McComb, M.; Costello, C.E.; Newnam, G.P.; Ganti, R.; Romanova, N.V.; Shanmugasundaram, M.; et al. RuvbL1 and RuvbL2 enhance aggresome formation and disaggregate amyloid fibrils. EMBO J. 2015, 34, 2363–2382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vashist, S.; Cushman, M.; Shorter, J. Applying Hsp104 to protein-misfolding disorders. Biochem. Cell Biol. 2010, 88, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Shorter, J. Engineering therapeutic protein disaggregases. Mol. Biol. Cell 2016, 27, 1556–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poepsel, S.; Sprengel, A.; Sacca, B.; Kaschani, F.; Kaiser, M.; Gatsogiannis, C.; Raunser, S.; Clausen, T.; Ehrmann, M. Determinants of amyloid fibril degradation by the PDZ protease HTRA1. Nat. Chem. Biol. 2015, 11, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ritz, B. The Search for Environmental Causes of Parkinson’s Disease: Moving Forward. J. Parkinsons Dis. 2018, 8, S9–S17. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Mayeux, R. Alzheimer disease: Epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharm. 2014, 88, 640–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prusiner, S.B. Biology and genetics of prions causing neurodegeneration. Annu. Rev. Genet. 2013, 47, 601–623. [Google Scholar] [CrossRef] [PubMed]
- Novati, A.; Hentrich, T.; Wassouf, Z.; Weber, J.J.; Yu-Taeger, L.; Deglon, N.; Nguyen, H.P.; Schulze-Hentrich, J.M. Environment-dependent striatal gene expression in the BACHD rat model for Huntington disease. Sci. Rep. 2018, 8, 5803. [Google Scholar] [CrossRef] [Green Version]
- Mo, C.; Hannan, A.J.; Renoir, T. Environmental factors as modulators of neurodegeneration: Insights from gene-environment interactions in Huntington’s disease. Neurosci. Biobehav. Rev. 2015, 52, 178–192. [Google Scholar] [CrossRef]
- Ascherio, A.; Chen, H.; Weisskopf, M.G.; O’Reilly, E.; McCullough, M.L.; Calle, E.E.; Schwarzschild, M.A.; Thun, M.J. Pesticide exposure and risk for Parkinson’s disease. Ann. Neurol. 2006, 60, 197–203. [Google Scholar] [CrossRef]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, paraquat, and Parkinson’s disease. Env. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Kanthasamy, A.; Jin, H.; Anantharam, V.; Kanthasamy, A.G. Paraquat induces epigenetic changes by promoting histone acetylation in cell culture models of dopaminergic degeneration. Neurotoxicology 2011, 32, 586–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbas, M.M.; Xu, Z.; Tan, L.C.S. Epidemiology of Parkinson’s Disease-East Versus West. Mov. Disord. Clin. Pr. 2018, 5, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Bihaqi, S.W. Early life exposure to lead (Pb) and changes in DNA methylation: Relevance to Alzheimer’s disease. Rev. Env. Health 2019, 34, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Orsini, N. Coffee Consumption and Risk of Dementia and Alzheimer’s Disease: A Dose-Response Meta-Analysis of Prospective Studies. Nutrients 2018, 10, 1501. [Google Scholar] [CrossRef] [PubMed]
- Ritz, B.; Lee, P.C.; Lassen, C.F.; Arah, O.A. Parkinson disease and smoking revisited: Ease of quitting is an early sign of the disease. Neurology 2014, 83, 1396–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davanipour, Z.; Sobel, E. Long-term exposure to magnetic fields and the risks of Alzheimer’s disease and breast cancer: Further biological research. Pathophysiology 2009, 16, 149–156. [Google Scholar] [CrossRef]
- Arendash, G.W.; Mori, T.; Dorsey, M.; Gonzalez, R.; Tajiri, N.; Borlongan, C. Electromagnetic treatment to old Alzheimer’s mice reverses β-amyloid deposition, modifies cerebral blood flow, and provides selected cognitive benefit. PLoS ONE 2012, 7, e35751. [Google Scholar] [CrossRef]
- Banaceur, S.; Banasr, S.; Sakly, M.; Abdelmelek, H. Whole body exposure to 2.4 GHz WIFI signals: Effects on cognitive impairment in adult triple transgenic mouse models of Alzheimer’s disease (3xTg-AD). Behav. Brain Res. 2013, 240, 197–201. [Google Scholar] [CrossRef]
- Schuz, J.; Waldemar, G.; Olsen, J.H.; Johansen, C. Risks for central nervous system diseases among mobile phone subscribers: A Danish retrospective cohort study. PLoS ONE 2009, 4, e4389. [Google Scholar] [CrossRef]
- Grova, N.; Schroeder, H.; Olivier, J.L.; Turner, J.D. Epigenetic and Neurological Impairments Associated with Early Life Exposure to Persistent Organic Pollutants. Int. J. Genom. 2019, 2019, 19. [Google Scholar] [CrossRef] [PubMed]
- Borenstein, A.R.; Copenhaver, C.I.; Mortimer, J.A. Early-life risk factors for Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2006, 20, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.B.; O’Callaghan, J.P. Do early-life insults contribute to the late-life development of Parkinson and Alzheimer diseases? Metabolism 2008, 57 (Suppl. 2), S44–S49. [Google Scholar] [CrossRef]
- Kennedy, B.K.; Berger, S.L.; Brunet, A.; Campisi, J.; Cuervo, A.M.; Epel, E.S.; Franceschi, C.; Lithgow, G.J.; Morimoto, R.I.; Pessin, J.E.; et al. Geroscience: Linking aging to chronic disease. Cell 2014, 159, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Fu, Z.; Meng, L.; He, M.; Zhang, Z. The Early Events That Initiateβ-Amyloid Aggregation in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 359. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.N.; Bondy, S.C. Oxidative and Inflammatory Events in Prion Diseases: Can They Be Therapeutic Targets? Curr. Aging Sci. 2019, 11, 216–225. [Google Scholar] [CrossRef] [Green Version]
- Walker, L.C.; Lynn, D.G.; Chernoff, Y.O. A standard model of Alzheimer’s disease? Prion 2018, 12, 261–265. [Google Scholar] [CrossRef]
- Chernoff, Y.O.; Derkach, I.L.; Inge-Vechtomov, S.G. Multicopy SUP35 gene induces de-novo appearance of psi-like factors in the yeast Saccharomyces cerevisiae. Curr. Genet. 1993, 24, 268–270. [Google Scholar] [CrossRef]
- Masison, D.C.; Wickner, R.B. Prion-inducing domain of yeast Ure2p and protease resistance of Ure2p in prion-containing cells. Science 1995, 270, 93–95. [Google Scholar] [CrossRef]
- Derkatch, I.L.; Chernoff, Y.O.; Kushnirov, V.V.; Inge-Vechtomov, S.G.; Liebman, S.W. Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics 1996, 144, 1375–1386. [Google Scholar]
- Derkatch, I.L.; Bradley, M.E.; Hong, J.Y.; Liebman, S.W. Prions affect the appearance of other prions: The story of [PIN+]. Cell 2001, 106, 171–182. [Google Scholar] [CrossRef]
- Osherovich, L.Z.; Weissman, J.S. Multiple Gln/Asn-rich prion domains confer susceptibility to induction of the yeast [PSI+] prion. Cell 2001, 106, 183–194. [Google Scholar] [CrossRef]
- Tyedmers, J.; Madariaga, M.L.; Lindquist, S. Prion switching in response to environmental stress. PLoS Biol. 2008, 6, e294. [Google Scholar] [CrossRef]
- Morano, K.A.; Grant, C.M.; Moye-Rowley, W.S. The response to heat shock and oxidative stress in Saccharomyces cerevisiae. Genetics 2012, 190, 1157–1195. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.M. Sup35 methionine oxidation is a trigger for de novo [PSI+] prion formation. Prion 2015, 9, 257–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doronina, V.A.; Staniforth, G.L.; Speldewinde, S.H.; Tuite, M.F.; Grant, C.M. Oxidative stress conditions increase the frequency of de novo formation of the yeast [PSI+] prion. Mol. Microbiol. 2015, 96, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, N.; Kritsiligkou, P.; Grant, C.M. ER stress causes widespread protein aggregation and prion formation. J. Cell Biol. 2017, 216, 2295–2304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derkatch, I.L.; Bradley, M.E.; Zhou, P.; Chernoff, Y.O.; Liebman, S.W. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics 1997, 147, 507–519. [Google Scholar]
- Lian, H.Y.; Lin, K.W.; Yang, C.; Cai, P. Generation and propagation of yeast prion [URE3] are elevated under electromagnetic field. Cell Stress Chaperones 2018, 23, 581–594. [Google Scholar] [CrossRef]
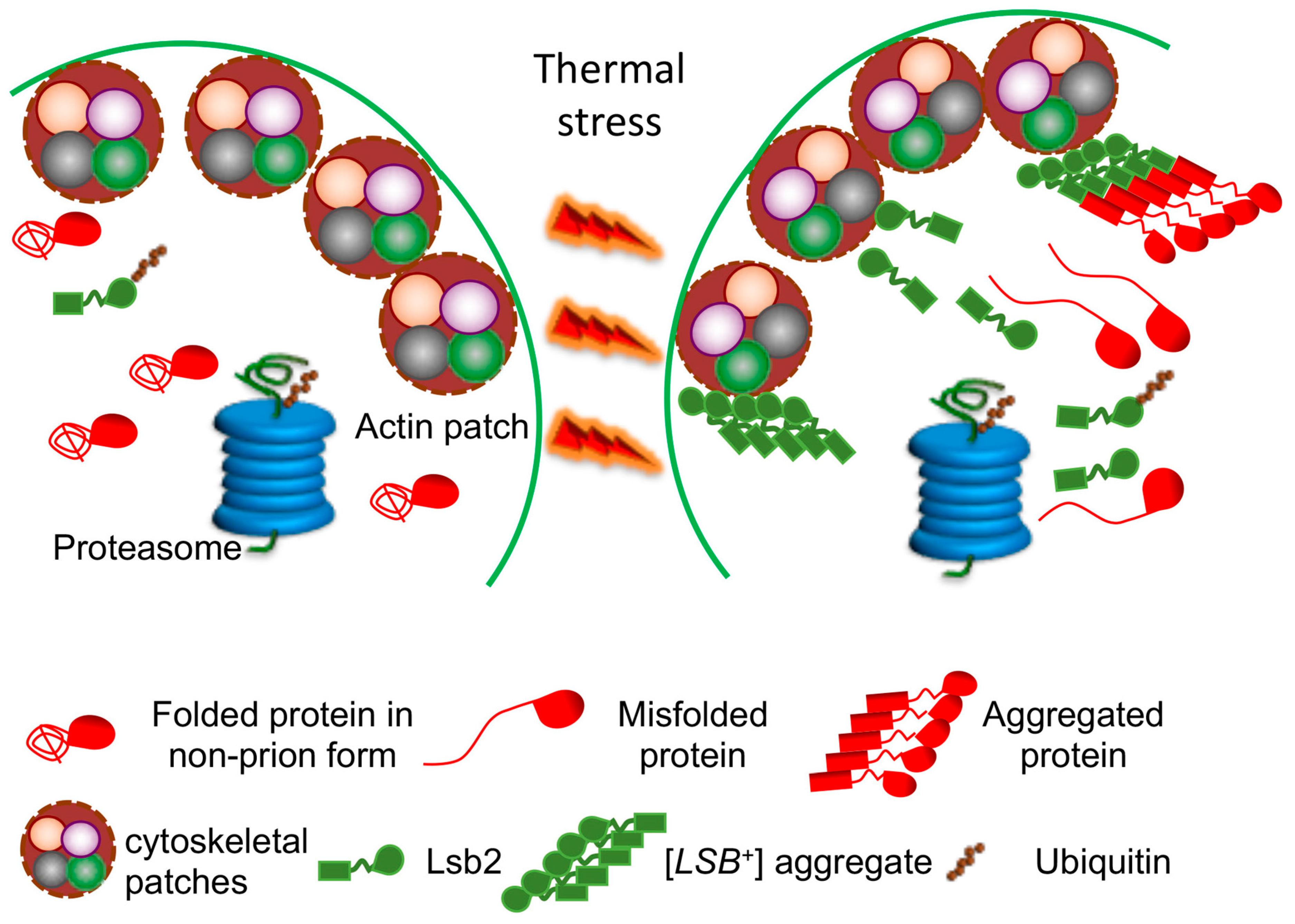
- Chernova, T.A.; Kiktev, D.A.; Romanyuk, A.V.; Shanks, J.R.; Laur, O.; Ali, M.; Ghosh, A.; Kim, D.; Yang, Z.; Mang, M.; et al. Yeast Short-Lived Actin-Associated Protein Forms a Metastable Prion in Response to Thermal Stress. Cell Rep. 2017, 18, 751–761. [Google Scholar] [CrossRef]
- Chernova, T.A.; Romanyuk, A.V.; Karpova, T.S.; Shanks, J.R.; Ali, M.; Moffatt, N.; Howie, R.L.; O’Dell, A.; McNally, J.G.; Liebman, S.W.; et al. Prion induction by the short-lived, stress-induced protein Lsb2 is regulated by ubiquitination and association with the actin cytoskeleton. Mol. Cell 2011, 43, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Chernova, T.A.; Chernoff, Y.O.; Wilkinson, K.D. Prion-based memory of heat stress in yeast. Prion 2017, 11, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Holmes, D.L.; Lancaster, A.K.; Lindquist, S.; Halfmann, R. Heritable remodeling of yeast multicellularity by an environmentally responsive prion. Cell 2013, 153, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, G.; Shimazu, N.; Tanaka, M. A yeast prion, Mod5, promotes acquired drug resistance and cell survival under environmental stress. Science 2012, 336, 355–359. [Google Scholar] [CrossRef]
- Du, Z.; Park, K.W.; Yu, H.; Fan, Q.; Li, L. Newly identified prion linked to the chromatin-remodeling factor Swi1 in Saccharomyces cerevisiae. Nat. Genet. 2008, 40, 460–465. [Google Scholar] [CrossRef] [Green Version]
- Alberti, S.; Halfmann, R.; King, O.; Kapila, A.; Lindquist, S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 2009, 137, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Hatin, I.; Bidou, L.; Cullin, C.; Rousset, J.P. Translational errors as an early event in prion conversion. Cell Mol. Biol. 2001, 47, OL23-8. [Google Scholar]
- Brown, J.C.; Lindquist, S. A heritable switch in carbon source utilization driven by an unusual yeast prion. Genes Dev. 2009, 23, 2320–2332. [Google Scholar] [CrossRef] [Green Version]
- Jarosz, D.F.; Lancaster, A.K.; Brown, J.C.; Lindquist, S. An evolutionarily conserved prion-like element converts wild fungi from metabolic specialists to generalists. Cell 2014, 158, 1072–1082. [Google Scholar] [CrossRef]
- Jarosz, D.F.; Brown, J.C.S.; Walker, G.A.; Datta, M.S.; Ung, W.L.; Lancaster, A.K.; Rotem, A.; Chang, A.; Newby, G.A.; Weitz, D.A.; et al. Cross-kingdom chemical communication drives a heritable, mutually beneficial prion-based transformation of metabolism. Cell 2014, 158, 1083–1093. [Google Scholar] [CrossRef]
- Garcia, D.M.; Dietrich, D.; Clardy, J.; Jarosz, D.F. A common bacterial metabolite elicits prion-based bypass of glucose repression. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Tapia, H.; Koshland, D.E. Trehalose is a versatile and long-lived chaperone for desiccation tolerance. Curr. Biol. Cb. 2014, 24, 2758–2766. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Brundin, P. The ubiquitin proteasome system in neurodegenerative diseases: Sometimes the chicken, sometimes the egg. Neuron 2003, 40, 427–446. [Google Scholar] [CrossRef]
- Ohsumi, Y. Historical landmarks of autophagy research. Cell Res. 2014, 24, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Nystrom, T.; Liu, B. The mystery of aging and rejuvenation-a budding topic. Curr. Opin. Microbiol. 2014, 18, 61–67. [Google Scholar] [CrossRef]
- Tuite, M.F.; Mundy, C.R.; Cox, B.S. Agents that cause a high frequency of genetic change from [psi+] to [psi-] in Saccharomyces cerevisiae. Genetics 1981, 98, 691–711. [Google Scholar] [PubMed]
- Ferreira, P.C.; Ness, F.; Edwards, S.R.; Cox, B.S.; Tuite, M.F. The elimination of the yeast [PSI+] prion by guanidine hydrochloride is the result of Hsp104 inactivation. Mol. Microbiol. 2001, 40, 1357–1369. [Google Scholar] [CrossRef]
- Ness, F.; Ferreira, P.; Cox, B.S.; Tuite, M.F. Guanidine hydrochloride inhibits the generation of prion seeds but not prion protein aggregation in yeast. Mol. Cell. Biol. 2002, 22, 5593–5605. [Google Scholar] [CrossRef]
- Jung, G.; Jones, G.; Masison, D.C. Amino acid residue 184 of yeast Hsp104 chaperone is critical for prion-curing by guanidine, prion propagation, and thermotolerance. Proc. Natl. Acad. Sci. USA 2002, 99, 9936–9941. [Google Scholar] [CrossRef] [Green Version]
- Grimminger, V.; Richter, K.; Imhof, A.; Buchner, J.; Walter, S. The prion curing agent guanidinium chloride specifically inhibits ATP hydrolysis by Hsp104. J. Biol. Chem. 2004, 279, 7378–7383. [Google Scholar] [CrossRef]
- Toyama, B.H.; Kelly, M.J.; Gross, J.D.; Weissman, J.S. The structural basis of yeast prion strain variants. Nature 2007, 449, 233–237. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.; Chernova, T.A.; Newnam, G.P.; Yin, L.; Shanks, J.; Karpova, T.S.; Lee, A.; Laur, O.; Subramanian, S.; Kim, D.; et al. Stress-dependent proteolytic processing of the actin assembly protein Lsb1 modulates a yeast prion. J. Biol. Chem. 2014, 289, 27625–27639. [Google Scholar] [CrossRef] [PubMed]
- Klaips, C.L.; Hochstrasser, M.L.; Langlois, C.R.; Serio, T.R. Spatial quality control bypasses cell-based limitations on proteostasis to promote prion curing. eLife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Howie, R.L.; Jay-Garcia, L.M.; Kiktev, D.A.; Faber, Q.L.; Murphy, M.; Rees, K.A.; Sachwani, N.; Chernoff, Y.O. Role of the Cell Asymmetry Apparatus and Ribosome-Associated Chaperones in the Destabilization of a Saccharomyces cerevisiae Prion by Heat Shock. Genetics 2019, 212, 757–771. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Larsson, L.; Caballero, A.; Hao, X.; Oling, D.; Grantham, J.; Nystrom, T. The polarisome is required for segregation and retrograde transport of protein aggregates. Cell 2010, 140, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Kiktev, D.A.; Melomed, M.M.; Lu, C.D.; Newnam, G.P.; Chernoff, Y.O. Feedback control of prion formation and propagation by the ribosome-associated chaperone complex. Mol. Microbiol. 2015, 96, 621–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernoff, Y.O.; Kiktev, D.A. Dual role of ribosome-associated chaperones in prion formation and propagation. Curr. Genet. 2016, 62, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, H.; Conz, C.; Otto, H.; Wolfle, T.; Fitzke, E.; Mayer, M.P.; Rospert, S. The chaperone network connected to human ribosome-associated complex. Mol. Cell. Biol. 2011, 31, 1160–1173. [Google Scholar] [CrossRef]
- Ciechanover, A. Intracellular protein degradation: From a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Best Pr. Res. Clin. Haematol. 2017, 30, 341–355. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Dennissen, F.J.; Kholod, N.; van Leeuwen, F.W. The ubiquitin proteasome system in neurodegenerative diseases: Culprit, accomplice or victim? Prog. Neurobiol. 2012, 96, 190–207. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Schwartz, D.; Elias, J.E.; Thoreen, C.C.; Cheng, D.; Marsischky, G.; Roelofs, J.; Finley, D.; Gygi, S.P. A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 2003, 21, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Thellung, S.; Corsaro, A.; Nizzari, M.; Barbieri, F.; Florio, T. Autophagy Activator Drugs: A New Opportunity in Neuroprotection from Misfolded Protein Toxicity. Int. J. Mol. Sci. 2019, 20, 901. [Google Scholar] [CrossRef] [PubMed]
- Speldewinde, S.H.; Grant, C.M. Spermidine cures yeast of prions. Microb. Cell 2015, 3, 46–48. [Google Scholar] [CrossRef] [PubMed]
- Kachroo, A.H.; Laurent, J.M.; Yellman, C.M.; Meyer, A.G.; Wilke, C.O.; Marcotte, E.M. Evolution. Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science 2015, 348, 921–925. [Google Scholar] [CrossRef] [PubMed]
- Truong, D.M.; Boeke, J.D. Resetting the Yeast Epigenome with Human Nucleosomes. Cell 2017, 171, 1508–1519. [Google Scholar] [CrossRef] [PubMed]
- Bach, S.; Talarek, N.; Andrieu, T.; Vierfond, J.M.; Mettey, Y.; Galons, H.; Dormont, D.; Meijer, L.; Cullin, C.; Blondel, M. Isolation of drugs active against mammalian prions using a yeast-based screening assay. Nat. Biotechnol. 2003, 21, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Bach, S.; Tribouillard, D.; Talarek, N.; Desban, N.; Gug, F.; Galons, H.; Blondel, M. A yeast-based assay to isolate drugs active against mammalian prions. Methods 2006, 39, 72–77. [Google Scholar] [CrossRef] [Green Version]
- Voisset, C.; Saupe, S.J.; Galons, H.; Blondel, M. Procedure for identification and characterization of drugs efficient against mammalian prion: From a yeast-based antiprion drug screening assay to in vivo mouse models. Infect. Disord. Drug Targets 2009, 9, 31–39. [Google Scholar] [CrossRef]
- Oumata, N.; Nguyen, P.H.; Beringue, V.; Soubigou, F.; Pang, Y.; Desban, N.; Massacrier, C.; Morel, Y.; Paturel, C.; Contesse, M.A.; et al. The toll-like receptor agonist imiquimod is active against prions. PLoS ONE 2013, 8, e72112. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, D.; Sanyal, S. Protein folding activity of the ribosome (PFAR)—A target for antiprion compounds. Viruses 2014, 6, 3907–3924. [Google Scholar] [CrossRef] [PubMed]
- Blondel, M.; Soubigou, F.; Evrard, J.; Nguyen, P.H.; Hasin, N.; Chedin, S.; Gillet, R.; Contesse, M.A.; Friocourt, G.; Stahl, G.; et al. Protein Folding Activity of the Ribosome is involved in Yeast Prion Propagation. Sci. Rep. 2016, 6, 32117. [Google Scholar] [CrossRef] [PubMed]
- Jennings, L.K.; Ahmed, I.; Munn, A.L.; Carroll, A.R. Yeast-based screening of natural product extracts results in the identification of prion inhibitors from a marine sponge. Prion 2018, 12, 234–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seynnaeve, D.; Vecchio, M.D.; Fruhmann, G.; Verelst, J.; Cools, M.; Beckers, J.; Mulvihill, D.P.; Winderickx, J.; Franssens, V. Recent Insights on Alzheimer’s Disease Originating from Yeast Models. Int. J. Mol. Sci. 2018, 19, 1947. [Google Scholar] [CrossRef] [PubMed]
- Klein, W.L.; Stine, W.B., Jr.; Teplow, D.B. Small assemblies of unmodified amyloid beta-protein are the proximate neurotoxin in Alzheimer’s disease. Neurobiol. Aging 2004, 25, 569–580. [Google Scholar] [CrossRef]
- Bagriantsev, S.; Liebman, S. Modulation of Abeta42 low-n oligomerization using a novel yeast reporter system. BMC Biol. 2006, 4, 32. [Google Scholar] [CrossRef] [PubMed]
- Macreadie, I.; Lotfi-Miri, M.; Mohotti, S.; Shapira, D.; Bennett, L.; Varghese, J. Validation of folate in a convenient yeast assay suited for identification of inhibitors of Alzheimer’s amyloid-β aggregation. J. Alzheimers Dis. 2008, 15, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Park, S.K.; Ratia, K.; Ba, M.; Valencik, M.; Liebman, S.W. Inhibition of Aβ42 oligomerization in yeast by a PICALM ortholog and certain FDA approved drugs. Microb. Cell 2016, 3, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Von der Haar, T.; Josse, L.; Wright, P.; Zenthon, J.; Tuite, M.F. Development of a novel yeast cell-based system for studying the aggregation of Alzheimer’s disease-associated Aβ peptides in vivo. Neurodegener. Dis. 2007, 4, 136–147. [Google Scholar] [CrossRef]
- Park, S.K.; Pegan, S.D.; Mesecar, A.D.; Jungbauer, L.M.; LaDu, M.J.; Liebman, S.W. Development and validation of a yeast high-throughput screen for inhibitors of Aβ42 oligomerization. Dis. Model. Mech. 2011, 4, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Rubel, A.A.; Ryzhova, T.A.; Antonets, K.S.; Chernoff, Y.O.; Galkin, A. Identification of PrP sequences essential for the interaction between the PrP polymers and Abeta peptide in a yeast-based assay. Prion 2013, 7, 469–476. [Google Scholar] [CrossRef]
- Caine, J.; Sankovich, S.; Antony, H.; Waddington, L.; Macreadie, P.; Varghese, J.; Macreadie, I. Alzheimer’s Abeta fused to green fluorescent protein induces growth stress and a heat shock response. FEMS Yeast Res. 2007, 7, 1230–1236. [Google Scholar] [CrossRef]
- Lauren, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Salazar, S.V.; Gallardo, C.; Kaufman, A.C.; Herber, C.S.; Haas, L.T.; Robinson, S.; Manson, J.C.; Lee, M.K.; Strittmatter, S.M. Conditional Deletion of Prnp Rescues Behavioral and Synaptic Deficits after Disease Onset in Transgenic Alzheimer’s Disease. J. Neurosci. 2017, 37, 9207–9221. [Google Scholar] [CrossRef]
- Rajasekhar, K.; Suresh, S.N.; Manjithaya, R.; Govindaraju, T. Rationally designed peptidomimetic modulators of abeta toxicity in Alzheimer’s disease. Sci. Rep. 2015, 5, 8139. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Gavrilova, S.I.; Sano, M.; Thomas, R.G.; Aisen, P.S.; Bachurin, S.O.; Seely, L.; Hung, D.; Dimebon, I. Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer’s disease: A randomised, double-blind, placebo-controlled study. Lancet 2008, 372, 207–215. [Google Scholar] [CrossRef]
- Bharadwaj, P.R.; Verdile, G.; Barr, R.K.; Gupta, V.; Steele, J.W.; Lachenmayer, M.L.; Yue, Z.; Ehrlich, M.E.; Petsko, G.; Ju, S.; et al. Latrepirdine (dimebon) enhances autophagy and reduces intracellular GFP-Aβ42 levels in yeast. J. Alzheimers Dis. 2012, 32, 949–967. [Google Scholar] [CrossRef]
- Steele, J.W.; Lachenmayer, M.L.; Ju, S.; Stock, A.; Liken, J.; Kim, S.H.; Delgado, L.M.; Alfaro, I.E.; Bernales, S.; Verdile, G.; et al. Latrepirdine improves cognition and arrests progression of neuropathology in an Alzheimer’s mouse model. Mol. Psychiatry 2013, 18, 889–897. [Google Scholar] [CrossRef]
- Matlack, K.E.; Tardiff, D.F.; Narayan, P.; Hamamichi, S.; Caldwell, K.A.; Caldwell, G.A.; Lindquist, S. Clioquinol promotes the degradation of metal-dependent amyloid-beta (Aβ) oligomers to restore endocytosis and ameliorate Aβ toxicity. Proc. Natl. Acad. Sci. USA 2014, 111, 4013–4018. [Google Scholar] [CrossRef]
- D’Angelo, F.; Vignaud, H.; Di Martino, J.; Salin, B.; Devin, A.; Cullin, C.; Marchal, C. A yeast model for amyloid-β aggregation exemplifies the role of membrane trafficking and PICALM in cytotoxicity. Dis. Model. Mech. 2013, 6, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Cherny, R.A.; Atwood, C.S.; Xilinas, M.E.; Gray, D.N.; Jones, W.D.; McLean, C.A.; Barnham, K.J.; Volitakis, I.; Fraser, F.W.; Kim, Y.; et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 2001, 30, 665–676. [Google Scholar] [CrossRef]
- Lannfelt, L.; Blennow, K.; Zetterberg, H.; Batsman, S.; Ames, D.; Harrison, J.; Masters, C.L.; Targum, S.; Bush, A.I.; Murdoch, R.; et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Aβ as a modifying therapy for Alzheimer’s disease: A phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008, 7, 779–786. [Google Scholar] [CrossRef]
- Treusch, S.; Hamamichi, S.; Goodman, J.L.; Matlack, K.E.; Chung, C.Y.; Baru, V.; Shulman, J.M.; Parrado, A.; Bevis, B.J.; Valastyan, J.S.; et al. Functional links between Aβ toxicity, endocytic trafficking, and Alzheimer’s disease risk factors in yeast. Science 2011, 334, 1241–1245. [Google Scholar] [CrossRef]
- Hofer, S.; Kainz, K.; Zimmermann, A.; Bauer, M.A.; Pendl, T.; Poglitsch, M.; Madeo, F.; Carmona-Gutierrez, D. Studying Huntington’s Disease in Yeast: From Mechanisms to Pharmacological Approaches. Front. Mol. Neurosci. 2018, 11, 318. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Aylward, E.H.; Wild, E.J.; Langbehn, D.R.; Long, J.D.; Warner, J.H.; Scahill, R.I.; Leavitt, B.R.; Stout, J.C.; Paulsen, J.S.; et al. Huntington disease: Natural history, biomarkers and prospects for therapeutics. Nat. Rev. Neurol. 2014, 10, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Krobitsch, S.; Lindquist, S. Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc. Natl. Acad. Sci. USA 2000, 97, 1589–1594. [Google Scholar] [CrossRef] [Green Version]
- Meriin, A.B.; Zhang, X.; He, X.; Newnam, G.P.; Chernoff, Y.O.; Sherman, M.Y. Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J. Cell Biol. 2002, 157, 997–1004. [Google Scholar] [CrossRef]
- Gokhale, K.C.; Newnam, G.P.; Sherman, M.Y.; Chernoff, Y.O. Modulation of prion-dependent polyglutamine aggregation and toxicity by chaperone proteins in the yeast model. J. Biol. Chem. 2005, 280, 22809–22818. [Google Scholar] [CrossRef]
- Hageman, J.; Rujano, M.A.; van Waarde, M.A.; Kakkar, V.; Dirks, R.P.; Govorukhina, N.; Oosterveld-Hut, H.M.; Lubsen, N.H.; Kampinga, H.H. A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol. Cell 2010, 37, 355–369. [Google Scholar] [CrossRef]
- Kakkar, V.; Mansson, C.; de Mattos, E.P.; Bergink, S.; van der Zwaag, M.; van Waarde, M.; Kloosterhuis, N.J.; Melki, R.; van Cruchten, R.T.P.; Al-Karadaghi, S.; et al. The S/T-Rich Motif in the DNAJB6 Chaperone Delays Polyglutamine Aggregation and the Onset of Disease in a Mouse Model. Mol. Cell 2016, 62, 272–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, H.; Romanova, N.V.; Allen, K.D.; Chandramowlishwaran, P.; Gokhale, K.; Newnam, G.P.; Mieczkowski, P.; Sherman, M.Y.; Chernoff, Y.O. Polyglutamine toxicity is controlled by prion composition and gene dosage in yeast. PLoS Genet. 2012, 8, e1002634. [Google Scholar] [CrossRef] [PubMed]
- Vis, J.C.; Schipper, E.; de Boer-van Huizen, R.T.; Verbeek, M.M.; de Waal, R.M.; Wesseling, P.; ten Donkelaar, H.J.; Kremer, B. Expression pattern of apoptosis-related markers in Huntington’s disease. Acta Neuropathol 2005, 109, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Meriin, A.B.; Zhang, X.; Miliaras, N.B.; Kazantsev, A.; Chernoff, Y.O.; McCaffery, J.M.; Wendland, B.; Sherman, M.Y. Aggregation of expanded polyglutamine domain in yeast leads to defects in endocytosis. Mol. Cell. Biol. 2003, 23, 7554–7565. [Google Scholar] [CrossRef] [PubMed]
- Meriin, A.B.; Zhang, X.; Alexandrov, I.M.; Salnikova, A.B.; Ter-Avanesian, M.D.; Chernoff, Y.O.; Sherman, M.Y. Endocytosis machinery is involved in aggregation of proteins with expanded polyglutamine domains. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 21, 1915–1925. [Google Scholar] [CrossRef] [PubMed]
- Solans, A.; Zambrano, A.; Rodriguez, M.; Barrientos, A. Cytotoxicity of a mutant huntingtin fragment in yeast involves early alterations in mitochondrial OXPHOS complexes II and III. Hum. Mol. Genet. 2006, 15, 3063–3081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgini, F.; Guidetti, P.; Nguyen, Q.; Bennett, S.C.; Muchowski, P.J. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nat. Genet. 2005, 37, 526–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, R.E.; Lo, R.S.; Davis, C.; Strand, A.D.; Neal, C.L.; Olson, J.M.; Fields, S. Altered transcription in yeast expressing expanded polyglutamine. Proc. Natl. Acad. Sci. USA 2001, 98, 13201–13206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Smith, D.L.; Meriin, A.B.; Engemann, S.; Russel, D.E.; Roark, M.; Washington, S.L.; Maxwell, M.M.; Marsh, J.L.; Thompson, L.M.; et al. A potent small molecule inhibits polyglutamine aggregation in Huntington’s disease neurons and suppresses neurodegeneration in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Chopra, V.; Fox, J.H.; Lieberman, G.; Dorsey, K.; Matson, W.; Waldmeier, P.; Housman, D.E.; Kazantsev, A.; Young, A.B.; Hersch, S. A small-molecule therapeutic lead for Huntington’s disease: Preclinical pharmacology and efficacy of C2-8 in the R6/2 transgenic mouse. Proc. Natl. Acad. Sci. USA 2007, 104, 16685–16689. [Google Scholar] [CrossRef]
- Wang, N.; Lu, X.H.; Sandoval, S.V.; Yang, X.W. An independent study of the preclinical efficacy of C2-8 in the R6/2 transgenic mouse model of Huntington’s disease. J. Huntingt. Dis. 2013, 2, 443–451. [Google Scholar]
- Colby, D.W.; Chu, Y.; Cassady, J.P.; Duennwald, M.; Zazulak, H.; Webster, J.M.; Messer, A.; Lindquist, S.; Ingram, V.M.; Wittrup, K.D. Potent inhibition of huntingtin aggregation and cytotoxicity by a disulfide bond-free single-domain intracellular antibody. Proc. Natl. Acad. Sci. USA 2004, 101, 17616–17621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Southwell, A.L.; Ko, J.; Patterson, P.H. Intrabody gene therapy ameliorates motor, cognitive, and neuropathological symptoms in multiple mouse models of Huntington’s disease. J. Neurosci. 2009, 29, 13589–13602. [Google Scholar] [CrossRef] [PubMed]
- Southwell, A.L.; Khoshnan, A.; Dunn, D.E.; Bugg, C.W.; Lo, D.C.; Patterson, P.H. Intrabodies binding the proline-rich domains of mutant huntingtin increase its turnover and reduce neurotoxicity. J. Neurosci. 2008, 28, 9013–9020. [Google Scholar] [CrossRef] [PubMed]
- Manogaran, A.L.; Fajardo, V.M.; Reid, R.J.; Rothstein, R.; Liebman, S.W. Most, but not all, yeast strains in the deletion library contain the [PIN+] prion. Yeast 2010, 27, 159–166. [Google Scholar] [CrossRef]
- Jacobs, K.R.; Castellano-Gonzalez, G.; Guillemin, G.J.; Lovejoy, D.B. Major Developments in the Design of Inhibitors along the Kynurenine Pathway. Curr. Med. Chem. 2017, 24, 2471–2495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wild, E.J.; Tabrizi, S.J. Targets for future clinical trials in Huntington’s disease: what’s in the pipeline? Mov. Disord. 2014, 29, 1434–1445. [Google Scholar] [CrossRef] [PubMed]
- Campesan, S.; Green, E.W.; Breda, C.; Sathyasaikumar, K.V.; Muchowski, P.J.; Schwarcz, R.; Kyriacou, C.P.; Giorgini, F. The kynurenine pathway modulates neurodegeneration in a Drosophila model of Huntington’s disease. Curr. Biol. CB 2011, 21, 961–966. [Google Scholar] [CrossRef]
- Zwilling, D.; Huang, S.Y.; Sathyasaikumar, K.V.; Notarangelo, F.M.; Guidetti, P.; Wu, H.Q.; Lee, J.; Truong, J.; Andrews-Zwilling, Y.; Hsieh, E.W.; et al. Kynurenine 3-monooxygenase inhibition in blood ameliorates neurodegeneration. Cell 2011, 145, 863–874. [Google Scholar] [CrossRef]
- Ehrnhoefer, D.E.; Duennwald, M.; Markovic, P.; Wacker, J.L.; Engemann, S.; Roark, M.; Legleiter, J.; Marsh, J.L.; Thompson, L.M.; Lindquist, S.; et al. Green tea (-)-epigallocatechin-gallate modulates early events in huntingtin misfolding and reduces toxicity in Huntington’s disease models. Hum. Mol. Genet. 2006, 15, 2743–2751. [Google Scholar] [CrossRef]
- Niu, Y.; Na, L.; Feng, R.; Gong, L.; Zhao, Y.; Li, Q.; Li, Y.; Sun, C. The phytochemical, EGCG, extends lifespan by reducing liver and kidney function damage and improving age-associated inflammation and oxidative stress in healthy rats. Aging Cell 2013, 12, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.E.; Piegholdt, S.; Rabe, D.; Baenas, N.; Schloesser, A.; Eggersdorfer, M.; Stocker, A.; Rimbach, G. Epigallocatechin gallate affects glucose metabolism and increases fitness and lifespan in Drosophila melanogaster. Oncotarget 2015, 6, 30568–30578. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Deng, J.; Man, Y.; Qu, Y. Green Tea Extracts Epigallocatechin-3-gallate for Different Treatments. Biomed Res. Int. 2017, 2017, 5615647. [Google Scholar] [CrossRef] [PubMed]
- Pallauf, K.; Duckstein, N.; Rimbach, G. A literature review of flavonoids and lifespan in model organisms. Proc. Nutr. Soc. 2017, 76, 145–162. [Google Scholar] [CrossRef] [PubMed]
- Walter, G.M.; Raveh, A.; Mok, S.A.; McQuade, T.J.; Arevang, C.J.; Schultz, P.J.; Smith, M.C.; Asare, S.; Cruz, P.G.; Wisen, S.; et al. High-throughput screen of natural product extracts in a yeast model of polyglutamine proteotoxicity. Chem. Biol. Drug Des. 2014, 83, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Kallscheuer, N.; Menezes, R.; Foito, A.; Henriques da Silva, M.D.; Braga, A.; Dekker, W.; Mendez Sevillano, D.; Rosado-Ramos, R.; Jardim, C.; Oliveira, J.; et al. Identification and microbial production of the raspberry phenol salidroside that is active against Huntington’s disease. Plant Physiol. 2018, 179, 969–985. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Looking at the recent advances in understanding alpha-synuclein and its aggregation through the proteoform prism. F1000 Res. 2017, 6, 525. [Google Scholar] [CrossRef]
- Outeiro, T.F.; Lindquist, S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 2003, 302, 1772–1775. [Google Scholar] [CrossRef]
- Tenreiro, S.; Franssens, V.; Winderickx, J.; Outeiro, T.F. Yeast models of Parkinson’s disease-associated molecular pathologies. Curr. Opin. Genet. Dev. 2017, 44, 74–83. [Google Scholar] [CrossRef]
- Tardiff, D.F.; Lindquist, S. Phenotypic screens for compounds that target the cellular pathologies underlying Parkinson’s disease. Drug Discov. Today Technol. 2013, 10, e121–e128. [Google Scholar] [CrossRef]
- Griffioen, G.; Duhamel, H.; Van Damme, N.; Pellens, K.; Zabrocki, P.; Pannecouque, C.; van Leuven, F.; Winderickx, J.; Wera, S. A yeast-based model of α-synucleinopathy identifies compounds with therapeutic potential. Biochimica Biophysica Acta 2006, 1762, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Fleming, J.; Outeiro, T.F.; Slack, M.; Lindquist, S.L.; Bulawa, C.E. Detection of compounds that rescue Rab1-synuclein toxicity. Methods Enzym. 2008, 439, 339–351. [Google Scholar]
- Su, L.J.; Auluck, P.K.; Outeiro, T.F.; Yeger-Lotem, E.; Kritzer, J.A.; Tardiff, D.F.; Strathearn, K.E.; Liu, F.; Cao, S.; Hamamichi, S.; et al. Compounds from an unbiased chemical screen reverse both ER-to-Golgi trafficking defects and mitochondrial dysfunction in Parkinson’s disease models. Dis. Model. Mech. 2010, 3, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Kritzer, J.A.; Hamamichi, S.; McCaffery, J.M.; Santagata, S.; Naumann, T.A.; Caldwell, K.A.; Caldwell, G.A.; Lindquist, S. Rapid selection of cyclic peptides that reduce α-synuclein toxicity in yeast and animal models. Nat. Chem. Biol. 2009, 5, 655–663. [Google Scholar] [CrossRef]
- Tardiff, D.F.; Jui, N.T.; Khurana, V.; Tambe, M.A.; Thompson, M.L.; Chung, C.Y.; Kamadurai, H.B.; Kim, H.T.; Lancaster, A.K.; Caldwell, K.A.; et al. Yeast reveal a druggable Rsp5/Nedd4 network that ameliorates α-synuclein toxicity in neurons. Science 2013, 342, 979–983. [Google Scholar] [CrossRef] [PubMed]
- Faria, C.; Jorge, C.D.; Borges, N.; Tenreiro, S.; Outeiro, T.F.; Santos, H. Inhibition of formation of α-synuclein inclusions by mannosylglycerate in a yeast model of Parkinson’s disease. Biochimica Biophysica Acta 2013, 1830, 4065–4072. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.T.; Tenreiro, S.; Gameiro, A.; Chu, V.; Outeiro, T.F.; Conde, J.P. Modulation of α-synuclein toxicity in yeast using a novel microfluidic-based gradient generator. Lab Chip 2014, 14, 3949–3957. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chernova, T.A.; Chernoff, Y.O.; Wilkinson, K.D. Yeast Models for Amyloids and Prions: Environmental Modulation and Drug Discovery. Molecules 2019, 24, 3388. https://doi.org/10.3390/molecules24183388
Chernova TA, Chernoff YO, Wilkinson KD. Yeast Models for Amyloids and Prions: Environmental Modulation and Drug Discovery. Molecules. 2019; 24(18):3388. https://doi.org/10.3390/molecules24183388
Chicago/Turabian StyleChernova, Tatiana A., Yury O. Chernoff, and Keith D. Wilkinson. 2019. "Yeast Models for Amyloids and Prions: Environmental Modulation and Drug Discovery" Molecules 24, no. 18: 3388. https://doi.org/10.3390/molecules24183388
APA StyleChernova, T. A., Chernoff, Y. O., & Wilkinson, K. D. (2019). Yeast Models for Amyloids and Prions: Environmental Modulation and Drug Discovery. Molecules, 24(18), 3388. https://doi.org/10.3390/molecules24183388