Discovery of Novel Integrase Inhibitors Acting outside the Active Site Through High-Throughput Screening


Abstract
:
1. Introduction
2. Results
2.1. Screening for IN Strand Transfer Inhibitors
2.2. Biochemical Characterization of NSC34931 and Derivatives
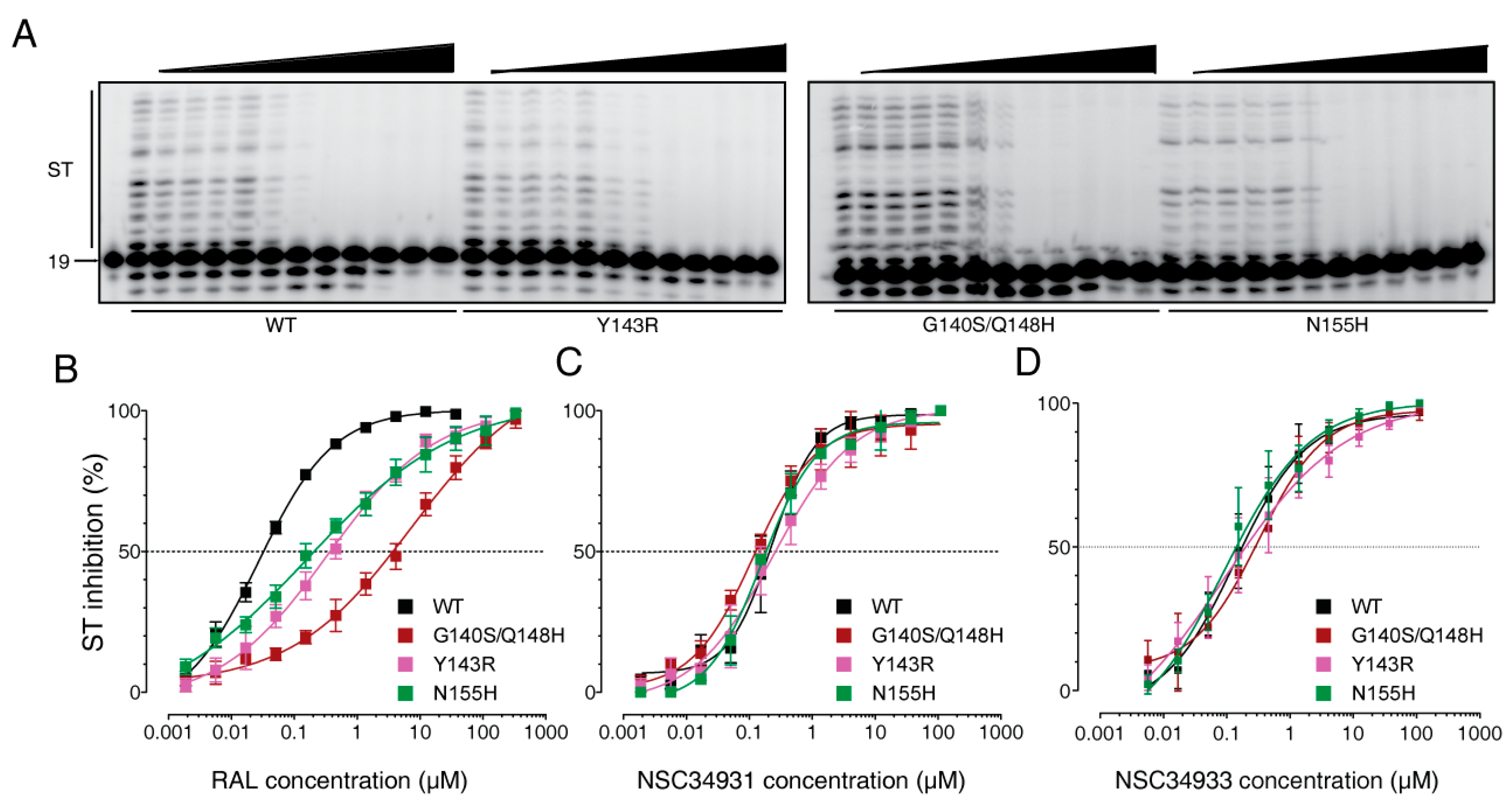
2.3. Stilbene Disulfonic Acid Derivatives Overcome Resistance to IN Strand Transfer Inhibitors
2.4. Molecular Mechanism of Action of Stilbene Disulfonic Acid Derivatives
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Oligonucleotides
4.3. Integrase Enzymes
4.4. Electrochemiluminescent Integrase Strand Transfer Assay
4.5. Integrase Reactions
4.6. Shiff Base Cross-Linking Assay
4.7. DNA-Binding Experiments
4.8. Antiviral Assays
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Engelman, A.N.; Singh, P.K. Cellular and molecular mechanisms of HIV-1 integration targeting. Cell. Mol. Life Sci. 2018, 75, 2491–2507. [Google Scholar] [CrossRef]
- Métifiot, M.; Marchand, C.; Pommier, Y. HIV integrase inhibitors: 20-year landmark and challenges. Adv. Pharmacol. 2013, 67, 75–105. [Google Scholar] [PubMed]
- Brooks, K.M.; Sherman, E.M.; Egelund, E.F.; Brotherton, A.; Durham, S.; Badowski, M.E.; Cluck, D.B. Integrase Inhibitors: After 10 Years of Experience, Is the Best Yet to Come? Pharmacotherapy 2019, 39, 576–598. [Google Scholar] [CrossRef] [PubMed]
- Marchand, C.; Johnson, A.A.; Karki, R.G.; Pais, G.C.G.; Zhang, X.; Cowansage, K.; Patel, T.A.; Nicklaus, M.C.; Burke, T.R.; Pommier, Y. Metal-dependent inhibition of HIV-1 integrase by beta-diketo acids and resistance of the soluble double-mutant (F185K/C280S). Mol. Pharmacol. 2003, 64, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Svarovskaia, E.S.; Barr, R.; Zhang, X.; Pais, G.C.G.; Marchand, C.; Pommier, Y.; Burke, T.R.; Pathak, V.K. Azido-containing diketo acid derivatives inhibit human immunodeficiency virus type 1 integrase in vivo and influence the frequency of deletions at two-long-terminal-repeat-circle junctions. J. Virol. 2004, 78, 3210–3222. [Google Scholar] [CrossRef] [PubMed]
- Pannecouque, C.; Pluymers, W.; Van Maele, B.; Tetz, V.; Cherepanov, P.; De Clercq, E.; Witvrouw, M.; Debyser, Z. New Class of HIV Integrase Inhibitors that Block Viral Replication in Cell Culture. Curr. Biol. 2002, 12, 1169–1177. [Google Scholar] [CrossRef] [Green Version]
- Weislow, O.S.; Kiser, R.; Fine, D.L.; Bader, J.; Shoemaker, R.H.; Boyd, M.R. New soluble-formazan assay for HIV-1 cytopathic effects: Application to high-flux screening of synthetic and natural products for AIDS-antiviral activity. J. Natl. Cancer Inst. 1989, 81, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Métifiot, M.; Maddali, K.; Naumova, A.; Zhang, X.; Marchand, C.; Pommier, Y. Biochemical and pharmacological analyses of HIV-1 integrase flexible loop mutants resistant to raltegravir. Biochemistry 2010, 49, 3715–3722. [Google Scholar] [CrossRef]
- Métifiot, M.; Vandegraaff, N.; Maddali, K.; Naumova, A.; Zhang, X.; Rhodes, D.; Marchand, C.; Pommier, Y. Elvitegravir overcomes resistance to raltegravir induced by integrase mutation Y143. AIDS 2011, 25, 1175–1178. [Google Scholar] [CrossRef]
- Pommier, Y.; Marchand, C. Interfacial inhibitors: Targeting macromolecular complexes. Nat. Rev. Drug Discov. 2011, 11, 25–36. [Google Scholar] [CrossRef]
- Chow, S.A.; Vincent, K.A.; Ellison, V.; Brown, P.O. Reversal of integration and DNA splicing mediated by integrase of human immunodeficiency virus. Science 1992, 255, 723–726. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.A.; Marchand, C.; Patil, S.S.; Costi, R.; Di Santo, R.; Burke, T.R.; Pommier, Y. Probing HIV-1 integrase inhibitor binding sites with position-specific integrase-DNA cross-linking assays. Mol. Pharmacol. 2007, 71, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Marchand, C.; Neamati, N.; Pommier, Y. In vitro human immunodeficiency virus type 1 integrase assays. Meth. Enzymol. 2001, 340, 624–633. [Google Scholar] [PubMed]
- Mazumder, A.; Neamati, N.; Pilon, A.A.; Sunder, S.; Pommier, Y. Chemical trapping of ternary complexes of human immunodeficiency virus type 1 integrase, divalent metal, and DNA substrates containing an abasic site. Implications for the role of lysine 136 in DNA binding. J. Biol. Chem. 1996, 271, 27330–27338. [Google Scholar] [CrossRef]
- National Toxicology Program. Toxicology and Carcinogenesis Studies of 4,4′-Diamino-2,2′-Stilbenedisulfonic Acid Disodium Salt (CAS No. 7336-20-1) in F344 Rats and B6C3F1 Mice (Feed Studies). Natl. Toxicol. Program Tech. Rep. Ser. 1992, 412, 1–244. [Google Scholar]
- Brasch, J.; Kreiselmaier, I.; Christophers, E. Inhibition of dermatophytes by optical brighteners. Mycoses 2003, 46, 120–125. [Google Scholar] [CrossRef]
- Liantonio, A.; Pusch, M.; Picollo, A.; Guida, P.; De Luca, A.; Pierno, S.; Fracchiolla, G.; Loiodice, F.; Tortorella, P.; Conte Camerino, D. Investigations of pharmacologic properties of the renal CLC-K1 chloride channel co-expressed with barttin by the use of 2-(p-Chlorophenoxy)propionic acid derivatives and other structurally unrelated chloride channels blockers. J. Am. Soc. Nephrol. 2004, 15, 13–20. [Google Scholar] [CrossRef]
- Himi, T.; Ishizaki, Y.; Murota, S.-I. 4,4′-diisothiocyano-2,2′-stilbenedisulfonate protects cultured cerebellar granule neurons from death. Life Sci. 2002, 70, 1235–1249. [Google Scholar] [CrossRef]
- Shami, Y.; Carver, J.; Ship, S.; Rothstein, A. Inhibition of C1- binding to anion transport protein of the red blood cell by DIDS (4, 4′-diisothiocyano-2, 2′-stilbene disulfonic acid) measured by [35C1]NMR. Biochem. Biophys. Res. Commun. 1976, 76, 429–436. [Google Scholar] [CrossRef]
- Tomaskova, Z.; Gaburjakova, J.; Brezova, A.; Gaburjakova, M. Inhibition of anion channels derived from mitochondrial membranes of the rat heart by stilbene disulfonate--DIDS. J. Bioenerg. Biomembr. 2007, 39, 301–311. [Google Scholar] [CrossRef]
- Métifiot, M.; Johnson, B.C.; Kiselev, E.; Marler, L.; Zhao, X.Z.; Burke, T.R.; Marchand, C.; Hughes, S.H.; Pommier, Y. Selectivity for strand-transfer over 3′-processing and susceptibility to clinical resistance of HIV-1 integrase inhibitors are driven by key enzyme-DNA interactions in the active site. Nucleic Acids Res. 2016, 44, 6896–6906. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.N. Multifaceted HIV integrase functionalities and therapeutic strategies for their inhibition. J. Biol. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Dharmarajan, V.; Serrao, E.; Hoyte, A.; Larue, R.C.; Slaughter, A.; Sharma, A.; Plumb, M.R.; Kessl, J.J.; Fuchs, J.R.; et al. The Competitive Interplay between Allosteric HIV-1 Integrase Inhibitor BI/D and LEDGF/p75 during the Early Stage of HIV-1 Replication Adversely Affects Inhibitor Potency. ACS Chem. Biol. 2016, 11, 1313–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenwick, C.; Amad, M.; Bailey, M.D.; Bethell, R.; Bös, M.; Bonneau, P.; Cordingley, M.; Coulombe, R.; Duan, J.; Edwards, P.; et al. Preclinical profile of BI 224436, a novel HIV-1 non-catalytic-site integrase inhibitor. Antimicrob. Agents Chemother. 2014, 58, 3233–3244. [Google Scholar] [CrossRef]
- Cardin, A.D.; Smith, P.L.; Hyde, L.; Blankenship, D.T.; Bowlin, T.L.; Schroeder, K.; Stauderman, K.A.; Taylor, D.L.; Tyms, A.S. Stilbene disulfonic acids. CD4 antagonists that block human immunodeficiency virus type-1 growth at multiple stages of the virus life cycle. J. Biol. Chem. 1991, 266, 13355–13363. [Google Scholar] [CrossRef]
- Witvrouw, M.; Fikkert, V.; Pluymers, W.; Matthews, B.; Mardel, K.; Schols, D.; Raff, J.; Debyser, Z.; De Clercq, E.; Holan, G.; et al. Polyanionic (i.e., polysulfonate) dendrimers can inhibit the replication of human immunodeficiency virus by interfering with both virus adsorption and later steps (reverse transcriptase/integrase) in the virus replicative cycle. Mol. Pharmacol. 2000, 58, 1100–1108. [Google Scholar] [CrossRef]
- Zhao, X.Z.; Métifiot, M.; Kiselev, E.; Kessl, J.J.; Maddali, K.; Marchand, C.; Kvaratskhelia, M.; Pommier, Y.; Burke, T.R. HIV-1 Integrase-Targeted Short Peptides Derived from a Viral Protein R Sequence. Molecules 2018, 23, 1858. [Google Scholar] [CrossRef]
- Van Loock, M.; Meersseman, G.; Van Acker, K.; Van Den Eynde, C.; Jochmans, D.; Van Schoubroeck, B.; Dams, G.; Heyndrickx, L.; Clayton, R.F. A novel high-throughput cellular screening assay for the discovery of HIV-1 integrase inhibitors. J. Virol. Methods 2012, 179, 396–401. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available through the NIH AIDS reagent program and/or to the DTP. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| In Vitro (µM, IC50 +/−SD) | Ex Vivo (µM) | |||||
|---|---|---|---|---|---|---|
| NSC # | R | 3’-P | ST | Antiviral Activity | Cytotoxicity | Selectivity Index |
| EC50 | CC50 | SI | ||||
| 34931 |  | 0.32 ± 0.12 | 0.18 ± 0.07 | 3.07 | 59.0 | 19.2 |
| 34933 |  | 1.1 ± 0.3 | 0.5 ± 0.2 | 0.60 | >100 | >166 |
| 47745 |  | 3.2 ± 1.5 | 3.1 ± 1.7 | 15.3 | >100 | >6.5 |
| 163175 |  | >111 | >111 | ND | ND | ND |
| 163 |  | >111 | >111 | ND | ND | ND |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aknin, C.; Smith, E.A.; Marchand, C.; Andreola, M.-L.; Pommier, Y.; Metifiot, M. Discovery of Novel Integrase Inhibitors Acting outside the Active Site Through High-Throughput Screening. Molecules 2019, 24, 3675. https://doi.org/10.3390/molecules24203675
Aknin C, Smith EA, Marchand C, Andreola M-L, Pommier Y, Metifiot M. Discovery of Novel Integrase Inhibitors Acting outside the Active Site Through High-Throughput Screening. Molecules. 2019; 24(20):3675. https://doi.org/10.3390/molecules24203675
Chicago/Turabian StyleAknin, Cindy, Elena A. Smith, Christophe Marchand, Marie-Line Andreola, Yves Pommier, and Mathieu Metifiot. 2019. "Discovery of Novel Integrase Inhibitors Acting outside the Active Site Through High-Throughput Screening" Molecules 24, no. 20: 3675. https://doi.org/10.3390/molecules24203675