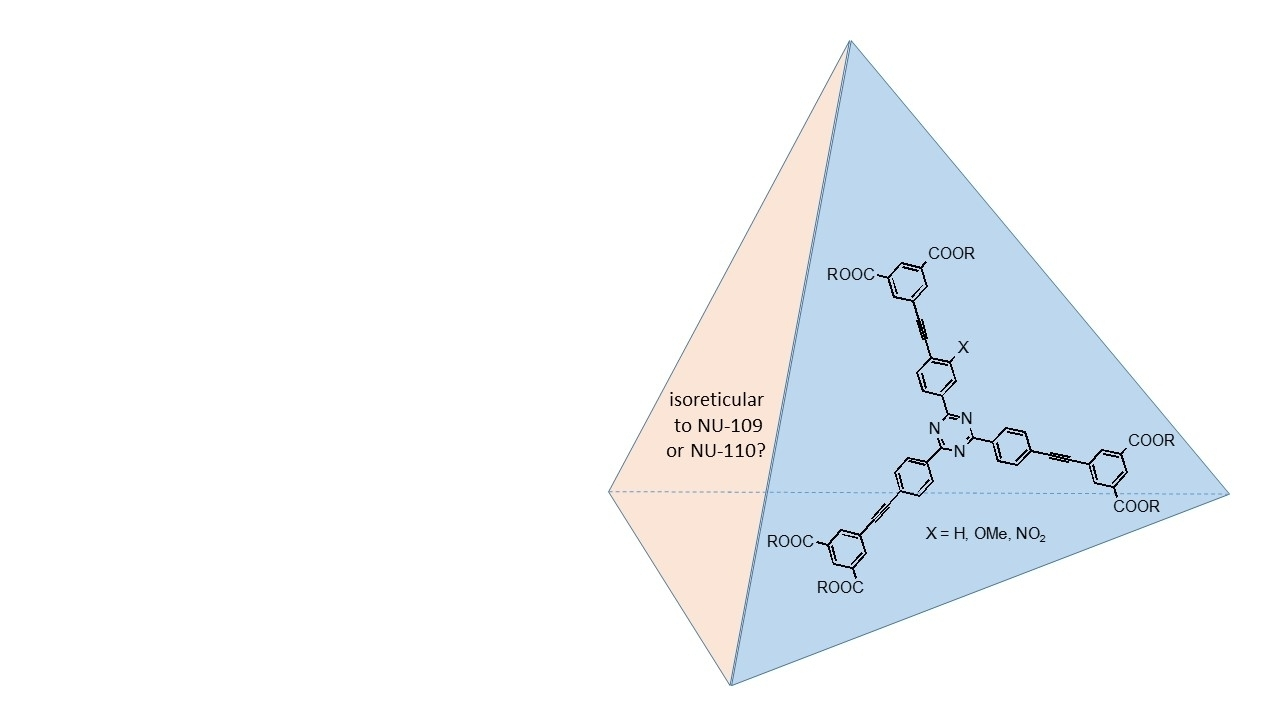
3. Experimental Section
General Remarks: 1,1′-Bis(diphenylphosphine)ferrocene-palladium(II) chloride (99.9%, ABCR, Karlsruhe, Germany), bis(triphenylphosphine)palladium(II) dichloride (98%, ABCR), tetrakis(triphenylphosphine)palladium(0) (99%, ABCR), and trimethylsilylethyne (
8, 98%, ABCR) were purchased and used without further purification. Dry solvents were obtained using suitable desiccants. Other solvents were distilled before use. Melting points were measured with a Gallenkamp MPD350.BM2.5 instrument. NMR spectra were recorded with a Bruker DRX 500 or Avance 600 instrument at 300 K (Billerica, MA, USA). Assignments are supported by COSY, HSQC, and HMBC. Even when obtained by DEPT, the type of
13C signal is always listed as singlet, doublet, etc. All chemical shifts are referenced to tetramethylsilane or the residual proton or carbon signals of the solvent.
1H and
13C-NMR spectra of compounds
6,
7,
9,
10, and
11 can be found in the
Supplementary Materials. HRMS-EI mass spectra were recorded with JEOL AccuTOF GCV 4G (Tokyo, Japan). MALDI-TOF mass spectra were recorded with a Bruker-Daltronics Biflex III (Billerica, MA, USA) with Cl-CCA (4-chloro-α-cyanocinnamic acid) as matrix. IR spectra were recorded with a Perkin-Elmer Spectrum 100 spectrometer (Waltham, MA, USA) equipped with a Golden Gate Diamond ATR unit A-531-G. Elemental analyses were carried out with a Euro EA 3000 Elemental Analyzer from Euro Vector (Pavia, Italy). Traces of solvent originated from the purification step. The analytical sample of
11a was obtained from an NMR solution.
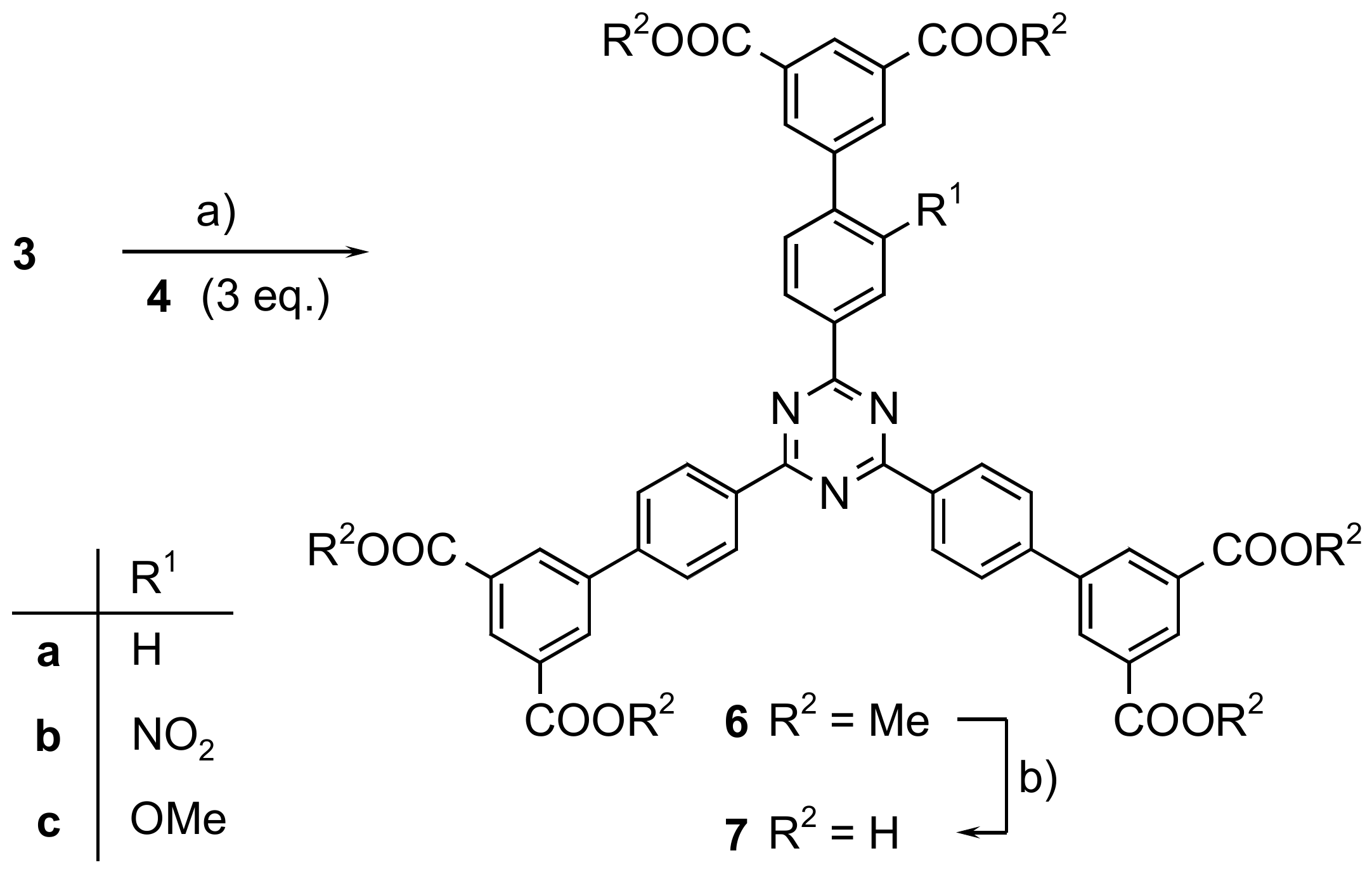
3.1. 2,4,6-Tris[3′,5′-bis(methoxycarbonyl)-biphenyl-4-yl]-1,3,5-triazine (6a)
![Molecules 24 03480 i001]()
Under nitrogen, a solution of 2,4,6-tris(4-bromophenyl)-1,3,5-triazine (
3a, 803 mg, 1.47 mmol) [
20], dimethyl 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-isophthalate (
4, 1.69 g, 5.28 mmol), potassium acetate (1.30 g, 13.2 mmol), and 1,1′-bis(diphenylphosphine)ferrocene-palladium(II) chloride (120 mg, 164 µmol) in a 10:1 mixture (90 mL) of 1,4-dioxane and water was heated to reflux for 48 h. After evaporation of the dioxane, deionized water (100 mL) was added, and the aq. layer was extracted with chloroform (3 × 100 mL). The combined organic extract was washed with brine (100 mL), dried with magnesium sulfate, and filtered. Activated charcoal was added, and the mixture was heated and filtered while hot. After reduction of the volume, the crude product was recrystallized from chloroform/petrol ether, yielding 1.14 g (1.29 mmol, 88%) of a colorless solid. M. p.: >300 °C.
1H NMR (600 MHz, CDCl
3): δ = 8.88 (m
c(d), 6 H,
J = 8.1 Hz, Ar-
H-3,5), 8.69 (s, 3 H, Ar-
H-4′), 8.56 (s, 6 H, Ar-
H-2′,6′), 7.87 (m
c(d), 6 H,
J = 8.1 Hz, Ar-
H-2,6), 4.01 (s, 18 H, CO
2C
H3) ppm.
13C NMR (150 MHz, CDCl
3): δ = 171.2 (s, tri-
C-2,4,6), 166.1 (s,
CO
2Me), 142.9 (s, Ar-
C-1), 141.1 (s, Ar-
C-1′), 136.0 (s, Ar-
C-4), 132.4 (d, Ar-
C-2′,6′), 131.3 (s, Ar-
C-3′,5′), 129.9 (d, Ar-
C-4′), 129.7 (d, Ar-
C-3,5), 127.4 (d, Ar-
C-2,6), 52.6 (q, CO
2CH
3) ppm. MS (MALDI, Cl-CCA):
m/
z = 886 [M + H]
+. IR (ATR):
= 3005 (aryl-H), 2993 (C-H-val.), 1690 (C=O), 1609, 1581, 1509 (arom. C=C, arom. C=N), 1435 (CH-def.), 1349 (C-N-val.), 813 (1,4-disubst. aryl, 1,3,5-trisubst. aryl) cm
−1. Elemental analysis (C
51H
39N
3O
12) (885.87): calcd. C 69.15 H 4.44 N 4.74; (C
51H
39N
3O
12·0.1 CHCl
3) (897.81): calcd. C 68.36 H 4.39 N 4.68; found C 68.26 H 4.28 N 4.81.
3.2. 2-[3′,5′-Bis(methoxycarbonyl)-2-nitrobiphenyl-4-yl]-4,6-bis[3′,5′-bis(methoxycarbonyl)-biphenyl-4-yl]-1,3,5-triazine (6b)
![Molecules 24 03480 i002]()
Under nitrogen, a mixture of 2-(4-bromo-3-nitrophenyl)-4,6-bis(4-bromophenyl)-1,3,5-triazine (
3b, 100 mg, 170 µmol) [
13], dimethyl 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-isophthalate (
4, 245 mg, 765 µmol), tetrakis(triphenylphosphine)-palladium(0) (30 mg, 26 µmol), and potassium phosphate (234 mg, 1.10 mmol) in a mixture of 1,4-dioxane (10 mL) and deionized water (1 mL) was heated to reflux for 48 h. After evaporation of the dioxane in vacuo, the residue was dissolved in water (25 mL) and extracted with chloroform (3 × 25 mL). The combined organic layer was washed with brine (25 mL), dried with magnesium sulfate, and filtered. Activated charcoal was added to the filtrate, and the mixture was heated and filtered through celite while hot. The solvent was evaporated in vacuo and the residue was recrystallized from a hot mixture of toluene and
n-heptane yielding 131 mg (141 µmol, 83%) of a colorless solid, m. p.: >300 °C.
1H NMR (500 MHz, CDCl
3): δ = 9.33 (s, 1 H, Ar-
H-3), 9.02 (d, 1 H,
J = 7.0 Hz, Ar-
H-5), 8.86 (d, 4 H,
J = 8.0 Hz, Ar′-
H-3,5), 8.75 (s, 1 H, Ar-
H-4′), 8.69 (s, 2
H, Ar-
H-2′,6′), 8.53 (s, 4 H, Ar′-
H-2′,6′), 8.23 (s, 2 H, Ar′-
H-4′), 7.87 (d, 4 H,
J = 8.0 Hz, Ar′-
H-2,6), 7.65 (d, 1 H,
J = 7.0 Hz, Ar-
H-6), 4.01 (s, 12 H, Ar′-CO
2C
H3), 3.98 (s, 6 H, Ar-CO
2C
H3) ppm.
13C NMR (125 MHz, CDCl
3): δ = 171.6 (s, tri-
C-4,6), 169.3 (s, tri-
C-2), 166.0 (s, Ar-
CO
2Me), 166.0 (s, Ar′-
CO
2Me), 149.1 (s, Ar-
C-2), 143.3 (s, Ar′-
C-1), 140.8 (s, Ar′-
C-1′), 138.0 (s, Ar-
C-1), 137.2 (Ar-
C-1′), 135.2 (s, Ar′-
C-4), 133.1 (d, Ar′-
C-4′), 132.5 (d, Ar-
C-5), 132.4 (d, Ar′-
C-2′,6′), 132.4 (d, Ar-
C-6), 131.4 (s, Ar′-
C-3′,5′), 131.3 (s, Ar-
C-3′,5′), 130.6 (d, Ar-
C-4′), 130.0 (s, Ar-
C-4), 129.9 (d, Ar-
C-2′,6′), 129.7 (d, Ar′-
C-3,5), 127.5 (d, Ar′-
C-2,6), 124.8 (d, Ar-
C-3), 52.6 (q, Ar-CO
2CH
3), 52.6 (q, Ar′-CO
2CH
3) ppm. MS (MALDI, Cl-CCA):
m/
z = 931 [M + H]
+. IR (ATR):
= 3005 (aryl-H), 2992, 2952 (C-H-val.), 1719 (C=O), 1607, 1575, 1506 (arom. C=C, arom. C=N), 1515 (NO
2), 1431 (CH-def.), 1341 (C-N-val.), 817 (1,4-disubst. aryl, 1,3,4-trisubst. aryl) cm
−1. Elemental analysis (C
51H
38N
4O
14) (930.87): calcd. C 65.80 H 4.11 N 6.02; found C 65.50 H 4.12 N 5.91.
3.3. 2-[2-Methoxy-3′,5′-bis(methoxycarbonyl)-biphenyl-4-yl]-4,6-bis[3′,5′-bis(methoxycarbonyl)-biphenyl-4-yl]-1,3,5-triazine (6c)
![Molecules 24 03480 i003]()
Under nitrogen, 2-(4-bromo-3-methoxyphenyl)-4,6-bis(4-bromophenyl)-1,3,5-triazine (
3c, 100 mg, 175 µmol) [
13], dimethyl 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-isophthalate (
4, 252 mg, 788 µmol) and potassium phosphate (234 mg, 1.10 mmol) were mixed with a mixture of 1,4-dioxane (10 mL), and deionized water (1 mL). Tetrakis(triphenylphosphine)-palladium(0) (30 mg, 26 µmol) was added, and the mixture was stirred for 48 h at 100 °C. After evaporation of the dioxane in vacuo, chloroform (25 mL) was added to the residue, and the resulting mixture was washed with deionized water (3 × 25 mL). The organic layer was dried with magnesium sulfate, filtered, and heated with little activated charcoal. After filtration through celite while hot, the solvent was evaporated in vacuo until the residue turned turbide. Recrystallization from a boiling mixture of chloroform and petrol ether (b. p. 40–60 °C) yielded 130 mg (142 µmol, 81 %) of a colorless solid, m. p.: >300 °C.
1H NMR (500 MHz, CDCl
3): δ = 8.88 (d, 4 H,
J = 8.5 Hz, Ar′-
H-3,5), 8.70 (t, 2 H,
J = 1.5 Hz, Ar′-
H-4′), 8.67 (t, 1 H,
J = 1.6 Hz, Ar-
H-4′), 8.55 (d, 4 H,
J = 1.5 Hz, Ar′-
H-2′,6′), 8.49 (dd, 1 H,
J = 7.9 Hz,
J = 1.4 Hz, Ar-
H-5), 8.47 (d, 2 H,
J = 1.6 Hz, Ar-
H-2′,6′), 8.42 (d, 1 H,
J = 1.4 Hz, Ar-
H-3), 7.88 (d, 4 H,
J = 8.5 Hz, Ar′-
H-2,6), 7.56 (d, 1 H,
J = 7.9 Hz, Ar-
H-6), 4.07 (s, 3 H, Ar-OC
H3), 4.01 (s, 12 H, Ar′-CO
2C
H3), 3.99 (s, 6 H, Ar-CO
2C
H3) ppm.
13C NMR (125 MHz, CDCl
3): δ = 171.2 (s, tri-
C-4,6), 171.1 (s, tri-
C-2), 166.3 (s, Ar-
CO
2Me), 166.1 (s, Ar′-
CO
2Me), 156.7 (s, Ar-
C-2), 143.0 (s, Ar′-
C-1), 141.0 (s, Ar′-
C-1′), 138.7 (s, Ar-
C-1′), 137.4 (s, Ar-
C-4), 135.9 (s, Ar′-
C-4), 134.9 (d, Ar-
C-2′,6′), 132.7 (s, Ar-
C-1), 132.3 (d, Ar′-
C-2′,6′), 131.4 (s, Ar-
C-3′,5′). 131.0 (d, Ar-
C-6), 130.5 (s, Ar′-
C-3′,5′), 129.9 (d, Ar′-
C-4′), 129.7 (d, Ar′-
C-3,5), 129.6 (d, Ar-
C-4′), 127.4 (d, Ar′-
C-2,6), 121.9 (d, Ar-
C-5), 111.2 (d, Ar-
C-3), 55.9 (q, Ar-O
CH
3), 52.5 (q, Ar′-CO
2CH
3), 52.4 (q, Ar-CO
2CH
3) ppm. MS (MALDI, Cl-CCA):
m/
z = 916 [M + H]
+. IR (ATR):
~ = 3005 (aryl-H), 2954 (C-H-val.), 1728 (C=O), 1606, 1578, 1517 (arom. C=C, arom. C=N), 1429 (CH-def.), 1371 (OCH
3), 1342 (C-N-val.), 809 (1,4-disubst. aryl, 1,3,4-trisubst. aryl) cm
−1. Elemental analysis (C
52H
41N
3O
13) (915.89): calcd. C 68.19 H 4.51 N 4.59; (C
52H
41N
3O
13·0.2 CHCl
3) (939.71): calcd. C 66.71 H 4.42 N 4.47; found C 66.34 H 4.74 N 4.86.
3.4. 2,4,6-Tris[3′,5′-dicarboxybiphenyl-4-yl]-1,3,5-triazine (7a)
![Molecules 24 03480 i004]()
A mixture of 2,4,6-tris[3′,5′-bis(methoxycarbonyl)-biphenyl-4-yl]-1,3,5-triazine (6a, 1.04 g, 1.17 mmol) and lithium hydroxide monohydrate (1.38 g, 32.8 mmol) in tetrahydrofuran (130 mL) and deionized water (15 mL) was heated to 60 °C for 48 h. After evaporation of the solvent in vacuo, a small amount of deionized water was added, and the mixture was acidified with hydrochloric acid (6 M). The precipitate was filtered off, washed with deionized water and chloroform, and dried in vacuo, yielding 939 mg (1.17 mmol, > 99%) of a yellow solid, m. p.: >300 °C. 1H NMR (600 MHz, DMSO-d6): δ = 8.76 (d, 6 H, J = 8.3 Hz, Ar-H-3,5), 8.47 (t, 3 H, J = 1.4 Hz, Ar-H-4′), 8.41 (d, 6 H, J = 1.4 Hz, Ar-H-2′,6′), 7.94 (d, 6 H, J = 8.3 Hz, Ar-H-2,6) ppm. 13C NMR (150 MHz, DMSO-d6): δ = 170.6 (s, tri-C-2,4,6), 166.4 (s, CO2H), 142.5 (s, Ar-C-1), 140.0 (s, Ar-C-1′), 135.0 (s, Ar-C-4), 132.2 (s, Ar-C-3′,5′), 131.4 (d, Ar-C-2′,6′), 129.5 (d, Ar-C-4′), 128.8 (d, Ar-C-3,5), 127.4 (d, Ar-C-2,6) ppm. MS (MALDI, Cl-CCA): m/z = 802 [M + H]+. IR (ATR): = 3018 (br., OH), 1690 (C=O), 1603, 1578, 1508, 1412 (arom. C=C, arom. C=N), 1367 (C-N-val.), 815 (1,3,5-trisubst. aryl) cm−1. Elemental analysis (C45H27N3O12) (801.71): calcd. C 67.42 H 3.39 N 4.87; (C45H27N3O12·1.4 H2O·0.2 CHCl3) (850.80): calcd. C 63.81 H 3.55 N 4.94; found C 63.87 H 3.67 N 4.87.
3.5. 2-[3′,5′-Dicarboxy-2-nitrobiphenyl-4-yl]-4,6-bis[3′,5′-dicarboxybiphenyl-4-yl]-1,3,5-triazine (7b)
![Molecules 24 03480 i005]()
A suspension of 2-[3′,5′-bis(methoxycarbonyl)-2-nitrobiphenyl-4-yl]-4,6-bis[3′,5′-bis(methoxycarbonyl)-biphenyl-4-yl]-1,3,5-triazine (6b, 44 mg, 47 µmol) and lithium hydroxide monohydrate (101 mg, 2.41 mmol) in a mixture of tetrahydrofuran (10 mL) and deionized water (1.5 mL) was stirred at 60 °C for 24 h. After evaporation of the tetrahydrofuran in vacuo, some deionized water was added to the residue. Acidification with hydrochloric acid (6 M) produced a precipitate which was filtered off and washed thoroughly with deionized water and chloroform yielding 39 mg (46 µmol, >99%) of a yellow solid, m. p.: >300 °C. 1H NMR (500 MHz, DMSO-d6): δ = 9.21 (d, 1 H, J = 1.6 Hz, Ar-H-3), 9.01 (dd, 1 H, J = 8.0 Hz, J = 1.6 Hz, Ar-H-5), 8.81 (d, 4 H, J = 8.4 Hz, Ar′-H-3,5), 8.53 (t, 1 H, J = 1.5 Hz, Ar-H-4′), 8.48 (t, 2 H, J = 1.5 Hz, Ar′-H-4′), 8.43 (d, 4 H, J = 1.5 Hz, Ar′-H-2′,6′), 8.15 (d, 2 H, J = 1.5 Hz, Ar-H-2′,6′), 7.98 (d, 4 H, J = 8.4 Hz, Ar′-H-2,6), 7.88 (d, 1 H, J = 8.0 Hz, Ar-H-6) ppm. 13C NMR (125 MHz, DMSO-d6): δ = 171.4 (s, tri-C-4,6), 169.4 (s, tri-C-2), 166.8 (s, Ar′-CO2H), 166.5 (s, Ar-CO2H), 149.3 (s, Ar-C-2), 143.3 (s, Ar′-C-1), 140.9 (s, Ar-C-4), 140.4 (s, Ar′-C-4), 137.7 (s, Ar-C-1), 137.1 (s, Ar-C-4), 136.8 (s, Ar-C-1′), 135.1 (s, Ar′-C-1′), 133.4 (d, Ar-C-6), 133.0 (d, Ar-C-5), 132.9 (d, Ar-C-2′,6′), 132.6 (s, Ar-C-3′,5′), 132.4 (s, Ar′-C-3′,5′), 131.9 (d, Ar′-C-2′,6′), 130.2 (d, Ar-C-4′), 130.1 (d, Ar′-C-3,5), 129.99 (s, Ar-C-1′), 129.95 (Ar′-C-4′), 127.9 (d, Ar′-C-2,6), 124.5 (d, Ar-C-3) ppm. MS (MALDI, Cl-CCA): m/z = 847 [M + H]+. IR (ATR): = 3080 (OH), 2921, 2854 (CO2H), 1696 (C=O), 1608, 1576, 1515, 1456 (arom. C=C, arom. C=N), 1360 (C-N-val.), 1243 (NO2), 813 (1,4-disubst. aryl, 1,3,5-trisubst. aryl) cm−1. Elemental analysis (C45H26N4O14) (846.71): calcd. C 63.83 H 3.10 N 6.62; (C45H26N4O14·0.95 CHCl3·0.05 H2O) (960.95): calcd. C 57.43 H 2.84 N 5.83; found C 57.80 H 3.04 N 5.43.
3.6. 2,4-Bis[3′,5′-dicarboxybiphenyl-4-yl]-6-[3′,5′-dicarboxy-2-methoxybiphenyl-4-yl]-1,3,5-triazine (7c)
![Molecules 24 03480 i006]()
A suspension of 2-[2-methoxy-3′,5′-bis(methoxycarbonyl)-biphenyl-4-yl]-4,6-bis[3′,5′-bis(methoxycarbonyl)-biphenyl-4-yl]-1,3,5-triazine (6c, 50 mg, 54 µmol) and lithium hydroxide monohydrate (101 mg, 2.41 mmol) in a mixture of tetrahydrofuran (10 mL) and deionized water (1.5 mL) was heated to 60 °C for 24 h. After evaporation of tetrahydrofuran in vacuo, the aqueous residue was diluted slightly with deionized water and acidified with hydrochloric acid (6 M). The precipitate was washed with deionized water and chloroform, yielding 40 mg (48 µmol, 89%) of a yellow solid, m. p.: >300 °C. 1H NMR (600 MHz, DMSO-d6): δ = 13.39 (br. s, 6 H, CO2H), 8.84 (d, 4 H, J = 8.2 Hz, Ar′-H-3,5), 8.51–8.50 (m, 2 H, Ar′-H-4′), 8.48–8.47 (m, 5 H, Ar′-H-2′,6′, Ar-H-4′), 8.44 (mc(d), 1 H, J = 7.8 Hz, Ar-H-5), 8.41 (s, 1 H, Ar-H-3), 8.34 (mc(d), 2 H, J = 1.1 Hz, Ar-H-2′,6′), 8.01 (d, 4 H, J = 8.2 Hz, Ar′-H-2,6), 7.66 (d, 1 H, J = 7.8 Hz, Ar-H-6), 4.04 (s, 3 H, OCH3) ppm. 13C NMR (150 MHz, DMSO-d6): δ = 170.7(s, tri-C-4,6), 170.4 (s, tri-C-2), 166.5 (s, Ar-CO2H), 166.4 (s, Ar′-CO2H), 156.4 (s, Ar-C-2), 142.6 (s, Ar′-C-1), 140.0 (s, Ar′-C-1′), 137.9 (s, Ar-C-1′), 136.7 (s, Ar-C-4), 135.1 (s, Ar′-C-4), 134.0 (d, Ar-C-2′,6′), 132.3 (s, Ar-C-1), 132.2 (s, Ar′-C-3′,5′), 131.5 (d, Ar-C-5), 131.3 (s, Ar-C-3′,5′), 131.0 (d, Ar-C-6), 129.6 (d, Ar′-C-3,5), 129.3 (d, Ar′-C-2′,6′), 129.0 (d, Ar′-C-4′), 127.5 (d, Ar′-C-2,6), 121.6 (d, Ar-C-4′), 111.2 (d, Ar-C-3), 55.8 (q, OCH3) ppm. MS (MALDI, Cl-CCA): m/z = 832 [M + H]+. IR (ATR): = 3100 (br., OH), 1692 (C=O), 1603, 1578, 1509, 1405 (arom. C=C, arom. C=N), 1360 (C-N-val.), 1221 (aryl-OCH3), 810 (1,3,5-trisubst. aryl) cm−1. Elemental analysis (C46H29N3O13) (831.17): calcd. C 67.42 H 3.39 N 4.87; (C46H29N3O13·0.45 H2O·0.9 CHCl3) (947.28): calcd. C 59.47 H 3.28 N 4.44; found C 59.58 H 3.40 N 4.48.
3.7. 2,4,6-Tris{4-[(trimethylsilyl)ethynyl]-phenyl}-1,3,5-triazine (9a)
![Molecules 24 03480 i007]()
Under nitrogen, trimethylsilylethyne (
8, 972 μL, 6.88 mmol) was added to a mixture of 2,4,6-tris(4-bromophenyl)-1,3,5-triazine (
3a, 1.00 g, 1.83 mmol) [
20] in tetrahydrofuran (200 mL) and triethylamine (120 mL). After the addition of tetrakis(triphenylphosphine)-palladium(0) (240 mg, 208 µmol), copper(I) iodide (40 mg, 208 µmol), and triethylamine (120 mL), the mixture was stirred for 48 h at 40 °C (TLC control, silica gel, cyclohexane,
Rf = 0.23). Solvents were distilled off in vacuo, and the residue was mixed with deionized water (100 mL) and extracted with ethyl acetate (3 × 100 mL). The combined organic layer was washed with brine (100 mL), dried with magnesium sulfate, and filtered. The solvent was distilled off in vacuo, and the residue was dissolved in toluene and filtered through neutral aluminium oxide. After evaporation of the solvent in vacuo, the residue was recrystallized from boiling
n-heptane yielding 826 mg (1.38 mmol, 75%) of a colorless solid, m. p.: 274 °C (ref. [
19]: no m.p. given).
1H NMR (600 MHz, CDCl
3): δ = 8.67 (m
c(d), 6 H,
J = 8.4 Hz, Ar-
H-3,5), 7.64 (m
c(d), 6 H,
J = 8.4 Hz, Ar-
H-2,6), 0.30 (s, 27 H, Si(C
H3)
3) ppm.
13C NMR (150 MHz, CDCl
3): δ = 171.0 (s, tri-
C-2,4,6), 135.7 (s, Ar-
C-1), 132.2 (d, Ar-
C-2,6), 128.7 (d, Ar-
C-3,5), 127.4 (s, Ar-
C-4), 104.7 (s, Ar-
C≡C), 97.5 (s, Ar-C≡
C), 0.0 (s, Si(
CH
3)
3) ppm. HRMS (EI):
m/
z = calcd. 597.2451; found 597.2441 (Δ 1.78 ppm). Elemental analysis (C
36H
39N
3Si
3) (597.97): calcd. C 72.31 H 6.57 N 7.03; found C 72.17 H 6.52 N 6.93.
3.8. 2-{3-Nitro-4-[(trimethylsilyl)ethynyl]-phenyl}-4,6-bis{4-[(trimethylsilyl)ethynyl]-phenyl}-1,3,5-triazine (9b)
![Molecules 24 03480 i008]()
Under nitrogen, tetrakis(triphenylphosphine)-palladium(0) (88 mg, 77 µmol) and copper(I) iodide (15 mg, 77 µmol) were added to a mixture of 2-(4-bromo-3-nitrophenyl)-4,6-bis(4-bromophenyl)-1,3,5-triazine (
3b, 500 mg, 850 µmol) [
13] in tetrahydrofuran (100 mL) and triethylamine (60 mL). After the addition of trimethylsilylethyne (
8, 726 µL, 5.10 mmol), the mixture was stirred for 48 h at 55 °C (TLC control, silica gel, cyclohexane/ethyl acetate, 10:1,
Rf = 0.76). The solvent was evaporated in vacuo, and chloroform (150 mL) was added to the residue. The organic layer was washed with deionized water (3 × 100 mL), dried with magnesium sulfate, and filtered. Activated charcoal was added, and the mixture was heated and filtered through celite and silica gel. The solvent was evaporated in vacuo, and the crude product was recrystallized from boiling
n-heptane yielding 391 mg (608 µmol, 72%) of a colorless solid, m. p.: 273–275 °C.
1H NMR (500 MHz, CDCl
3): δ = 9.31 (d, 1 H,
J = 1.4 Hz, Ar-
H-2), 8.88 (dd, 1 H,
J = 8.1 Hz,
J = 1.4 Hz, Ar-
H-6), 8.67 (d, 4 H,
J = 8.4 Hz, Ar′-
H-2,6), 7.82 (d, 1 H,
J = 8.1 Hz, Ar-
H-5), 7.65 (d, 4 H,
J = 8.4 Hz, Ar′-
H-3,5), 0.32 (s, 9 H, Ar-C≡C-Si(C
H3)
3), 0.30 (s, 18 H, Ar′-C≡C-Si(C
H3)
3) ppm.
13C NMR (125 MHz, CDCl
3): δ = 171.4 (s, tri-
C-4,6), 169.2 (s, tri-
C-2), 150.7 (s, Ar-
C-3), 136.9 (s, Ar-
C-1), 135.4 (d, Ar-
C-5), 135.2 (s, Ar′-
C-1), 132.3 (d, Ar′-
C-3,5), 132.2 (d, Ar-
C-6), 128.8 (d, Ar-
C-2,6), 127.9 (s, Ar′-
C-4), 124.7 (d, Ar-
C-2), 121.7 (s, Ar-
C-4), 107.2 (s, Ar-
C≡C), 104.5 (s, Ar-C≡
C), 99.2 (s, Ar′-
C≡C), 98.0 (s, Ar′-C≡
C), 0.3 (q, Ar′-C≡C-Si(
CH
3)
3), 0.0 (q, Ar-C≡C-Si(
CH
3)
3) ppm. IR (ATR):
= 3005 (aryl-H), 2957, 2901 (C-H-val.), 2155 (C≡C), 1603, 1570, 1504, 1407 (arom. C=C, arom. C=N), 1537 (NO
2), 1355 (C-N-val.), 838 (1,4-disubst. aryl, 1,3,4-trisubst. aryl) cm
−1. HRMS (EI):
m/
z = calcd. 642.2302; found 642.2295 (Δ 1.11 ppm). Elemental analysis (C
36H
38N
4O
2Si
3) (642.97): calcd. C 67.25 H 5.96 N 8.71; found C 67.32 H 6.25 N 8.46.
3.9. 2-{3-Methoxy-4-[(trimethylsilyl)ethynyl]-phenyl}-4,6-bis{4-[(trimethylsilyl)ethynyl]-phenyl}-1,3,5-triazine (9c)
![Molecules 24 03480 i009]()
Under nitrogen, tetrakis(triphenylphosphine)-palladium(0) (45 mg, 39 µmol) and copper(I) iodide (8 mg, 0.04 mmol) were added to a mixture of 2-(4-bromo-3-methoxyphenyl)-4,6-bis(4-bromophenyl)-1,3,5-triazine (
3c, 250 mg, 434 µmol) [
13] in tetrahydrofuran (50 mL) and triethylamine (30 mL). Trimethylsilylethyne (
8, 370 µL, 2.60 mmol) was added and the mixture was stirred for 48 h at 55 °C. After removal of the volatiles in vacuo, the residue was dissolved in chloroform (50 mL) and washed with deionized water (3 × 50 mL). The organic layer was dried with magnesium sulfate, filtered, and heated with activated charcoal. After filtration through celite, the solvent was removed in vacuo. The residue was recrystallized from boiling
n-heptane yielding 255 mg (406 µmol, 94%) of a colorless solid, m. p. 212 °C.
1H NMR (500 MHz, CDCl
3): δ = 8.67 (m
c(d), 4 H,
J = 8.7 Hz, Ar′-
H-2,6), 8.31 (dd, 1 H,
J = 8.0 Hz,
J = 1.4 Hz, Ar-
H-6), 8.24 (d, 1 H,
J = 1.4 Hz, Ar-
H-2), 7.65 (m
c(d), 4 H,
J = 8.7 Hz, Ar′-
H-3,5), 7.61 (d, 1 H,
J = 8.0 Hz, Ar-
H-5), 4.08 (s, 3 H, OC
H3), 0.31 (s, 9 H, Ar-C≡C-Si(C
H3)
3), 0.29 (s, 18 H, Ar′-C≡C-Si(C
H3)
3) ppm.
13C NMR (125 MHz, CDCl
3): δ = 171.0 (s, tri-
C-4,6), 171.0 (s, tri-
C-2), 160.6 (s, Ar-
C-3), 137.4 (s, Ar-
C-1), 135.7 (s, Ar′-
C-1), 134.3 (d, Ar-
C-2), 132.3 (d, Ar′-
C-3,5), 128.7 (d, Ar′-
C-2,6), 127.5 (s, Ar′-
C-4), 121.2 (d, Ar-
C-6), 116.8 (s, Ar-
C-4), 110.5 (d, Ar-
C-5), 104.6 (s, Ar-
C≡C), 101.8 (s, Ar-C≡
C), 100.9 (s, Ar′-
C≡C), 97.6 (s, Ar′-C≡
C), 56.2 (q, O
CH
3), 0.00 (s, Ar-C≡C-Si(
CH
3)
3), 0.01 (q, Ar′-C≡C-Si(
CH
3)
3) ppm. IR (ATR):
= 3005 (aryl-H), 2972 (C-H-val.), 2903 (OCH
3), 2060 (C≡C), 1606. 1570, 1511, 1407 (arom. C=C, arom. C=N), 1357 (C-N-val.), 813 (1,4-disubst. aryl, 1,3,4-trisubst. aryl) cm
−1. HRMS (EI):
m/
z = calcd. 672.2557; found 672.2543 (Δ 2.38 ppm). Elemental analysis (C
37H
41N
3OSi
3) (628.00): calcd. C 70.76 H 6.58 N 6.69; (C
37H
41N
3OSi
3·0.3 C
7H
16·0.4 H
2O) (664.93): calcd. C 70.63 H 7.06 N 6.32; found C 70.97 H 6.78 N 5.99.
3.10. 2,4,6-Tris(4-ethynylphenyl)-1,3,5-triazine (10a)
![Molecules 24 03480 i010]()
A mixture of 2,4,6-tris{4-[(trimethylsilyl)ethynyl]-phenyl}-1,3,5-triazine (
9a, 500 mg, 835 µmol) and potassium carbonate (1.04 g, 7.50 mmol) in methanol (25 mL) was stirred for 24 h at room temp. (TLC control, silica gel, cyclohexane,
Rf = 0.15). After evaporation of the methanol, the residue was dissolved in deionized water (25 mL) and extracted with chloroform (3 × 25 mL). The combined organic layer was washed with brine (25 mL) and dried with magnesium sulfate. After filtration and removal of the solvent in vacuo, the crude product was recrystallized from a boiling mixture of toluene and
n-heptane, yielding 315 mg (827 µmol, >99%) of a yellowish solid, m. p. >300 °C (ref. [
19]: no m.p. given).
1H NMR (500 MHz, CDCl
3): δ = 8.63 (m
c(d), 6 H,
J = 8.9 Hz, Ar-
H-3,5), 7.61 (m
c(d), 6 H,
J = 8.9 Hz, Ar-
H-2,6), 3.24 (s, 3 H, C≡CH) ppm.
13C NMR (125 MHz, CDCl
3): δ = 170.9 (s, tri-
C-2,4,6), 135.9 (s, Ar-
C-1), 132.2 (d, Ar-
C-2,6), 128.6 (d, Ar-
C-3,5), 126.3 (s, Ar-
C-4), 83.10 (s,
C≡CH), 80.0 (d, C≡
CH) ppm. IR (ATR):
= 3236 (C≡C-H), 3002 (aryl-H), 2160 (C≡C), 1606, 1574, 1505, 1408 (arom. C=C, arom. C=N), 1357 (C-N-val.), 813 (1,4-disubst. aryl) cm
−1. HRMS (EI):
m/z = calcd. 381.1266; found 381.1251 (Δ 3.85 ppm). Elemental analysis (C
27H
15N
3) (381.43): calcd. C 85.02 H 3.96 N 11.02; found C 85.24 H 3.94 N 10.64.
3.11. 2,4-Bis(4-ethynylphenyl)-6-(3-methoxy-4-ethynylphenyl)-1,3,5-triazine (10c)
![Molecules 24 03480 i011]()
A mixture of 2-{3-methoxy-4-[(trimethylsilyl)ethynyl]-phenyl}-4,6-bis{4-[(trimethylsilyl)ethynyl]-phenyl}-1,3,5-triazine (9c, 150 mg, 239 µmol) and potassium carbonate (297 mg, 2.15 mmol) in methanol (7.5 mL) was stirred for 24 h at room temp. After evaporation of the solvent in vacuo, the residue was dissolved in chloroform (25 mL) and washed with water (3 × 25 mL). The organic layer was dried with magnesium sulfate, filtered, and the solvent was evaporated in vacuo. The crude product was recrystallized from a boiling mixture of toluene and n-heptane, yielding 98 mg (239 µmol, >99%) of 10c, m. p. >300 °C. 1H NMR (600 MHz, CDCl3): δ = 8.71 (d, 4 H, J = 8.3 Hz, Ar′-H-2,6), 8.35 (dd, 1 H, J = 7.8 Hz, J = 1.2 Hz, Ar-H-6), 8.29 (br. s, 1 H, Ar-H-2), 7.70 (d, 4 H, J = 8.3 Hz, Ar′-H-3,5), 7.66 (d, 1 H, J = 7.8 Hz, Ar-H-5), 4.12 (s, 3 H, OCH3), 3.50 (s, 1 H, Ar-C≡CH), 3.28 (s, 1 H, Ar′-C≡CH) ppm. 13C NMR (150 MHz, CDCl3): δ = 171.1 (s, tri-C-2), 171.1 (tri-C-4,6), 160.5 (s, Ar-C-3), 136.1 (s, Ar′-C-1), 134.3 (d, Ar-C-5), 132.4 (d, Ar′-C-3,5), 131.9 (s, Ar-C-1), 128.8 (d, Ar′-C-2,6), 126.4 (s, Ar′-C-4), 121.2 (d, Ar-C-6), 115.5 (s, Ar-C-4), 83.7 (d, Ar-C≡CH), 83.2 (s, Ar′-C≡CH), 79.7 (s, Ar-C≡CH), 79.7 (d, Ar′-C≡CH), 56.0 (q, OCH3) ppm. HRMS (EI): m/z = calcd. 411.1371; found 411.1367 (Δ 1.13 ppm). IR (ATR): = 3280, 3246 (C≡C-H), 3008 (aryl-H), 2970 (OCH3), 2926 (C-H-val.), 2160 (C≡C), 1606, 1574, 1504, 1408 (arom. C=C, arom. C=N), 1437 (C-H-def.), 1353 (C-N-val.), 812 (1,4-disubst. aryl, 1,3,4-trisubst. aryl) cm−1.
3.12. 2,4,6-Tris{4-[3,5-bis(ethoxycarbonyl)phenylethynyl]-phenyl}-1,3,5-triazine (11a)
![Molecules 24 03480 i012]()
Under nitrogen, 2,4,6-tris(4-ethynylphenyl)-1,3,5-triazine (10a, 50.0 mg, 131 µmol) and diethyl 5-iodoisophthalate (5, 171 mg, 491 µmol) were dissolved in a mixture of tetrahydrofuran (6 mL) and triethylamine (4 mL). Bis(triphenylphosphine)-palladium(II) dichloride (10 mg, 14 µmol) and copper(I) iodide (3.0 mg, 14 µmol) were added, and the mixture was stirred for 48 h at 50 °C. After evaporation of the solvents in vacuo, chloroform (25 mL) was added to the residue. The organic layer was washed with deionized water (3 × 25 mL) and brine (25 mL). After drying with magnesium sulfate and filtration, the solvent was removed in vacuo. The crude product was recrystallized from a boiling mixture of toluene and n-heptane, yielding 108 mg (104 umol, 79%) of a yellowish solid, m. p. >300 °C. 1H NMR (500 MHz, CDCl3): δ = 8.75 (d, 6 H, J = 8.4 Hz, Ar-H-2,6), 8.64 (t, 3 H, J = 1.6 Hz, Ar′-H-4), 8.39 (d, 6 H, J = 1.6 Hz, Ar′-H-2,6), 7.74 (d, 6 H, J = 8.4 Hz, Ar-H-3,5), 4.44 (q, 12 H, J = 7.1 Hz, CO2CH2CH3), 1.45 (t, 18 H, J = 7.1 Hz, CO2CH2CH3) ppm. 13C NMR (125 MHz, CDCl3): δ = 171.0 (s, tri-C-2,4,6), 165.1 (s, CO2Et), 136.5 (d, Ar′-C-2,6), 136.0 (s, Ar-C-1), 132.0 (d, Ar-C-3,5), 131.4 (s, Ar′-C-3,5), 130.3 (d, Ar′-C-4), 129.0 (d, Ar-C-2,6), 126.9 (s, Ar-C-4), 123.9 (s, Ar′-C-1), 90.9 (s, Ar-C≡C-Ar′), 90.3 (s, Ar-C≡C-Ar′), 61.6 (t, CO2CH2CH3), 14.3 (q, CO2CH2CH3) ppm. MS (MALDI, Cl-CCA): m/z = 1042 [M + H]+. IR (ATR): = 3005 (aryl-H), 2985 (C-H-val.), 1690 (C=O), 1607, 1509 (arom. C=C, arom. C=N), 1429 (CH-def.), 1356 (C-N-val.), 825 (1,4-disubst. aryl, 1,3,5-trisubst. aryl) cm−1. Elemental analysis (C63H51N3O12) (1042.04): calcd. C 72.61 H 4.93 N 4.03; (C63H51N3O12·0.3 CDCl3) (1077.85): calcd. C 70.53 H 4.80 N 3.90; found C 70.20 H 4.68 N 4.28.
3.13. 2-{4-[3,5-Bis(ethoxycarbonyl)phenylethynyl]-3-methoxy-phenyl}-4,6-bis{4-[3,5-bis(ethoxycarbonyl)phenylethynyl]-phenyl}-1,3,5-triazine (11c)
![Molecules 24 03480 i013]()
Under nitrogen, 2,4-bis(4-ethynylphenyl)-6-(3-methoxy-4-ethynylphenyl)-1,3,5-triazine (10c, 50.0 mg, 122 µmol) and diethyl 5-iodoisophthalate (5, 159 mg, 456 µmol) were dissolved in a mixture of tetrahydrofuran (6 mL) and triethylamine (4 mL). After the addition of bis(triphenylphosphine)-palladium(II) dichloride (10 mg, 14 µmol) and copper(I) iodide (3.0 mg, 14 µmol), the mixture was stirred at 50 °C for 48 h. After evaporation of the volatiles, chloroform (25 mL) was added, and the mixture was extracted with deionized water (3 × 25 mL) and brine (25 mL). The organic layer was dried with magnesium sulfate, filtered, and the solvent was evaporated in vacuo. The crude product was recrystallized from a boiling mixture of toluene n-heptane, yielding 106 mg (98.8 μmol, 81%) of a yellowish solid, m. p. >300 °C. 1H NMR (500 MHz, CDCl3): δ = 8.79 (d, 4 H, J = 8.2 Hz, Ar′-H-2,6), 8.66 (s, 2 H, Ar′-H-4′), 8.65 (s, 1 H, Ar-H-4′), 8.44 (d, 2 H, J = 1.2 Hz, Ar-H-2′,6′), 8.43–8.41 (m, 5 H, Ar′-H-2′,6′, Ar-H-6), 8.35 (s, 1 H, Ar-H-2), 7.77 (d, 4 H, J = 8.2 Hz, Ar′-H-3,5), 7.72 (d, 1 H, J = 7.9 Hz, Ar-H-5), 4.48–4.42 (m, 12 H, OCH2CH3), 4.18 (s, 3 H, OCH3), 1.48–1-39 (m, 18 H, OCH2CH3) ppm. 13C NMR (125 MHz, CDCl3): δ = 171.1 (s, tri-C-2), 171.0 (s, tri-C-4,6), 165.2 (s, Ar′-COEt), 165.1 (s, Ar-COEt), 161.0 (s, Ar-C-3), 136.5 (d, Ar-C-2′,6′), 136.5 (s, Ar-C-1), 136.0 (s, Ar′-C-1), 135.3 (d, Ar′-C-2′,6′), 133.9 (d, Ar-C-5), 132.1 (d, Ar′-C-3,5), 131.4 (s, Ar′-C-3′,5′), 131.3 (s, Ar-C-3′,5′), 130.3 (d, Ar′-C-4′), 130.0 (d, Ar-C-4), 129.0 (d, Ar′-C-2,6), 127.0 (s, Ar′-C-4), 121.4 (d, Ar-C-6), 116.2 (s, Ar-C-4), 110.9 (d, Ar-C-2), 94.0 (s, Ar-C≡C), 90.7 (s, Ar′-C≡C), 90.1 (s, Ar′-C≡C), 87.8 (Ar-C≡C), 61.7 (t, Ar′-CO-CH2CH3), 61.6 (t, Ar-CO-CH2CH3), 56.1 (q, OCH3), 14.4 (q, OCH2CH3) ppm. IR (ATR): = 3075 (aryl-H), 2985, 2941 (C-H-val.), 1721 (C=O), 1599, 1569 (arom. C=C, arom. C=N), 1444 (CH-def.), 1366 (C-N-val.), 810 (1,4-disubst. aryl, 1,3,5-trisubst. aryl) cm−1. MS (MALDI, Cl-CCA): m/z = 1073 [M + H]+.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
















