1. Introduction
The expanding use of positron emission tomography (PET) imaging in oncology to aid in early detection, more accurate diagnosis, and patient treatment stratification has led to an increased demand of novel peptide-based molecular imaging agents [
1]. Peptides are attractive as imaging agents for several reasons, including favorable pharmacokinetic properties, such as high target affinity, good tumor penetration, rapid non-specific clearance, and minimal immunogenicity [
2]. Additionally, once peptide sequences are identified, they can be readily synthesized to high purity and are amenable to chemical modifications to alter affinity, charge, hydrophobicity, solubility, and stability [
3]. The biological half-life of peptides ideally matches with many clinically used radionuclides, such as the short-lived positron emitting fluorine-18 (t
1/2 = 109.7 min) that possesses favorable nuclear properties (e.g., 97% positron emission), and the ease of production with high yield and molar activity for PET imaging [
4].
Combinatorial libraries, such as the one-bead one-compound (OBOC) and phage display, have provided indispensable tools for rapid identification of peptides for a given biological target, especially those expressed on the cell surface [
5]. OBOC libraries display randomized peptide sequences on solid-support beads, which can be screened against either purified proteins or whole cells to identify receptor-specific peptides [
6]. We, and others, have utilized the OBOC approach to identify peptides for the development of PET imaging agents [
6,
7]. However, to date, only a handful of OBOC-derived peptides, notably the α
5β
1–targeting OA02 peptide [
8] and the α
4β
1-targeting LLP2A peptidomimetic [
9,
10], have been evaluated for preclinical in vivo imaging. Clearly, the current translation of OBOC-derived peptides from bench to bedside remains a slow process [
11].
To ensure target-specificity of the identified peptides, on-bead library screening requires multiple rounds of screening to eliminate false positive (non-specific binders) [
12]. This stringent and iterative screening of OBOC libraries for target-selective peptides is a lengthy, time-consuming, and inefficient process. For OBOC-derived peptides to be utilized as PET imaging agents, a positron-emitting isotope, such as fluorine-18, needs to be attached to the peptide. The introduction of the radioisotope, typically via a prosthetic group post-identification from the OBOC library, can have a negative impact on the pharmacokinetic properties and binding affinity of the original peptide, as it adds mass, alters hydrophobicity, and changes the charge of the peptide [
13]. In addition, once a peptide sequence has been successfully identified, it typically requires further optimization before it can be used as an imaging agent, as peptides are susceptible to proteolytic degradation in serum in vivo, and are often eliminated rapidly from the system [
14]. Therefore, chemical modifications of the OBOC-derived peptides are carried out to improve their in vivo stability to make them suitable for PET imaging.
In this work, we proposed to address some of the aforementioned limitations and describe the developmental process for the identification and optimization of integrin α
vβ
6-targeting peptide PET imaging agents, using OBOC and on-bead two-color (OBTC) technologies. The integrin α
vβ
6 is an epithelial-specific cell surface receptor that is undetectable in healthy adult epithelium, but is upregulated in many aggressive cancers, such as ovarian, colon, pancreatic, and breast, and its expression level has been linked to poor prognosis, making it an interesting target for both diagnostic and therapeutic agents [
15]. Notable examples of integrin α
vβ
6-targeting peptides that have been used as imaging agents include the A20FMDV2 [
16] and Cystine Knot R
01 peptides [
17]. Both peptides contain the known α
vβ
6-targeting motif (DLXXL), originally identified from phage display [
18], and they have been radiolabeled with
18F and
64Cu for in vivo imaging in small animal models [
7,
16,
17,
19,
20]. In a proof of concept effort, Gagnon et al. reported the use of combinatorial libraries, solid-phase radiolabeling, and high throughput in vivo screening to evaluate an OBOC library featuring the DLXXL motif, and discovered 42 lead peptides in 11 consecutive days [
7]. However, the identified lead peptides still required substantial optimization.
Herein, we designed an OBOC library containing the known integrin α
vβ
6-binding motif (DLXXL) [
18] and the nonradioactive substitute of the commonly used
18F-radiolabeling prosthetic group, 4-fluorobenzoic acid ([
19F]FBA) at the C-terminal lysine (XXDLXXLXK([
19F]FB, 9-mer). To facilitate the direct identification of integrin α
vβ
6-binding peptides in a single step, an OBTC cell screen assay [
21], utilizing transfected fluorescent DX3puroβ
6mCherry (α
vβ
6+, red) and DX3puroEmGFP (α
vβ
6−, green) cell lines, was investigated. Competitive binding ELISAs with purified integrins (α
vβ
1, α
vβ
3, α
vβ
5, α
vβ
6, and α
vβ
8) were performed to evaluate the binding profiles of lead peptides. Furthermore, efforts focused on optimizing the most promising peptide via N-, C-, and bi-terminal chemical modifications such as methylation, acetylation, 4-fluorobenzoylation, and PEGylation, as well as C-terminal extension for improved binding affinity, binding selectivity, and metabolic stability. Peptides were radiolabeled on solid-phase using 4-[
18F]fluorobenzoic acid ([
18F]FBA) for evaluation of cell binding and serum stability.
3. Discussion
The OBOC technology has been successfully utilized to identify cancer-targeting peptides [
6,
7,
8], yet the approach remains painstakingly slow, with clear delays between on-bead ligand identification and subsequent optimization to a potential imaging agent [
7,
11]. The aim of this work was to combine multiple strategies to streamline the development of integrin α
vβ
6-targeting peptide-based imaging agents for PET. This included (1) OBOC chemistry to produce large peptide libraries, (2) elimination of setbacks due to post-screening modifications of peptides by incorporating 4-[
19F]fluorobenzoic acid (FBA) into the peptide library design, (3) utilizing the OBTC fluorescent cell screening assay to expedite the on-bead library screening process (combine three rounds of screening into a single step), and (4) structural-based modifications to optimize OBOC-derived peptides for improved serum stability and pharmacokinetics properties.
The incorporation of a prosthetic group or a chelator into a peptide sequence post-identification from a library can often have a negative impact on the binding properties [
13]. To avoid this, the OBOC library was designed to contain the nonradioactive prosthetic group [
19F]FBA pre-attached at the C-terminal lysine’s side chain. The placement of the prosthetic group at the
C-terminus was designed to allow for Edman sequencing of the beads identified as selective binders to the cells that express integrin α
vβ
6. In addition, the library contained the α
vβ
6-targeting motif DLXXL, a motif originally discovered from a phage display library as an important binding sequence for the integrin α
vβ
6 [
18,
23]. Stringent and iterative screening of OBOC libraries for target-selective peptides is lengthy, time-consuming, and not highly efficient. Traditionally, the OBOC libraries are screened against either purified proteins or whole cells, and positive beads (i.e., bound to protein or cells) are visualized under a microscope, and manually isolated [
24]. To ensure target-specificity of the peptides, this process is often carried out in multiple rounds [
24], with multiple washing cycles between subsequent rounds of screening, which often leads to broken or damaged beads. To streamline the screening, the OBTC cell-screening assay developed by Udagamasooriya et al. was adopted [
21]. Using a mixed pool of two-color fluorescent cells differing only in the expression of integrin α
vβ
6, beads that were recognized by the DX3puroβ
6mCherry (α
vβ
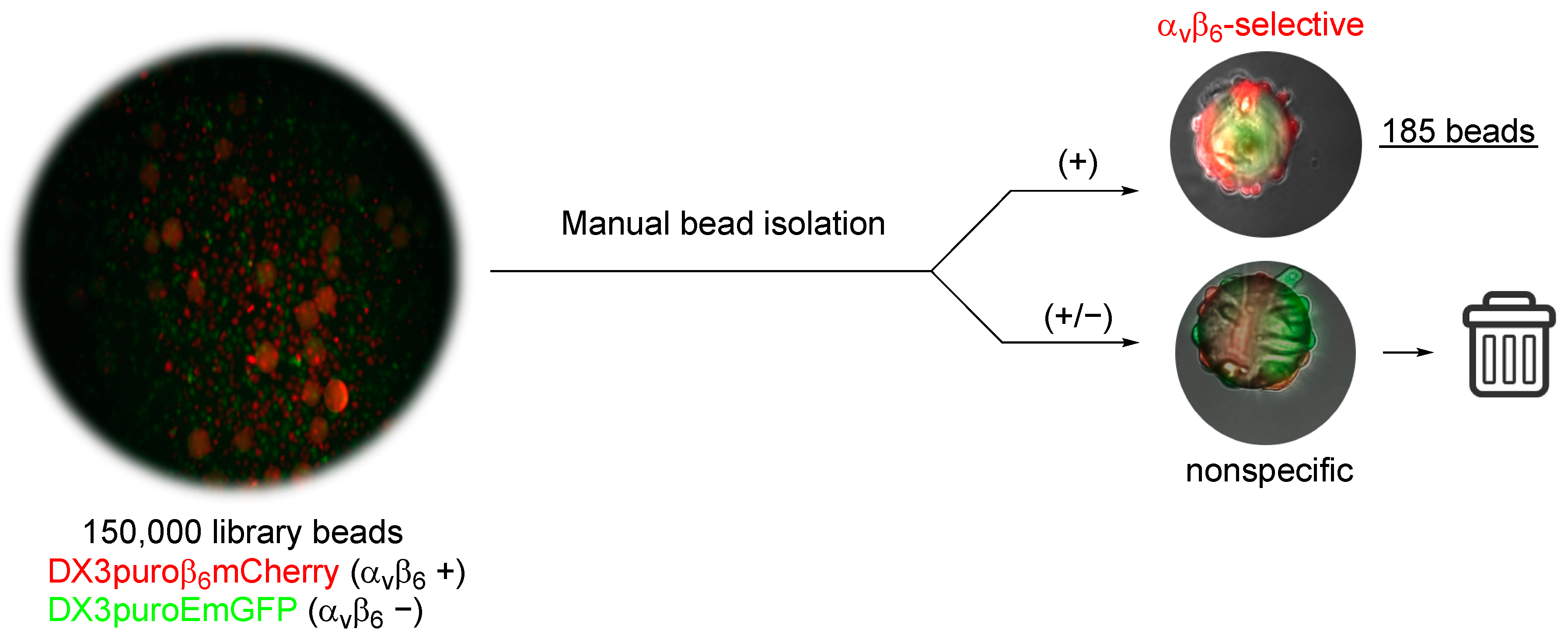
6+, red) cells were directly retrieved from the assay in one round of screening. This approach resulted in the rapid identification of 185 α
vβ
6-positive beads from the entire 4-fluorobenzoyl peptide library (~2.5 million compounds), which otherwise would be difficult to achieve using the traditional screening approach. Conventionally, sequence determination of on-resin peptide is commonly carried out with Edman degradation [
25,
26,
27]; however, this method is rather time consuming (hours to a day to sequence one peptide) and expensive (~
$300/peptide) [
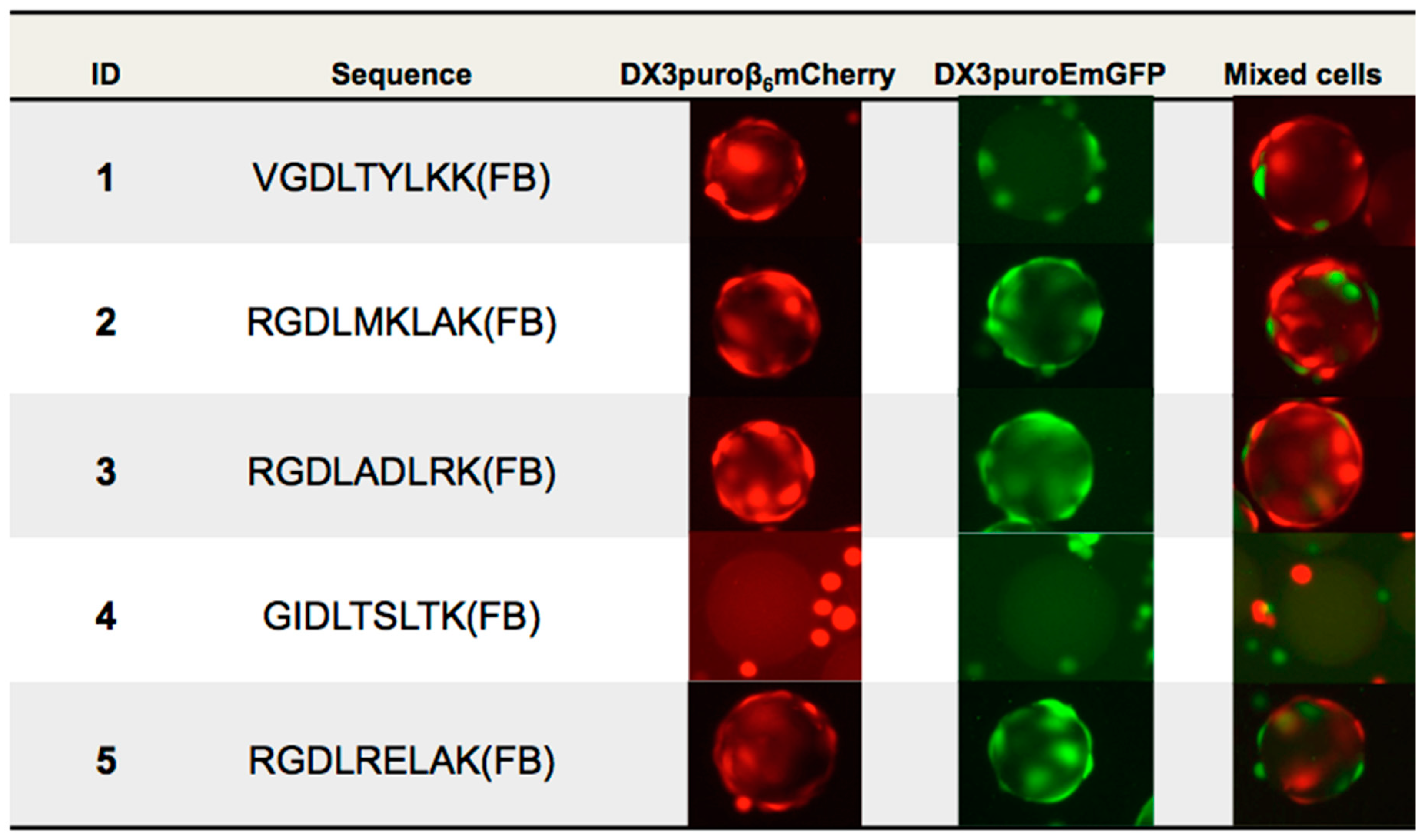
28]. With these limitations in mind for the purpose of this work only, the first five beads identified from the first aliquot of screening were sequenced by Edman degradation for further evaluations.
Of the five beads sequenced, three contained the three amino acids RGD sequence, a motif known to mediate cell adhesion and binds to α
v integrins, including integrin α
vβ
6 [
29]. The remaining two sequences did not share any homology and had the unique VGD and GID motifs. Qualitative on-bead cell binding with DX3puroβ
6mCherry (α
vβ
6+, red) cells confirmed binding of four of the five peptides. The three RGD containing peptides also showed some interactions with the negative cell line, which was likely due to recognition of the RGD motif by other RGD-binding integrins also expressed on the DX3puroEmGFP (α
vβ
6−, green) cell line. This was corroborated by the ELISA data, where binding to all α
v-integrins except for α
vβ
5 was observed. The GID containing peptide did not bind to either cell line or α
v-integrins by ELISA, and was possibly an outlier picked by mistake during the manual bead picking process.
Based on the initial in vitro data,
1 (VGDLTYLKK(FB)) was selected for further optimization. Modifications at the
N-terminus with various chemical moieties, including aliphatic (e.g., acetyl, methyl, PEG
12) and aromatic (e.g., FBA) groups, PEGylation (e.g., PEG
28) at the
C-terminus, as well as a combination of both, were performed. Unfortunately, none of the modifications resulted in a new analog with a better binding affinity or selectivity for the integrin α
vβ
6, as indicated by the ELISA and cell binding data. Interestingly, the
N-terminus of the peptide appeared to be more tolerant towards modifications with the aromatic FBA than the aliphatic methyl, acetyl, and PEG moieties, which all showed poor binding (IC
50 > 5–100 μM). These results suggest that the presence of the FB at the
N-terminus of the peptide could possibly have favorable interactions with the protein residues in an existing binding pocket. A similar observation has been recently reported for a series of
18F-radiolabeled phosphoamidate peptidomimetic inhibitors for prostate specific membrane antigen (PSMA), where the FB ring of one of the analogues in the series engaged in an “arene-binding” interaction with the surface of the protein, and thus contributing to its high affinity in vitro and in vivo [
30]. Future molecular docking studies or co-crystallization of these peptides with integrin α
vβ
6 are necessary to provide better understanding of the peptide-integrin interaction.
Although peptides possess many favorable pharmacokinetics properties, such as high target affinity, good tumor penetration, and rapid non-specific clearance, they are prone to proteolytic degradation in vivo. PEGylation has been a widely adopted strategy to improve the pharmacokinetics properties of both small molecules and biopharmaceuticals (e.g., peptides, proteins, and antibodies, etc.) [
31]. Based on our previous experience, the addition of PEG moieties to the
N- and
C-termini of the highly α
vβ
6-selective A20FMDV2 peptide successfully prolonged the biological half-life, improved metabolic stability and enhanced tumor retention of the [
18F]FB-PEG
28-A20FMDV2, [
18F]FB-(PEG
28)
2-A20FMDV2, and [
18F]FB-PEG
28-A20FMDV2-PEG
28 in human tumor xenograft models [
16,
19,
20]. Of the numerous peptides evaluated,
1 demonstrated the highest serum stability. To further improve the stability, C-terminal PEGylation of
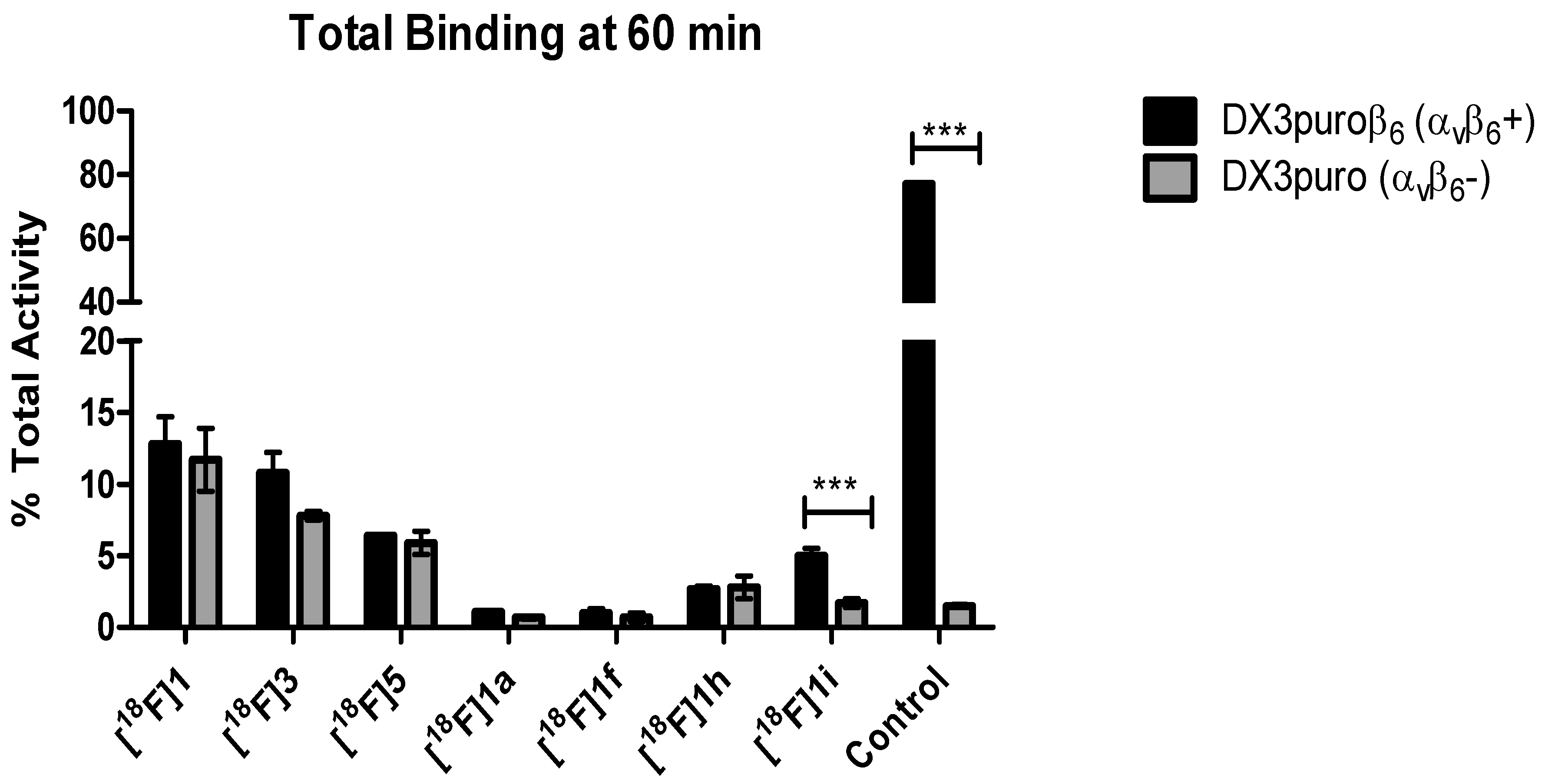
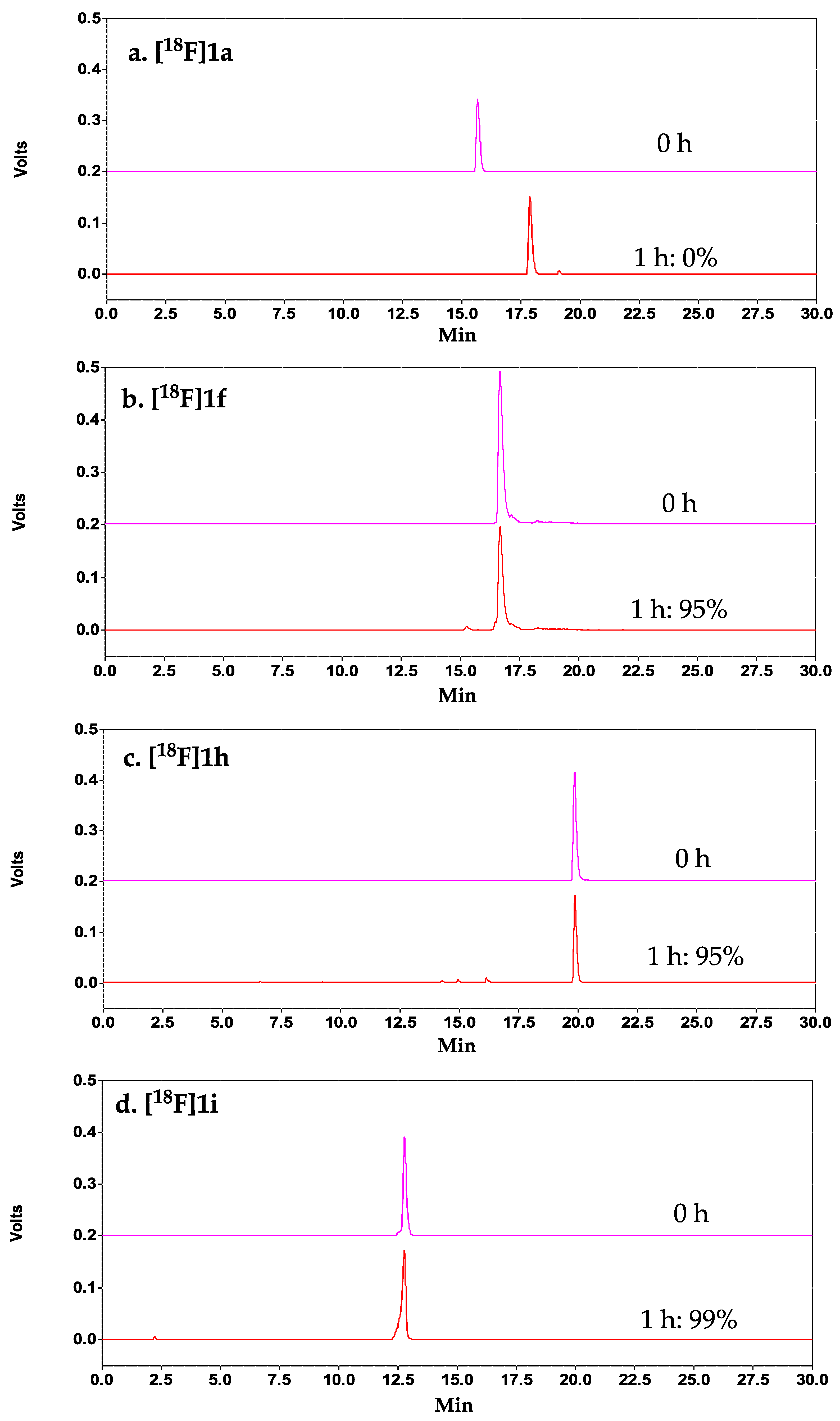
1 was explored. Our results demonstrated that C-terminal PEGylation of the peptide (with and without N-terminal FBA,
1h, and
1f, respectively) indeed did significantly improve the metabolic stability of the peptide, with >95% intact peptide observed at 1 h post-incubation in mouse serum. Despite having excellent serum stability, both analogs showed significant reduction in binding affinity (<2% binding to DX3puroβ
6 (α
vβ
6+) cells). Our data suggests that the introduction of a PEG moiety at the
C-terminus of the peptide could effectively shield the peptide from proteases, which is consistent with the trend observed for our [
18F]FB-A20FMDV2-PEG
28 (76% intact at 1 h) [
19], comparing to the first generation [
18F]FB-A20FMDV2 (0% intact) [
16]. However, the introduction of the relatively large PEG moiety (MW = 1.5 kDa) to the relatively short peptide (MW = 1.1 kDa) resulted in a drastic loss in binding of
1f and
1h to integrin α
vβ
6. A possible explanation is that the relatively large PEG chain length could have imposed some steric shielding effect on the peptide (i.e., PEG wrap around and shield the binding domain from interacting with the receptor), and in turn negatively affected the binding affinity of the resulting PEGylated peptides. This steric shielding effect of PEG on drug conjugates was previously reported by Mu et al. in a study investigating the impact of PEG chain length and PEGylation site on the bioactivity of the therapeutic protein Staphylokinase, using both experimental assays and computational simulations [
32]. Future work entails the investigation of C-terminal PEGylation of
1 with shorter PEG chain length to further test this hypothesis.
Since
1 (VGDLTYLKK(FB)) is a relatively small peptide, it is possible that modifications at either terminus of the peptide could negatively affect the binding affinity due to the close proximity to the binding domain. Furthermore, a co-crystal structure of the integrin α
vβ
6 with pro-TGFβ3 peptide revealed that integrin α
vβ
6 not only recognizes the RGD motif but also binds preferentially to the LXXL/I motif that folds into an amphipathic α-helix, fitting into a hydrophobic pocket composed solely from residues of the β
6 subunit [
33]. As reported previously, the α
vβ
6-binding A20FMDV2 peptide has been confirmed to have a post-RGD α-helix, contributing to its high affinity and selectivity for integrin α
vβ
6 [
34]. Additionally, other α
vβ
6-selective peptides reported in the literature, such as the R
01 peptide [
17] and the H2009.1 peptide [
23], are relatively larger, composing of 30- and 20-amino acids, respectively. To that end, we suspected that the lack of binding affinity and selectivity of our peptide
1 (9-mer) to integrin α
vβ
6 could be attributed to its inability to form a stabilized α-helix to fit the binding pocket tightly. This led us to explore extending the
C-terminus of
1 to improve its binding affinity and selectivity for integrin α
vβ
6. Indeed, extension of the
C-terminus of
1 with the KVART sequence from the known integrin α
vβ
6-binding A20FMDV2 (NAVPNLRGDLQVLAQKVART) resulted in a 14-mer peptide with improved binding affinity and a 3-fold increase in binding selectivity for DX3puroβ
6 (α
vβ
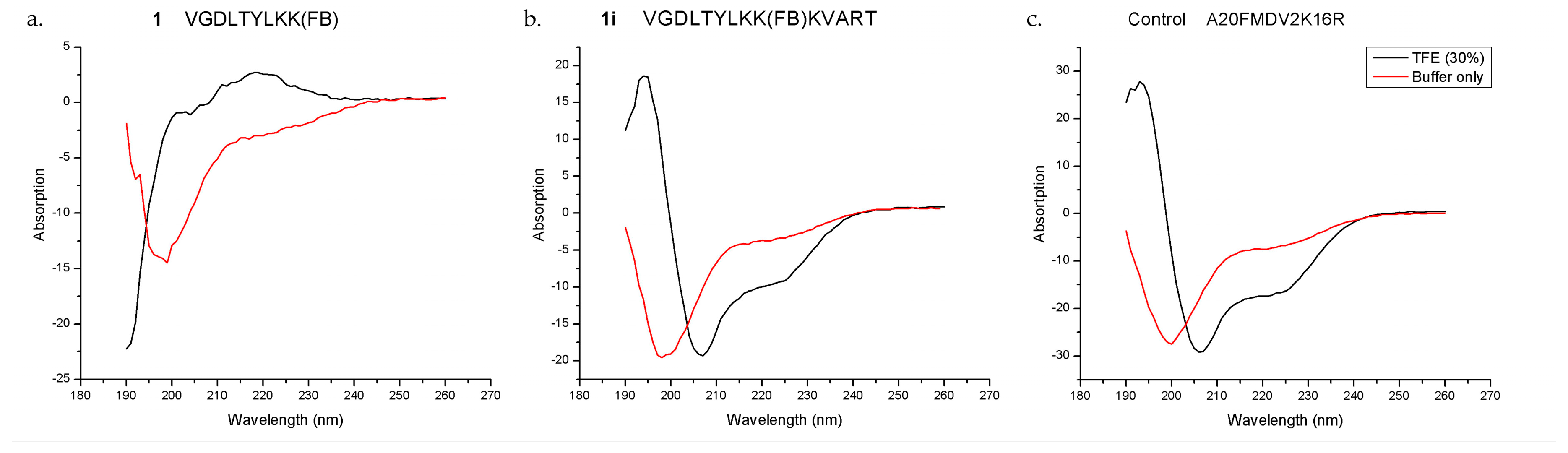
6+) cells, and excellent serum stability (>99% intact at 1 h). The CD results demonstrated that both the control A20FDMV2 and the 14-mer
1i peptides could form an α-helix in 30% TFE/buffer, while the parent peptide
1 could not under the same condition, which supported our hypothesis on the importance of peptide secondary structure on binding affinity and selectivity for integrin α
vβ
6.
4. Materials and Methods
General Experimental Information. All the Fmoc-protected amino acids were purchased from GL Biochem (Shanghai, China). TentaGel S NH2 resin (90 μm) was purchased from RAPP Polymere GmbH (Tuebingen, Germany) and NovaSyn TGR resin was purchased from NovaBiochem (Burlington, MA, USA). All other reagents were purchased from Sigma Aldrich (St. Louis, MO, USA) and Fluka (Mexico City, Mexico), unless otherwise specified. A reverse-phase high performance liquid chromatography (RP-HPLC) Beckman Coulter Gold system (Brea, CA, USA) was used for peptide characterization (Phenomenex Jupiter 4 μ Proteo 90 Å column, Torrance, CA, USA) and purification (Phenomenex Jupiter 10 μ C18 300 Å column). The mobile phase used was 0.05% trifluoroacetic acid (TFA) in water (v/v; solvent A) and 100% acetonitrile (solvent B); solvent B isocratic 9% for 2 min followed by a linear gradient to 81% over 30 min at a flow rate of 1.5 mL/min (analytical RP-HPLC) and 3 mL/min (semi-prep RP-HPLC). All peptides were characterized using a UltraFlextreme (Bruker, Billerica, MA, USA) Matrix-assisted laser desorption-ionization time of flight/time of flight (MALDI TOF/TOF) mass spectrometer with a matrix of sinapinic acid or α-cyano-4-hydroxycinnamic acid.
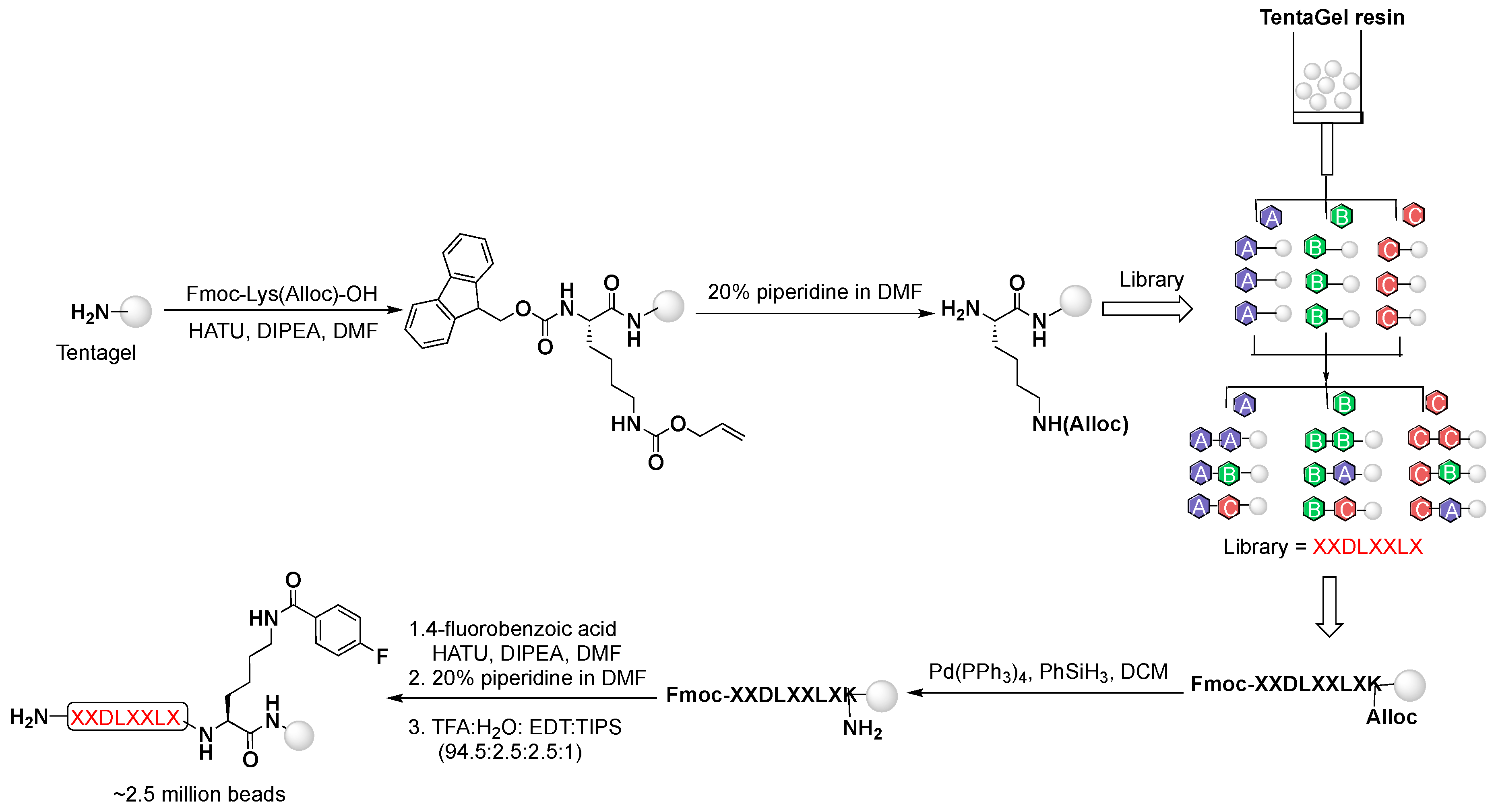
4.1. OBOC Library Synthesis
The peptide library was assembled on TentaGel resin using the “split-mix” technique and standard Fmoc-based solid phase peptide synthesis (SPPS), utilizing all l-amino acids except l-cysteine. Briefly, the resin was first divided into 19 aliquots; each was coupled with a single amino acid (3 equiv.) using O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU, 2.7 equiv.) as a coupling agent and N,N-diisopropylethylamine (DIPEA, 3 equiv.) as a base in N,N-dimethylformamide (DMF) at room temperature for 1.5 h. At the end of each coupling, the resin was re-pooled, mixed, and split for the next coupling step. Fluorenylmethyloxycarbonyl (Fmoc)-protecting group was removed with 20% piperidine in DMF, and allyloxycarbonyl (Alloc)-protecting group was removed with tetrakis(triphenylphosphine)palladium(0) (Pd(PPh3)4, 0.5 equiv.), and phenylsilane (PhSiH3, 20 equiv.) in dichloromethane (DCM). Global deprotection was achieved with trifluoroacetic acid (TFA) mixture with triisopropylsilane (TIPS) and water (95:2.5:2.5 v/v/v) for 3 h. Finally, the resin was washed thoroughly with DMF, water, and methanol (10× each) and stored in 70% ethanol at 4 °C until screening.
4.2. Peptide Synthesis
All peptides were synthesized manually on NovaSyn TGR resin (0.25 mmol/g, Burlington, MA, USA) using standard Fmoc-based SPPS yielding C-terminal amides. Starting with N-α-Fmoc-N-ε-1-(4,4-dimethyl-2,6-dioxocyclohex-1-ylidene)-3-methylbutyl-l-lysine (Fmoc-Lys(ivDde)-OH), orthogonal protecting group for site-specific labeling, each amino acid was coupled using HATU (2.7 equiv.) as a coupling agent and DIPEA (3 equiv.) as a base in DMF at room temperature for 1.5 h. The removal of the Fmoc- and ivDde-protecting groups was facilitated with 20% piperidine in DMF and 2% hydrazine in DMF, respectively. Peptides were cleaved off the resin using a mixture of TFA, water, and TIPS (95:2.5:2.5 v/v/v) for 3 h. The resin was washed with fresh TFA and the filtrate was collected then evaporated under reduced pressure. Crude peptides were precipitated in diethyl ether and extracted from the water/ether bilayer. All crude peptides were purified on the semi-prep HPLC. The purified peptides were analyzed on the analytical HPLC and characterized by MALDI-TOF/MS.
4.3. Cell Lines
DX3Puro (α
vβ
6−) and DX3Puroβ
6 (α
vβ
6+) have been described previously [
35,
36] and were a generous gift from Dr. John Marshall. To generate DX3puroEmGFP and DX3puroβ
6mCherry, DX3Puro and DX3Puroβ
6 cells were transduced with lentivirus carrying EmGFP or mCherry (under control of an EF1α promoter) and the blasticidin resistance gene (under control of a hybrid SV40/EM7 promoter). Transduced cells were cultured at ultra-low density in Dulbecco’s Modified Eagle Medium (DMEM, Gibco, Waltham, MA, USA), supplemented with 10% FBS and blasticidin (10 µg/mL) at 37 °C in the presence of 5% CO
2 for 21 days. Clonal populations of DX3puroEmGFP and DX3puroβ
6mCherry were then isolated, expanded, and screened for fluorescence, and in the case of DX3puroβ
6mCherry, retention of ITGβ
6 expression. Integrin expression was determined by flow cytometry.
4.4. Flow Cytometry
Subconfluent cells were washed with PBS and harvested with Trypsin-EDTA (0.25%, Phenol-red, Gibco). Cells were treated once with PBS containing 0.1% BSA and 0.1% sodium azide (wash buffer), counted by trypan blue exclusion and resuspended at 6 × 106 cells/mL. Aliquots of cells (50 μL) were treated with primary antibody clone 10D5 (Millipore Sigma, Burlington, MA, USA, at 10 µg/mL in serum free DMEM, 50 μL) for 60 min at 4 °C and washed twice with wash buffer (WB). AlexaFluor488 goat anti-mouse secondary antibody (ThermoFisher, Waltham, MA, USA, 1:50 dilution in serum free DMEM, 50 μL) was applied to the cells for 30 min at 4 °C. Cells were washed twice, resuspended in 0.5 mL WB, and scanned on the LSRFortesa Flow Cytometer (Becton Dickinson, Franklin Lakes, NJ, USA) and analyzed using FACSDiva software (8.02, San Jose, CA, USA), acquiring 1 × 104 events.
4.5. Library Screening
4.5.1. Library Beads Preparation
Prior to screening with cells, the beads were thoroughly washed to remove all traces of ethanol. An aliquot of beads (200 μL, equivalent to approximately 150,000 beads) was dried overnight, washed over vacuum with deionized water (10×), allowed to swell in water for 2 to 4 h at room temperature, followed by washing with PBS (5×), and suspended in PBS for 15 min.
4.5.2. OBTC Fluorescent Cell Screening
DX3puroEmGFP (αvβ6−) and DX3puroβ6mCherry (αvβ6+) cells were cultured in DMEM supplemented with 10% FBS, 1% PSG, and blasticidin (10 µg/mL) as adherent monolayers at 37 °C in the presence of 5% CO2 in a humidified incubator for 3 days. At 80% confluency, cells were harvested, counted, and re-suspended in 10% fetal bovine serum (FBS) phenol red–free DMEM for screening. Briefly, cells (500,000 cells/cell line) were mixed in 20 mL phenol red-free DMEM in a non-treated tissue culture petri dish (100 mm; Spectrum Chemicals and Laboratory Products) for 1 h. Library beads (150,000) were added to the mixed cells and incubated at 37 °C with gentle mixing for 3 h. After incubation, the beads were analyzed under a fluorescent microscope. Beads that were >80% covered in DX3puroβ6mCherry cells (αvβ6-specific) were manually selected with a micropipette. The αvβ6-specific beads isolated from the assay were sequenced using Edman degradation.
4.6. Competitive Binding ELISAs
Competitive binding ELISAs were performed to evaluate the binding affinity of each peptide relative to each integrin’s biotinylated natural ligand (transforming growth factor beta 1 latency-associated peptide or TGFβ1-LAP (G&P Biosciences, Santa Clara, CA, USA) for integrins α
vβ
6 and α
vβ
8, fibronectin (Thermo Fisher, Waltham, MA, USA) for integrin α
vβ
1, and vitronectin (Thermo Fisher) for integrins α
vβ
3 and α
vβ
5), as previously described [
22]. In brief, anti-α
v antibody (P2W7, 5 μg/mL, Abcam) was plated (50 μL/well) at 37 °C for 1 h, washed with PBS (3×), and blocked overnight with blocking buffer (300 μL/well, 5% bovine serum albumin (BSA), 1% Tween 20, in PBS), followed by washing with PBS (3×). Purified integrin (R&D Systems, Minneapolis, MN, USA) in conjugate buffer (50 μL/well, 20mM Tris, 1mM MnCl
2, 150 mM NaCl, 0.1% Tween, 1% BSA in water) was then added to each well, incubated at 37 °C for 1 h, followed by washing using wash buffer (20 mM Tris, 1 mM MnCl
2, 150 mM NaCl, 0.1% Tween in water, 3×). Serial dilutions of each peptide (10 μM–10 pM or 100 μM–100 pM in conjugate buffer) and each integrin’s respective biotinylated natural ligand was added to each well (a total volume of 50 μL/well). The plate was incubated at 37 °C for 1 h, then washed with wash buffer (3×). A 1:1000 dilution of ExtrAvidin Horseradish Peroxidase (HRP) was added to each well (50 μL/well), incubated at 37 °C for 1 h, and then washed with wash buffer (3×). The ExtrAvidin HRP was detected with 3,3′,5,5′-tetramethylbenzidine (50 μL/well, Promega, Madison, WI, USA) for 10–15 min at room temperature. The reaction was stopped by adding 1N H
2SO
4 (50 μL/well) and the absorbance was measured in a Multiscan Ascent plate reader (Thermo Fisher) at 450 nm. Half-maximal inhibitory concentration (IC
50) of peptides was determined fitting to sigmoidal dose-response model in GraphPad Prism 5.0 (San Diego, CA, USA).
4.7. Radiochemical Synthesis
All peptides were radiolabeled with [
18F]fluorobenzoic acid ([
18F]FBA) on resin following our well-established solid-phase radiolabeling protocols [
16,
20,
37]. Briefly, [
18F]fluoride was reacted with ethyl-4-(trimethylammonium triflate)benzoate in the presence of 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo [8.8.8]hexacosane (Kryptofix
TM [K2.2.2], (Sigma Aldrich, St. Louis, MO, USA) and potassium carbonate at 100 °C in dimethylsulfoxide (DMSO) for 15 min, followed by saponification of the ethyl ester protect group to yield [
18F]FBA. The [
18F]FBA was re-dissolved in DMF and coupled to the peptide (~5 mg resin) using HATU (5 mg)/DIPEA (10 μL). The
18F-radiolabeled peptide on resin was cleaved and globally deprotected with trifluoroacetic acid/triisopropylsilane/water 95:2.5:2.5 (
v/
v/
v) mixture for 30 min at room temperature. The crude peptide was purified by semi-prep radio-RP-HPLC (C18-column) and formulated in PBS for in vitro assays. The purity and molar activity of the
18F-radiolabeled peptide was confirmed via analytical radio-RP-HPLC. Identity of the
18F-radiolabeled peptide was confirmed by co-injection of the
19F-peptide, and all analytical radio-RP-HPLC analysis was performed for each
18F-radiolabeled peptide prior to use.
4.8. The 18F-Radiolabeled Peptide Cell Binding Assay
Cell binding assay was performed as previously described [
19,
20]. In brief, each cell line was suspended at a concentration of 75 × 10
6 cells/mL in serum-free medium and aliquots of each cell line (3.75 × 10
6 cells in 50 μL) was added to 1.5 mL microcentrifuge tubes previously blocked with 5% BSA in PBS for 10 min at room temperature. The
18F-radiolabeled peptide was prepared in a stock solution of 148 kBq in 1 mL PBS (pH 7.2). Aliquots of each
18F-radiolabeled peptide (50 μL, 7.4 kBq per sample) were added to each cell sample and incubated at 37 °C for 1 h with frequent suspension. After incubation, each sample was centrifuged and the supernatant was collected. The cell pellet was washed with 500 μL PBS, centrifuged, and the supernatant was combined with the previously collected supernatant samples. The cell pellet was resuspended in 600 μL PBS and transferred to gamma counter tubes. All pellet and supernatant samples were measured in a Wizard 1470 gamma counter (Perkin Elmer, Waltham, MA, USA). Cell binding percentage for each cell line was calculated as the radioactivity associated, with pellet divided by the total radioactivity associated with both the pellet and supernatant.
4.9. Serum Stability Assay
Peptide stability was evaluated in mouse serum as previously described [
19,
20]. In brief, formulated
18F-radiolabeled peptide (~3.7 MBq in PBS) was incubated with mouse serum (500 μL, Sigma Aldrich, St. Louis, MO) at 37 °C for 1 h. An aliquot of
18F-radiolabeled peptide in mouse serum (100 μL) was taken at 1 h, mixed with absolute ethanol (500 μL, 4 °C), and centrifuged to precipitate serum protein. The supernatant was diluted with HPLC solvent A before radio-HPLC analysis to determine the fraction of intact
18F-radiolabeled peptide.
4.10. Circular Dichroism (CD) Spectroscopy
CD spectroscopy was performed on a Jasco J-810 spectropolarimeter equipped with a Jasco CDF-426S Peltier set to 25 °C (Easton, MD, USA). Lyophilized peptide was diluted to 0.2 mg/mL in buffer (final concentration was 25 mM phosphate + 100 mM NaF in PBS, pH 7.4), with or without 30% TFE (v/v), placed in a quartz cuvette (1 mm), and after extensive purging with nitrogen, scanned in the region 190–260 nm (scan speed was 20 nm/min). An average of five scans were baseline-subtracted (buffer, 25 mM phosphate + 100 mM NaF in PBS).
4.11. Data and Statistical Analysis
Statistical analysis and comparisons between groups were performed using the student t test. A p value of 0.05 or less was considered statistically significant.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}