
Spray Congealing: An Emerging Technology to Prepare Solid Dispersions with Enhanced Oral Bioavailability of Poorly Water Soluble Drugs



Abstract
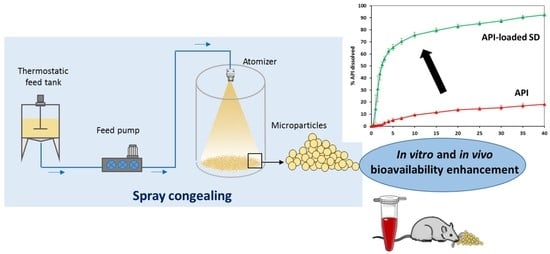
:
1. Introduction
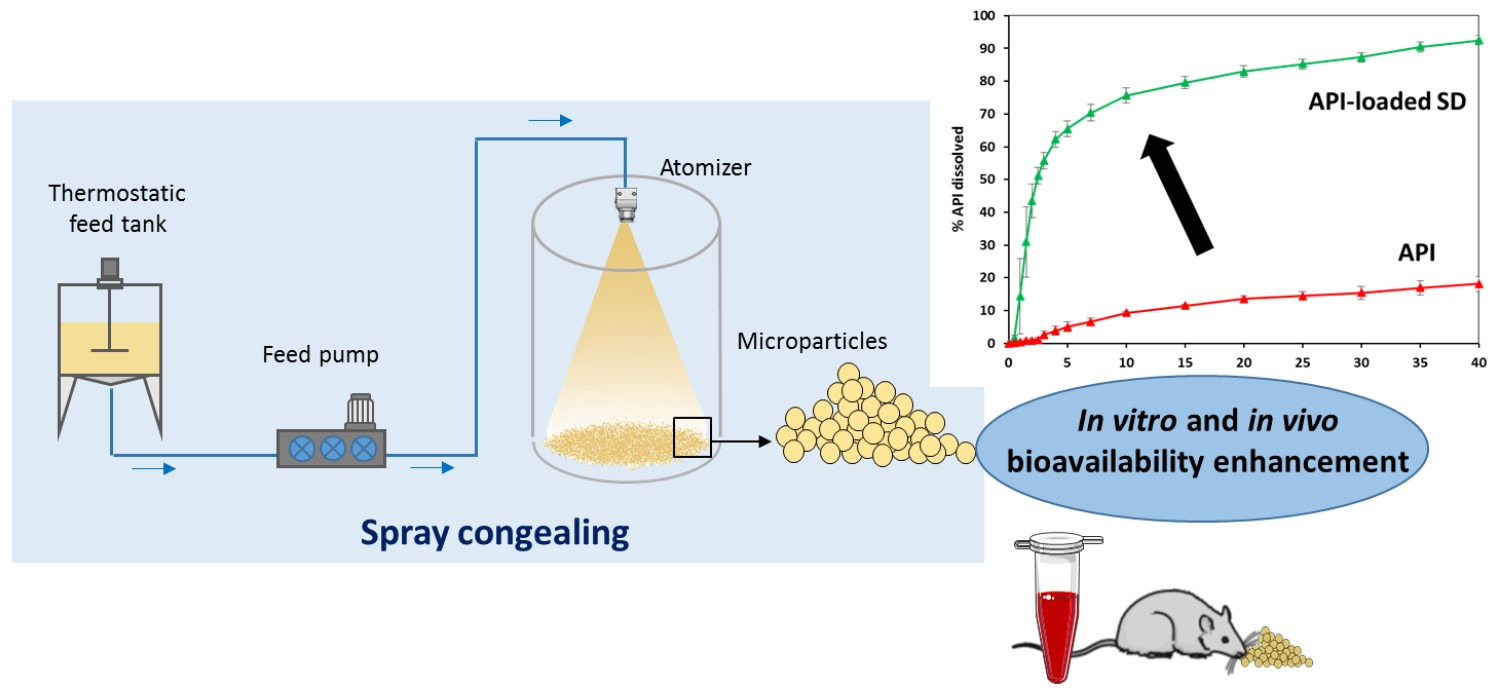
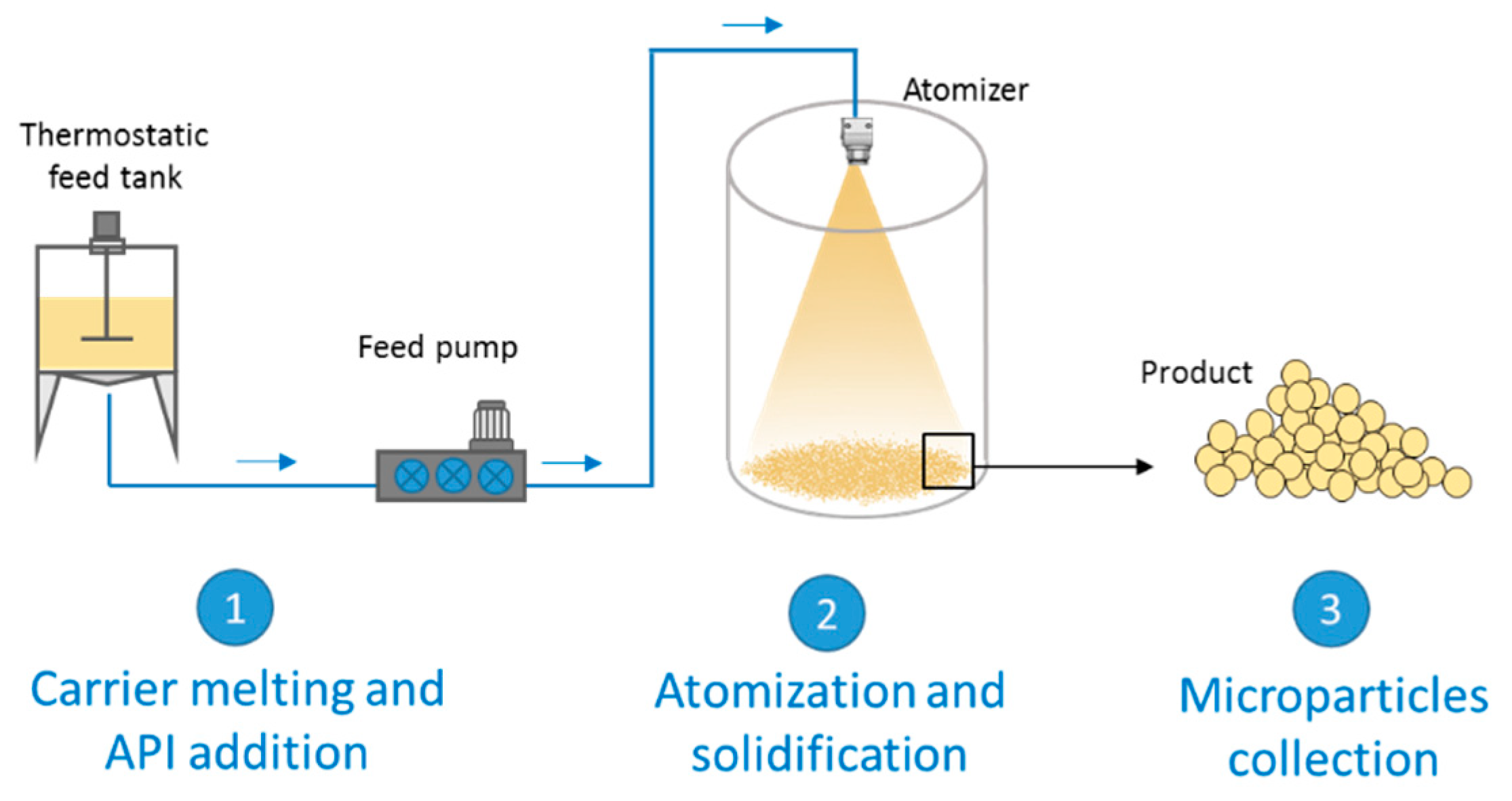
2. Spray Congealing Technology: General Aspects
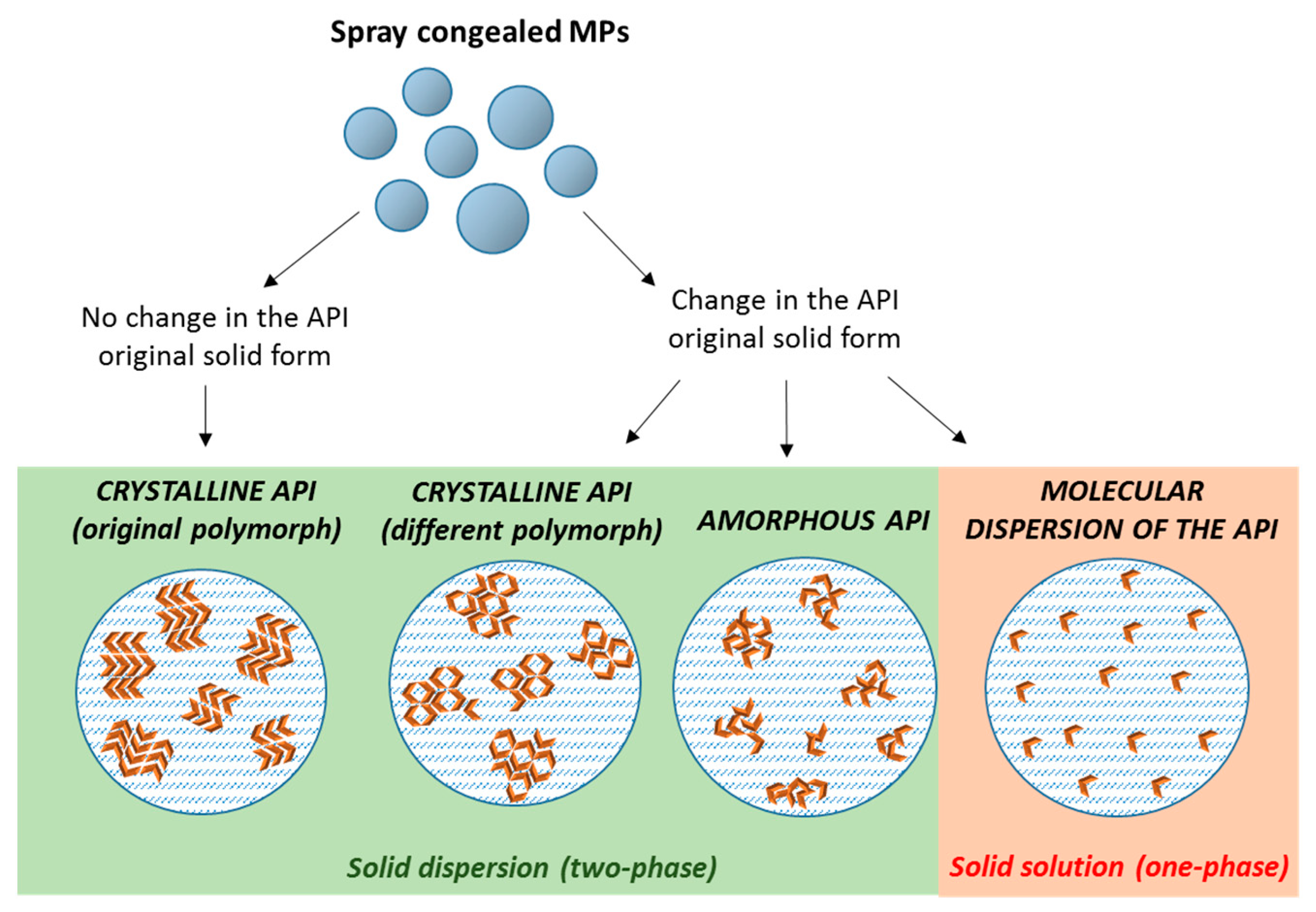
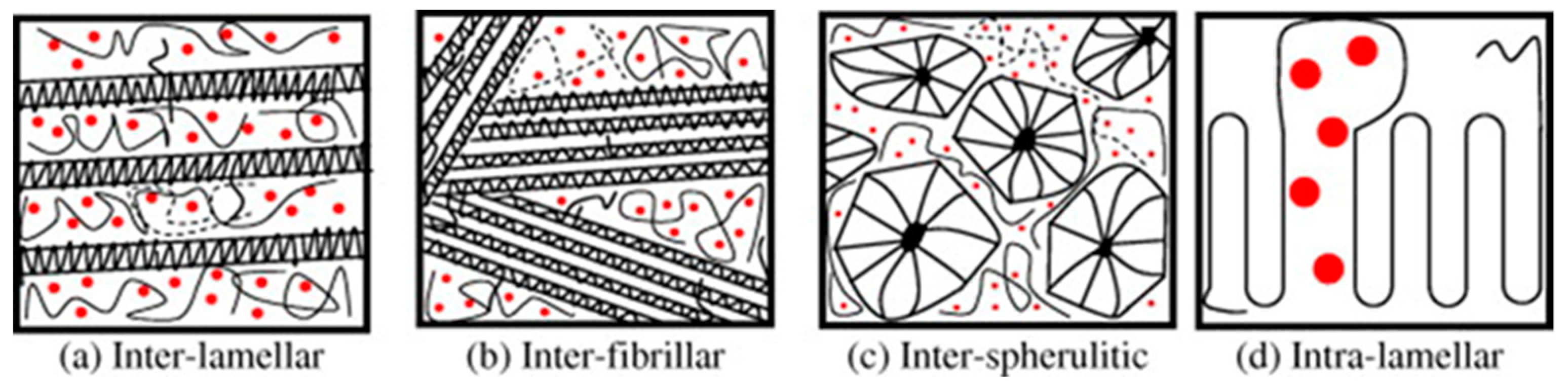
3. Structure and Composition of Spray Congealed SD
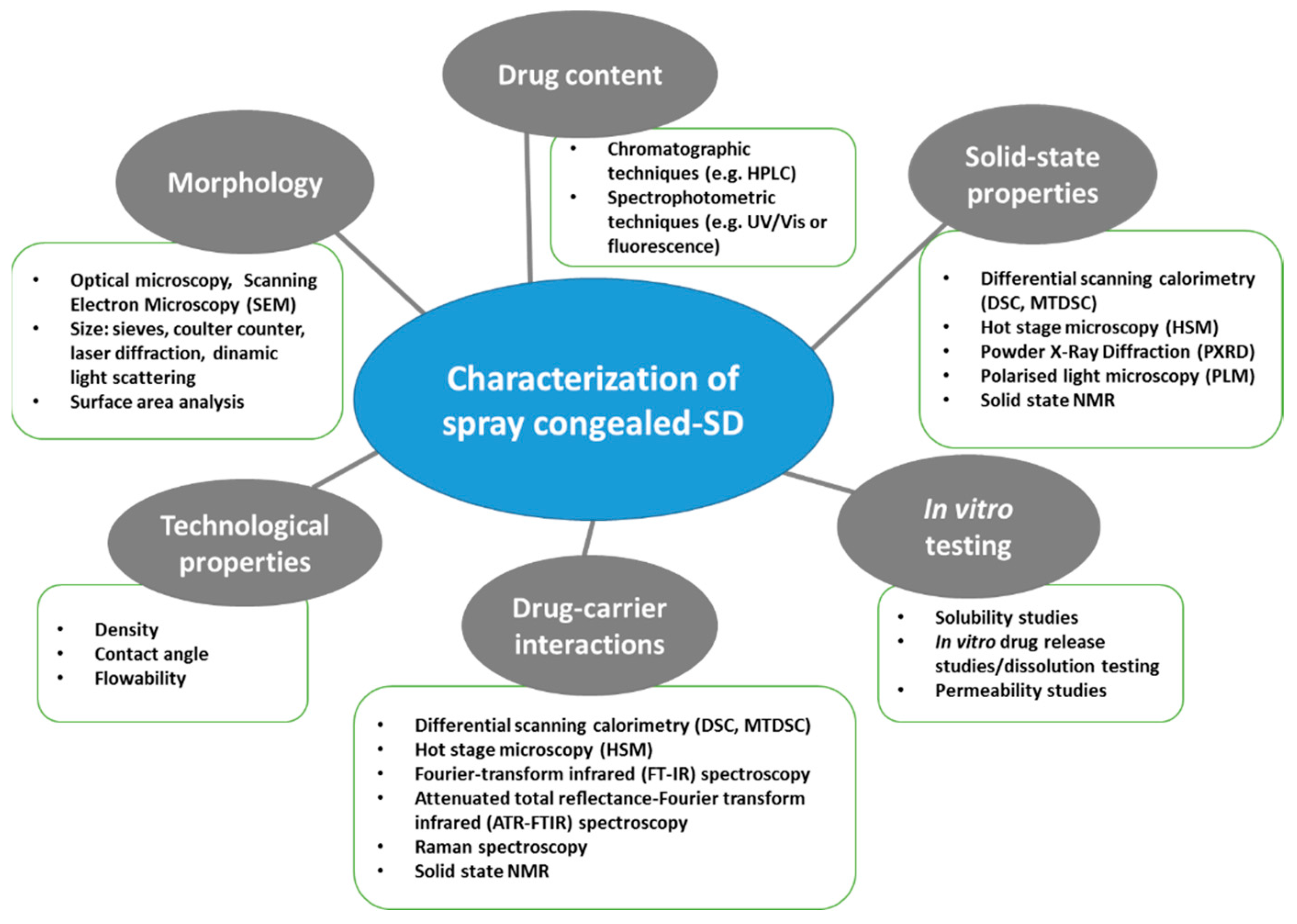
4. Properties and Characterization of Spray Congealed SD
4.1. Determination of the Drug Content
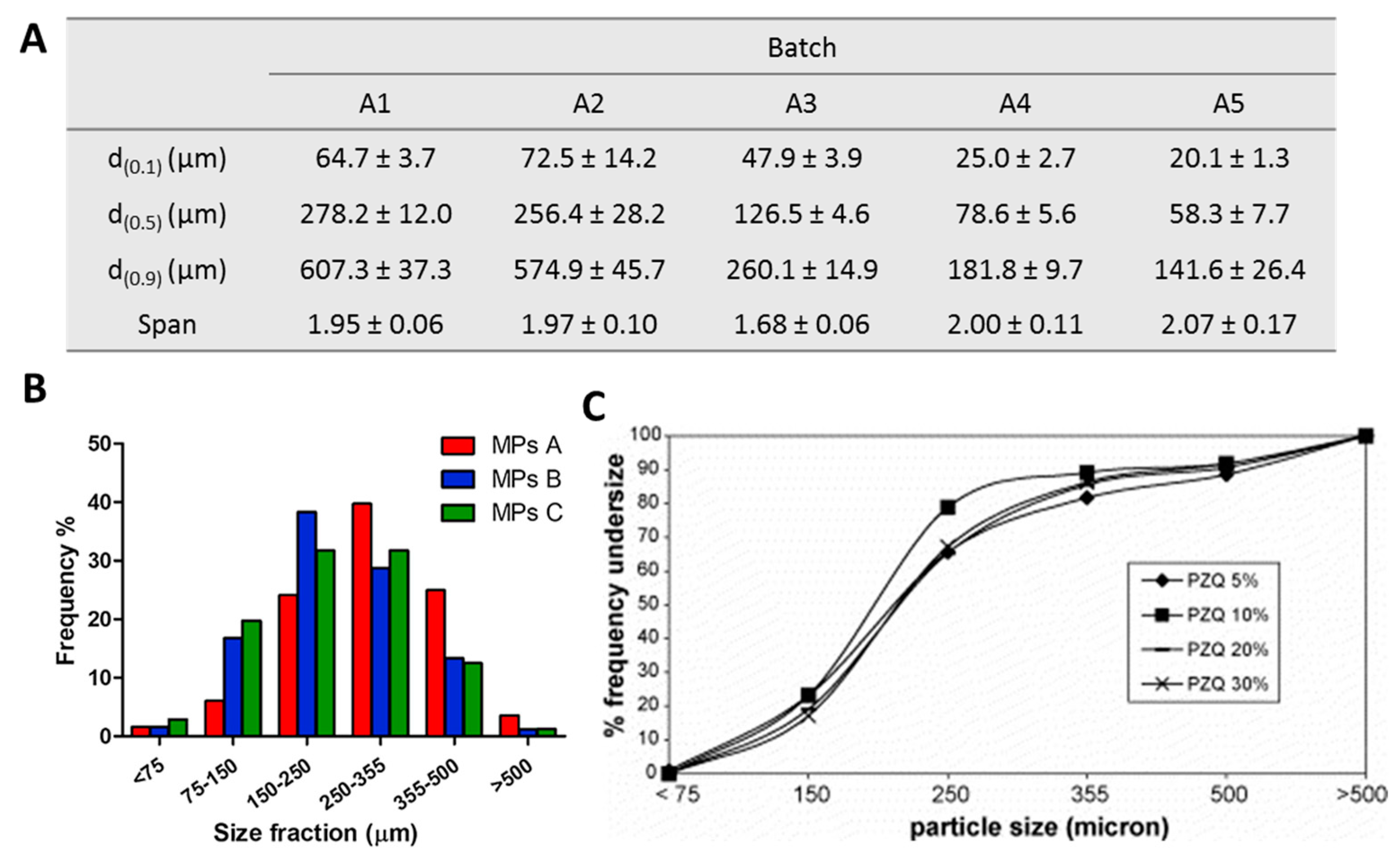
4.2. Particle Size
4.3. Shape and Surface Morphology
4.4. Drug-Carrier Interactions
4.5. Solid State Properties of Drug and Carrier
4.6. In Vitro Evaluation of the Biopharmaceutical Properties
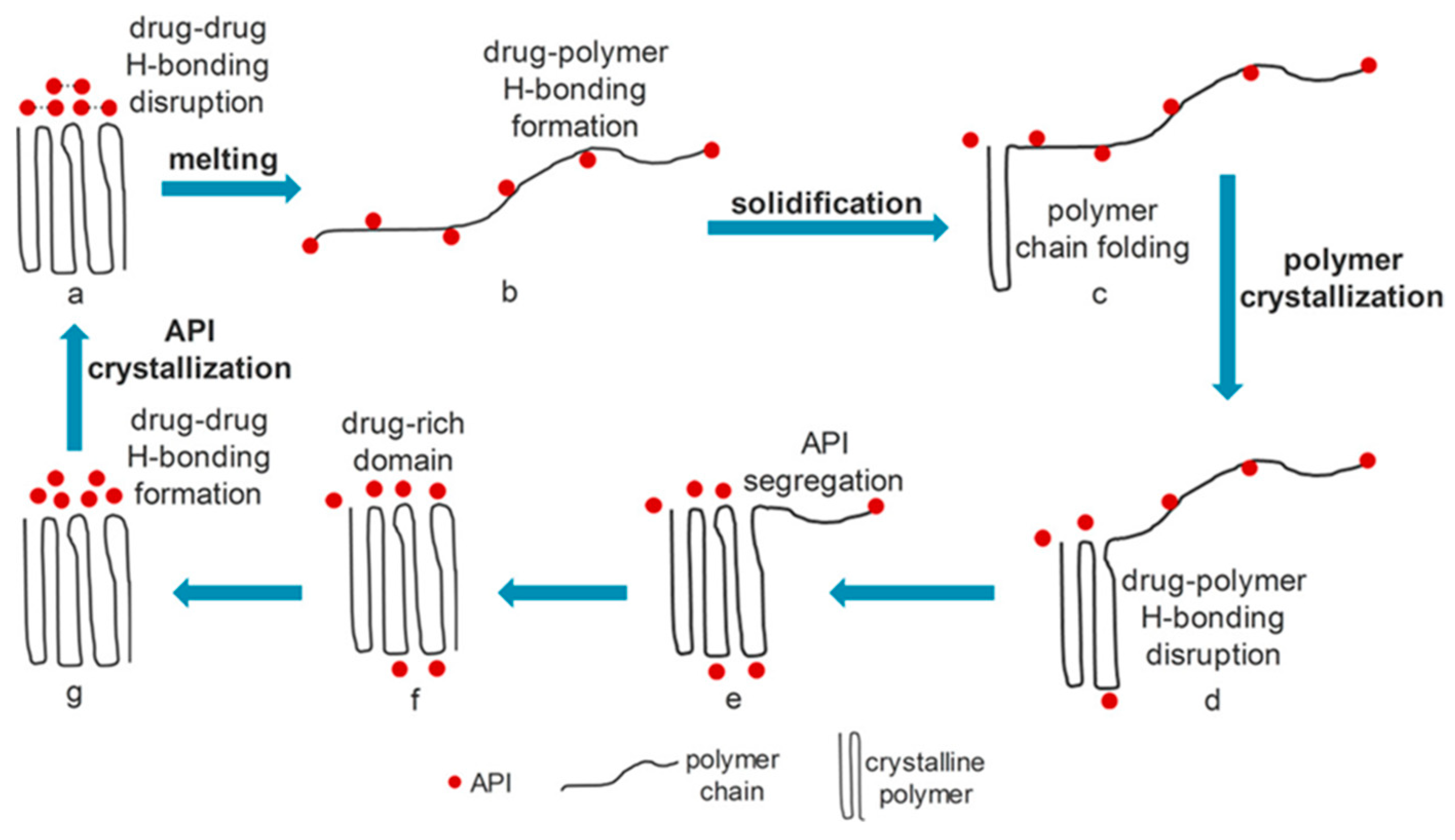
5. Mechanisms of Bioavailability Enhancement of Spray Congealed Systems
- MPs containing the drug used as received
- MPs containing pre-activated drug
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Chella, N.; Tadikonda, R. Melt dispersion granules: Formulation and evaluation to improve oral delivery of poorly soluble drugs—A case study with valsartan. Drug Dev. Ind. Pharm. 2015, 41, 888–897. [Google Scholar] [PubMed]
- Morishita, M.; Peppas, N.A. Advances in oral drug delivery: Improved bioavailability of poorly absorbed drugs by tissue and cellular optimization. Adv. Drug Deliv. Rev. 2012, 64, 479. [Google Scholar] [PubMed]
- Kawabata, Y.; Wada, K.; Nakatani, M.; Yamada, S.; Onoue, S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: Basic approaches and practical applications. Int. J. Pharm. 2011, 420, 1–10. [Google Scholar]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [PubMed]
- FDA. Guidance for Industry, Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms based on a Biopharmaceutics Classification System; FDA: Silver Spring, MD, USA, 2000. [Google Scholar]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric Amorphous Solid Dispersions: A Review of Amorphization, Crystallization, Stabilization, Solid-State Characterization, and Aqueous Solubilization of Biopharmaceutical Classification System Class II Drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [PubMed] [Green Version]
- Serajuddin, A.T.M. Solid dispersion of poorly water-soluble drugs: Early promises, subsequent problems, and recent breakthroughs. J. Pharm. Sci. 1999, 88, 1058–1066. [Google Scholar] [PubMed]
- Mishra, D.K.; Dhote, V.; Bhargava, A.; Jain, D.K.; Mishra, P.K. Amorphous solid dispersion technique for improved drug delivery: Basics to clinical applications. Drug Deliv. Transl. Res. 2015, 5, 552–565. [Google Scholar] [PubMed]
- Ellenberger, D.J.; Miller, D.A.; Williamsiii, R.O. Expanding the Application and Formulation Space of Amorphous Solid Dispersions with KinetiSol®: A Review. AAPS PharmSciTech 2018, 19, 1933–1956. [Google Scholar]
- Dedroog, S.; Huygens, C.; Van Den Mooter, G. Chemically identical but physically different: A comparison of spray drying, hot melt extrusion and cryo-milling for the formulation of high drug loaded amorphous solid dispersions of naproxen. Eur. J. Pharm. Biopharm. 2019, 135, 1–12. [Google Scholar]
- Zanolla, D.; Perissutti, B.; Passerini, N.; Invernizzi, S.; Voinovich, D.; Bertoni, S.; Melegari, C.; Millotti, G.; Albertini, B. Milling and comilling Praziquantel at cryogenic and room temperatures: Assessment of the process-induced effects on drug properties. J. Pharm. Biomed. Anal. 2018, 153, 82–89. [Google Scholar]
- Davis, M.; Walker, G. Recent strategies in spray drying for the enhanced bioavailability of poorly water-soluble drugs. J. Control. Release 2018, 269, 110–127. [Google Scholar] [PubMed] [Green Version]
- Patil, H.; Tiwari, R.V.; Repka, M.A. Hot-Melt Extrusion: From Theory to Application in Pharmaceutical Formulation. AAPS PharmSciTech 2016, 17, 20–42. [Google Scholar] [PubMed]
- Cordeiro, P.; Temtem, M.; Winters, C. Spray Congealing: Applications in the Pharmaceutical Industry. Chem. Today 2013, 31, 69–72. [Google Scholar]
- Bertoni, S.; Dolci, L.S.; Albertini, B.; Passerini, N. Spray congealing: A versatile technology for advanced drug delivery systems. Ther. Deliv. 2018, 9, 833–845. [Google Scholar] [PubMed]
- Oh, C.M.; Guo, Q.; Heng, P.W.S.; Chan, L.W. Spray-congealed microparticles for drug delivery—An overview of factors influencing their production and characteristics. Expert Opin. Drug Deliv. 2014, 11, 1047–1060. [Google Scholar] [PubMed]
- Kumar, S.; Jeet, K.; Ashish, B. Recent technological advancements in multiparticulate formulations: The smart drug delivery systems. Asian J. Pharm. 2018, 9, S13–S25. [Google Scholar]
- Van Duong, T.; Van den Mooter, G. The role of the carrier in the formulation of pharmaceutical solid dispersions. Part I: Crystalline and semi-crystalline carriers. Expert Opin. Drug Deliv. 2016, 13, 1583–1594. [Google Scholar]
- Ilić, I.; Dreu, R.; Burjak, M.; Homar, M.; Kerč, J.; Srčič, S. Microparticle size control and glimepiride microencapsulation using spray congealing technology. Int. J. Pharm. 2009, 381, 176–183. [Google Scholar]
- Zhu, Q.; Taylor, L.S.; Harris, M.T. Evaluation of the Microstructure of Semicrystalline Solid Dispersions. Mol. Pharm. 2010, 7, 1291–1300. [Google Scholar]
- Chiou, W.L.; Riegelman, S. Pharmaceutical Applications of Solid Dispersion Systems. J. Pharm. Sci. 1971, 60, 1281–1302. [Google Scholar]
- Bley, H.; Fussnegger, B.; Bodmeier, R. Characterization and stability of solid dispersions based on PEG/polymer blends. Int. J. Pharm. 2010, 390, 165–173. [Google Scholar] [PubMed]
- Faucher, J.A.; Koleske, J.V.; Santee, E.R.; Stratta, J.J.; Wilson, C.W. Glass Transitions of Ethylene Oxide Polymers. J. Appl. Phys. 1966, 37, 3962–3964. [Google Scholar]
- Yang, M.; Gogos, C. Crystallization of poly(ethylene oxide) with acetaminophen—A study on solubility, spherulitic growth, and morphology. Eur. J. Pharm. Biopharm. 2013, 85, 889–897. [Google Scholar] [PubMed]
- Elander, M.; Boll, J.B.; Hojman, A.S.; Rasmussen, A.D. Gelucire and Gelucire-PEG400 formulations; tolerability in species used for non-clinical safety testing after oral (gavage) dosing. J. Appl. Toxicol. 2016, 36, 1430–1436. [Google Scholar] [PubMed]
- Kawakami, K.; Miyoshi, K.; Ida, Y. Solubilization behavior of poorly soluble drugs with combined use of Gelucire 44/14 and cosolvent. J. Pharm. Sci. 2004, 93, 1471–1479. [Google Scholar] [PubMed]
- El Hadri, M.; Achahbar, A.; El Khamkhami, J.; Khelifa, B.; Faivre, V.; Abbas, O.; Bresson, S. Lyotropic behavior of Gelucire 50/13 by XRD, Raman and IR spectroscopies according to hydration. Chem. Phys. Lipids 2016, 200, 11–23. [Google Scholar] [PubMed]
- Dumortier, G.; Grossiord, J.L.; Agnely, F.; Chaumeil, J.C. A Review of Poloxamer 407 Pharmaceutical and Pharmacological Characteristics. Pharm. Res. 2006, 23, 2709–2728. [Google Scholar] [PubMed]
- Giuliano, E.; Paolino, D.; Fresta, M.; Cosco, D. Mucosal Applications of Poloxamer 407-Based Hydrogels: An Overview. Pharmaceutics 2018, 10, e159. [Google Scholar] [PubMed]
- Albertini, B.; Passerini, N.; Di Sabatino, M.; Monti, D.; Burgalassi, S.; Chetoni, P.; Rodriguez, L. Poloxamer 407 microspheres for orotransmucosal drug delivery. Part I: Formulation, manufacturing and characterization. Int. J. Pharm. 2010, 399, 71–79. [Google Scholar] [PubMed]
- Monti, D.; Burgalassi, S.; Rossato, M.S.; Albertini, B.; Passerini, N.; Rodriguez, L.; Chetoni, P. Poloxamer 407 microspheres for orotransmucosal drug delivery. Part II: In vitro/in vivo evaluation. Int. J. Pharm. 2010, 400, 32–36. [Google Scholar]
- Chen, W.; Zhou, S.; Ge, L.; Wu, W.; Jiang, X. Translatable High Drug Loading Drug Delivery Systems Based on Biocompatible Polymer Nanocarriers. Biomacromolecules 2018, 19, 1732–1745. [Google Scholar] [PubMed]
- Kulthe, V.V.; Chaudhari, P.D. Effectiveness of Spray Congealing to Obtain Physically Stabilized Amorphous Dispersions of a Poorly Soluble Thermosensitive API. AAPS PharmSciTech 2014, 15, 1370–1377. [Google Scholar] [PubMed] [Green Version]
- Kolašinac, N.; Kachrimanis, K.; Homšek, I.; Grujić, B.; Đurić, Z.; Ibrić, S. Solubility enhancement of desloratadine by solid dispersion in poloxamers. Int. J. Pharm. 2012, 436, 161–170. [Google Scholar] [PubMed]
- Medarevic, D.P.; Kachrimanis, K.; Mitric, M.; Djuris, J.; Djuric, Z.; Ibric, S. Dissolution rate enhancement and physicochemical characterization of carbamazepine-poloxamer solid dispersions. Pharm. Dev. Technol. 2015, 21, 268–276. [Google Scholar] [PubMed]
- Passerini, N.; Albertini, B.; Perissutti, B.; Rodriguez, L. Evaluation of melt granulation and ultrasonic spray congealing as techniques to enhance the dissolution of praziquantel. Int. J. Pharm. 2006, 318, 92–102. [Google Scholar] [PubMed]
- Passerini, N.; Perissutti, B.; Moneghini, M.; Voinovich, D.; Albertini, B.; Cavallari, C.; Rodriguez, L. Characterization of Carbamazepine-Gelucire 50/13 Microparticles Prepared by a Spray-Congealing Process Using Ultrasounds. 2002, 91, 699–707. J. pharm. Sci. 2002, 91, 699–707. [Google Scholar] [PubMed]
- Perissutti, B.; Rubessa, F.; Princivalle, F. Solid dispersions of carbamazepine with Gelucire 44/14 and 50/13. STP Pharm. Sci. J. 2000, 10, 479–484. [Google Scholar]
- Rumondor, A.C.F.; Taylor, L.S. Effect of Polymer Hygroscopicity on the Phase Behavior of Amorphous Solid Dispersions in the Presence of Moisture. Mol. Pharm. 2010, 7, 477–490. [Google Scholar] [PubMed]
- Scalia, S.; Young, P.M.; Traini, D. Solid lipid microparticles as an approach to drug delivery. Expert Opin. Drug Deliv. 2014, 12, 1–17. [Google Scholar]
- Maschke, A.; Becker, C.; Eyrich, D.; Kiermaier, J.; Blunk, T.; Gopferich, A. Development of a spray congealing process for the preparation of insulin-loaded lipid microparticles and characterization thereof. Eur. J. Pharm. Biopharm. 2007, 65, 175–187. [Google Scholar] [PubMed]
- Albertini, B.; Passerini, N.; Pattarino, F.; Rodriguez, L. New spray congealing atomizer for the microencapsulation of highly concentrated solid and liquid substances. Eur. J. Pharm. Biopharm. 2008, 69, 348–357. [Google Scholar] [PubMed]
- Adeyeye, C.M.; Price, J.C. Development and Evaluation of Sustained-Release Ibuprofen-Wax Microspheres. II. In vitro Dissolution Studies. Pharm. Res. 1994, 11, 575–579. [Google Scholar] [PubMed]
- Fini, A.; Rodriguez, L.; Cavallari, C.; Albertini, B.; Passerini, N. Ultrasound-compacted and spray-congealed indomethacin/polyethyleneglycol systems. Int. J. Pharm. 2002, 247, 11–22. [Google Scholar] [PubMed]
- Bertoni, S.; Albertini, B.; Ferraro, L.; Beggiato, S.; Dalpiaz, A.; Passerini, N. Exploring the use of spray congealing to produce solid dispersions with enhanced indomethacin bioavailability: In vitro characterization and in vivo study. Eur. J. Pharm. Biopharm. 2019, 139, 132–141. [Google Scholar]
- Champion, J.A.; Katare, Y.K.; Mitragotri, S. Particle Shape: A New Design Parameter for Micro- and Nanoscale Drug Delivery Carriers. J. Control. Release 2007, 121, 3–9. [Google Scholar] [PubMed]
- Hsieh, D.S.T.; Rhine, W.D.; Langer, R. Zero-Order Controlled-Release Polymer Matrices for Micro- and Macromolecules. J. Pharm. Sci. 1983, 72, 17–22. [Google Scholar]
- Bile, J.; Bolzinger, M.; Vigne, C.; Boyron, O.; Valour, J.; Fessi, H.; Chevalier, Y. The parameters influencing the morphology of poly (-e -caprolactone ) microspheres and the resulting release of encapsulated drugs. Int. J. Pharm. 2015, 494, 152–166. [Google Scholar]
- Oh, C.M.; Wan, P.; Heng, S.; Chan, L.W. Influence of Hydroxypropyl Methylcellulose on Metronidazole Crystallinity in Spray-Congealed Polyethylene Glycol Microparticles and Its Impact with Various Additives on Metronidazole Release. AAPS PharmSciTech 2015, 16, 1357–1367. [Google Scholar] [Green Version]
- Zuo, W.; Li, N.; Zhao, Y.; Fu, T.; Fei, W.; Yu, R. Synchronized release of bufadienolides in a stable Lutrol F127 based solid dispersion prepared with spray congealing dispersion prepared with spray congealing. Drug Dev. Ind. Pharm. 2018, 44, 1817–1825. [Google Scholar]
- Tomšik, A.; Šarić, L.; Bertoni, S.; Protti, M.; Albertini, B.; Mercolini, L.; Passerini, N. Encapsulations of wild garlic (Allium ursinum L.) extract using spray congealing technology. Food Res. Int. 2018, 119, 941–950. [Google Scholar]
- Liu, X.; Feng, X.; Williams, R.O.; Zhang, F. Characterization of amorphous solid dispersions. J. Pharm. Investig. 2018, 48, 19–41. [Google Scholar]
- Qin, W.; He, Y.; Guo, Z.; Zhang, L.; Wu, L.; Yin, X.; Shakya, S.; Maharjan, A.; Tang, Y.; Zhu, W.; et al. Optimization of taste-masking on ibuprofen microspheres with selected structure features. Asian J. Pharm. Sci. 2018, 14, 174–182. [Google Scholar]
- Khan, N.; Craig, D.Q.M. Role of blooming in determining the storage stability of lipid-based dosage forms. J. Pharm. Sci. 2004, 93, 2962–2971. [Google Scholar] [PubMed]
- Cavallari, C.; Gonzalez-Rodriguez, M.; Tarterini, F.; Fini, A. Image analysis of lutrol/gelucire/olanzapine microspheres prepared by ultrasound-assisted spray congealing. Eur. J. Pharm. Biopharm. 2014, 88, 909–918. [Google Scholar] [PubMed]
- Li, Y.; Pang, H.; Guo, Z.; Lin, L.; Dong, Y.; Li, G.; Lu, M.; Wu, C. Interactions between drugs and polymers influencing hot melt extrusion. J. Pharm. Pharmacol. 2014, 66, 148–166. [Google Scholar] [PubMed]
- Anderson, B.D. Predicting Solubility/Miscibility in Amorphous Dispersions: It Is Time to Move Beyond Regular Solution Theories. J. Pharm. Sci. 2018, 107, 24–33. [Google Scholar] [PubMed]
- Van Duong, T.; Lüdeker, D.; Van Bockstal, P.J.; De Beer, T.; Van Humbeeck, J.; Van Den Mooter, G. Polymorphism of Indomethacin in Semicrystalline Dispersions: Formation, Transformation, and Segregation. Mol. Pharm. 2018, 15, 1037–1051. [Google Scholar]
- Van Duong, T.; Reekmans, G.; Venkatesham, A.; Van Aerschot, A.; Adriaensens, P.; Van Humbeeck, J.; Van Den Mooter, G. Spectroscopic Investigation of the Formation and Disruption of Hydrogen Bonds in Pharmaceutical Semicrystalline Dispersions. Mol. Pharm. 2017, 14, 1726–1741. [Google Scholar]
- Van Duong, T.; Van den Mooter, G. The role of the carrier in the formulation of pharmaceutical solid dispersions. Part II: Amorphous carriers. Expert Opin. Drug Deliv. 2016, 13, 1681–1694. [Google Scholar]
- Censi, R.; Gigliobianco, M.; Casadidio, C.; Di Martino, P. Hot Melt Extrusion: Highlighting Physicochemical Factors to Be Investigated While Designing and Optimizing a Hot Melt Extrusion Process. Pharmaceutics 2018, 10, E89. [Google Scholar]
- Chieng, N.; Rades, T.; Aaltonen, J. An overview of recent studies on the analysis of pharmaceutical polymorphs. J. Pharm. Biomed. Anal. 2011, 55, 618–644. [Google Scholar] [PubMed]
- Zhang, X.; Xing, H.; Zhao, Y.; Ma, Z. Pharmaceutical Dispersion Techniques for Dissolution and Bioavailability Enhancement of Poorly Water-Soluble Drugs. Pharmaceutics 2018, 10, E74. [Google Scholar]
- Martins, R.M.; Siqueira, S.; Machado, M.O.; Freitas, L.A.P. The effect of homogenization method on the properties of carbamazepine microparticles prepared by spray congealing. J. Microencapsul. 2013, 30, 692–700. [Google Scholar] [PubMed]
- Craig, D.Q. The mechanisms of drug release from solid dispersions in water-soluble polymers. Int. J. Pharm. 2002, 231, 131–144. [Google Scholar] [PubMed]
- Fini, A.; Moyano, J.R.; Ginés, J.M.; Perez-Martinez, J.I.; Rabasco, A.M. Diclofenac salts, II. Solid dispersions in PEG6000 and Gelucire 50/13. Eur. J. Pharm. Biopharm. 2005, 60, 99–111. [Google Scholar] [PubMed]
- El-Badry, M.; Fetih, G.; Fathy, M. Improvement of solubility and dissolution rate of indomethacin by solid dispersions in Gelucire 50/13 and PEG4000. Saudi Pharm. J. 2009, 17, 217–225. [Google Scholar] [Green Version]
- Albertini, B.; Passerini, N.; Di Sabatino, M.; Vitali, B.; Brigidi, P.; Rodriguez, L. Polymer-lipid based mucoadhesive microspheres prepared by spray-congealing for the vaginal delivery of econazole nitrate. Eur. J. Pharm. Sci. 2009, 36, 591–601. [Google Scholar]
- Passerini, N.; Perissutti, B.; Albertini, B.; Franceschinis, E.; Lenaz, D.; Hasa, D.; Locatelli, I.; Voinovich, D. A new approach to enhance oral bioavailability of Silybum Marianum dry extract: Association of mechanochemical activation and spray congealing. Phytomedicine 2012, 19, 160–168. [Google Scholar]
- Albertini, B.; Perissutti, B.; Bertoni, S.; Zanolla, D.; Franceschinis, E.; Voinovich, D.; Lombardo, F.; Keiser, J.; Passerini, N. Combining Mechanochemistry and Spray Congealing for New Praziquantel Pediatric Formulations in Schistosomiasis Treatment. Int. J. Mol. Sci. 2019, 20, E1233. [Google Scholar]
- Karagianni, A.; Kachrimanis, K.; Nikolakakis, I. Co-Amorphous Solid Dispersions for Solubility and Absorption Improvement of Drugs: Composition, Preparation, Characterization and Formulations for Oral Delivery. Pharmaceutics 2018, 10, E98. [Google Scholar]
- Schmitt, P.D. Recent Advances in Nonlinear Optical Analyses of Pharmaceutical Materials in the Solid State. Mol. Pharm. 2017, 14, 555–565. [Google Scholar] [PubMed]
- Jaspart, S.; Piel, G.; Delattre, L.; Evrard, B. Solid lipid microparticles: Formulation, preparation, characterisation, drug release and applications. Expert Opin. Drug Deliv. 2005, 2, 75–87. [Google Scholar] [PubMed]
- Kostewicz, E.S.; Abrahamsson, B.; Brewster, M.; Brouwers, J.; Butler, J.; Carlert, S.; Dickinson, P.A.; Dressman, J.; Holm, R.; Klein, S.; et al. In vitro models for the prediction of in vivo performance of oral dosage forms. Eur. J. Pharm. Sci. 2014, 57, 342–366. [Google Scholar] [PubMed]
- Klein, S. The Use of Biorelevant Dissolution Media to Forecast the In Vivo Performance of a Drug. AAPS J. 2010, 12, 397–405. [Google Scholar] [PubMed]
- van Drooge, D.J.; Hinrichs, W.L.J.; Frijlink, H.W. Anomalous dissolution behaviour of tablets prepared from sugar glass-based solid dispersions. J. Control. Release 2004, 97, 441–452. [Google Scholar] [PubMed]
- Karavas, E.; Georgarakis, E.; Sigalas, M.P.; Avgoustakis, K.; Bikiaris, D. Investigation of the release mechanism of a sparingly water-soluble drug from solid dispersions in hydrophilic carriers based on physical state of drug, particle size distribution and drug-polymer interactions. Eur. J. Pharm. Biopharm. 2007, 66, 334–347. [Google Scholar] [PubMed]
- Sarmento, B.; Andrade, F.; da Silva, S.B.; Rodrigues, F.; das Neves, J.; Ferreira, D. Cell-based in vitro models for predicting drug permeability. Expert Opin. Drug Metab. Toxicol. 2012, 8, 607–621. [Google Scholar] [PubMed]
- Buckley, S.T.; Fischer, S.M.; Fricker, G.; Brandl, M. In vitro models to evaluate the permeability of poorly soluble drug entities: Challenges and perspectives. Eur. J. Pharm. Sci. 2012, 45, 235–250. [Google Scholar] [PubMed]
- Wikman-Larhed, A.; Artursson, P. Co-cultures of human intestinal goblet (HT29-H) and absorptive (Caco-2) cells for studies of drug and peptide absorption. Eur. J. Pharm. Sci. 1995, 3, 171–183. [Google Scholar]
- Avdeef, A. The rise of PAMPA. Expert Opin. Drug Metab. Toxicol. 2005, 1, 325–342. [Google Scholar]
- Flaten, G.E.; Dhanikula, A.B.; Luthman, K.; Brandl, M. Drug permeability across a phospholipid vesicle based barrier: A novel approach for studying passive diffusion. Eur. J. Pharm. Sci. 2006, 27, 80–90. [Google Scholar] [PubMed] [Green Version]
- Takano, R.; Kataoka, M.; Yamashita, S. Integrating drug permeability with dissolution profile to develop IVIVC. Biopharm. Drug Dispos. 2012, 33, 354–365. [Google Scholar] [PubMed]
- Ginski, M.J.; Polli, J.E. Prediction of dissolution–Absorption relationships from a dissolution/Caco-2 system. Int. J. Pharm. 1999, 177, 117–125. [Google Scholar] [PubMed]
- Kobayashi, M.; Sada, N.; Sugawara, M.; Iseki, K.; Miyazaki, K. Development of a new system for prediction of drug absorption that takes into account drug dissolution and pH change in the gastro-intestinal tract. Int. J. Pharm. 2001, 221, 87–94. [Google Scholar] [PubMed]
- Albertini, B.; Di Sabatino, M.; Melegari, C.; Passerini, N. Formulation of spray congealed microparticles with self-emulsifying ability for enhanced glibenclamide dissolution performance. J. Microencapsul. 2015, 32, 181–192. [Google Scholar]
- Qi, S.; Marchaud, D.; Craig, D.Q. An Investigation into the mechanism of dissolution rate enhancement of poorly water-soluble drugs from spray chilled Gelucire 50/13 microspheres. J. Pharm. Sci. 2010, 99, 262–274. [Google Scholar] [PubMed]
- Cavallari, C.; Rodriguez, L.; Albertini, B.; Passerini, N.; Rosetti, F.; Fini, A. Thermal and Fractal Analysis of Diclofenac/Gelucire 50/13 Microparticles Obtained by Ultrasound-Assisted Atomization. J. Pharm. Sci. 2005, 94, 1124–1134. [Google Scholar]
- Oh, C.M.; Ru Shan Siow, C.; Heng, P.V.; Chan, L.W. Impact of HPMC on the physical properties of spray-congealed PEG microparticles and its swelling effect on rifampicin dissolution. Drug Dev. Ind. Pharm. 2016, 42, 403–411. [Google Scholar]
- Fini, A.; Cavallari, C.; Ospitali, F. Raman and thermal analysis of indomethacin/PVP solid dispersion enteric microparticles. Eur. J. Pharm. Biopharm. 2008, 70, 409–420. [Google Scholar]
- Hasa, D.; Perissutti, B.; Grassi, M.; Chierotti, M.R.; Gobetto, R.; Ferrario, V.; Lenaz, D.; Voinovich, D. Mechanochemical activation of vincamine mediated by linear polymers: Assessment of some “critical” steps. Eur. J. Pharm. Sci. 2013, 50, 56–68. [Google Scholar]
- Duarte, Í.; Andrade, R.; Pinto, J.F.; Temtem, M. Green production of cocrystals using a new solvent-free approach by spray congealing. Int. J. Pharm. 2016, 506, 68–78. [Google Scholar] [PubMed]
- Qiao, N.; Li, M.; Schlindwein, W.; Malek, N.; Davies, A.; Trappitt, G. Pharmaceutical cocrystals: An overview. Int. J. Pharm. 2011, 419, 1–11. [Google Scholar] [PubMed]
- Gadade, D.D.; Pekamwar, S.S. Pharmaceutical Cocrystals: Regulatory and Strategic Aspects, Design and Development. Adv. Pharm. Bull. 2016, 6, 479–494. [Google Scholar] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Carrier + Additives | Type of SD | Achievement | Ref. |
|---|---|---|---|---|
| Carbamazepine | Gelucire® 50/13 | Crystalline drug (original polymorph) | Increased in vitro dissolution rate | [37] |
| Carbamazepine | Gelucire® 50/13 | Crystalline drug (change from β to α) | Increased in vitro dissolution rate | [64] |
| Piroxicam | Gelucire® 50/13 | Crystalline drug (original polymorph) | Increased in vitro dissolution rate | [87] |
| Praziquantel | Gelucire® 50/13 | Crystalline drug (original polymorph) | Increased in vitro dissolution rate | [36] |
| Olanzapine | Gelucire® 50/13, Lutrol F68 or Lutrol F127 | Reduced-sized drug particles | Increased in vitro dissolution rate | [55] |
| Acetazolamide | Poloxamer-237 | Conversion to amorphous form/ dispersion on molecular level | Increased in vitro dissolution rate and 9-fold solubility enhancement | [33] |
| Glibenclamide | Myverol, Myvatex, Gelucire® 50/13, Gelucire® 44/14, Poloxamer 188 and PEG 4000 | Crystalline drug | Five-fold increased drug solubilization (Gelucire 50/13 + PEG 4000) | [86] |
| Metronidazole | PEG 3350 + HPMC, Dicalcium phosphate, Magnesium stearate, Methylcellulose, Polyvinylpyrrolidone, Silicon dioxide, Sodium oleate/Citric acid | Marked reduction in drug crystallinity | Slightly increased in vitro dissolution rate with only carrier (different results depending on the type and amount of additive) | [49] |
| Rifampicin | PEG 3350 + HPMC (different grades) | No information provided | Increased in vitro dissolution rate, with intensity depending on the HPMC grade. | [89] |
| Glimepiride | Gelucire® 50/13, poloxamer 188, and PEG 6000 | Crystalline drug (original polymorph) | Increased in vitro dissolution rate (Gelucire the highest increase). 10-fold (Gelucire) and 5-fold (poloxamer, PEG) solubility enhancement | [19] |
| Diclofenac | Gelucire® 50/13 | Marked reduction in drug crystallinity | Increase in the in vitro dissolution rate | [88] |
| Indomethacin | Gelucire® 50/13, Gelucire® 48/16 | Conversion to amorphous form/dispersion on molecular level | Increase in the in vitro dissolution rate and solubility Increased oral bioavailability in rats | [45] |
| Bufadienolides (bufalin, cinobufagin, and resibufogenin) | Lutrol F127 | Formation of molecular dispersions | Four-fold increase in vitro dissolution rate | [50] |
| Wild garlic extract | Gelucire® 50/13 | No information provided | Increase in the in vitro dissolution rate | [51] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertoni, S.; Albertini, B.; Passerini, N. Spray Congealing: An Emerging Technology to Prepare Solid Dispersions with Enhanced Oral Bioavailability of Poorly Water Soluble Drugs. Molecules 2019, 24, 3471. https://doi.org/10.3390/molecules24193471
Bertoni S, Albertini B, Passerini N. Spray Congealing: An Emerging Technology to Prepare Solid Dispersions with Enhanced Oral Bioavailability of Poorly Water Soluble Drugs. Molecules. 2019; 24(19):3471. https://doi.org/10.3390/molecules24193471
Chicago/Turabian StyleBertoni, Serena, Beatrice Albertini, and Nadia Passerini. 2019. "Spray Congealing: An Emerging Technology to Prepare Solid Dispersions with Enhanced Oral Bioavailability of Poorly Water Soluble Drugs" Molecules 24, no. 19: 3471. https://doi.org/10.3390/molecules24193471
APA StyleBertoni, S., Albertini, B., & Passerini, N. (2019). Spray Congealing: An Emerging Technology to Prepare Solid Dispersions with Enhanced Oral Bioavailability of Poorly Water Soluble Drugs. Molecules, 24(19), 3471. https://doi.org/10.3390/molecules24193471