4.2. Chemical Syntheses
General Procedure A: Base mediated ester hydrolysis. Esters were hydrolysed according to the method of Theodorou and co-workers [
25]. Esters were dissolved in a mixture of methanol:dichloromethane (1:9) and treated with a 2 M methanolic solution of sodium hydroxide (1–5 equiv) to give a final 0.1 M concentration of ester. The solution was stirred for 3–20 h at which time a cloudy suspension had formed. The suspension was concentrated under reduced pressure and the residue was dissolved in water. The solution was acidified with concentrated HCl to pH 1 then the carboxylic acids were extracted with dichloromethane or ethyl acetate (×3). The combined organic layers were dried over magnesium sulfate then concentrated under reduced pressure. Unless otherwise stated, no purification was required.
General Procedure B: Preparation of amines via Buchwald–Hartwig coupling. t-Butyl alcohol (25 mL/mmol) was degassed by bubbling nitrogen for 20 min. The aryl bromide, amine (1.2 equiv) and potassium carbonate (2.3 equiv) were added and the reaction vessel was evacuated and refilled with nitrogen. This was repeated twice more before addition of XPhos (10 mol%) and Pd2(dba)3 (5 mol%). The reaction vessel was again purged thrice more before stirring at reflux under nitrogen for 16 h. The reaction mixture was partitioned between water and dichloromethane. The aqueous phase was extracted with dichloromethane (×3) then the combined organic phases were washed with brine (×3). The dichloromethane was removed under reduced pressure and the crude amine was purified by flash chromatography.
General Procedure C: Preparation of ethers/esters via Williamson ether synthesis. Compound 9, anhydrous potassium carbonate (3.3 equiv) and alkyl bromide (4.6 equiv) in dry acetone or acetonitrile were refluxed under an atmosphere of nitrogen for 16 h. The reaction was cooled to room temperature and concentrated to a residue which was partitioned between ethyl acetate and brine. The aqueous phase was washed twice with ethyl acetate and the combined organic phases were dried over magnesium sulfate. The solution was concentrated under reduced pressure to afford the product that was purified, if necessary, by flash chromatography.
General Procedure D: Preparation of ethers via Ullmann-type coupling. Compound 9 or 10, phenol (1.2 equiv), tri-potassium phosphate (3 equiv), copper(I) iodide (10 mol%), and picolinic acid (20 mol%) were added to an oven-dried Schlenk tube charged with a stir bar. Dry dimethyl sulfoxide was added via syringe under a positive pressure of nitrogen. The tube was purged with nitrogen (×3), then sealed and heated to 90 °C causing a colour change from green to brown. The reaction was stirred until analytical RP-HPLC indicated no starting material (24 h), and then, was partitioned between ethyl acetate and water. The aqueous phase was acidified with concentrated hydrochloric acid and was extracted with ethyl acetate (×3). The combined organic phases were dried over magnesium sulfate then concentrated under reduced pressure to give the crude compound. Purification was carried out by flash chromatography, semi-preparative RP-HPLC or recrystallization to afford the pure products.
General Procedure F: Preparation of amides via coupling of amines to acid/sulfonyl chlorides. A solution of amine 39 and triethylamine (3 equiv) in dichloromethane was treated with acid chloride (1.1 equiv) or sulfonyl chloride (1.1 equiv). The solution was refluxed under nitrogen until complete consumption of starting material by TLC (5–16 h). The reaction was quenched with sodium hydrogen carbonate then extracted with dichloromethane (×3). The organic phase was washed with brine (×3), and then concentrated by rotary evaporation to yield the amide. Purification by flash chromatography was undertaken if required.
General Procedure G: Preparation of amides via in-situ formation of acid/sulfonyl chloride and subsequent coupling to amine. A solution of carboxylic acid (1–1.2 equiv) or sulfonic acid (1–1.2 equiv) in dry dichloromethane (30 mL/mmol) was treated with phosphorous pentachloride (1.0–1.2 equiv) and refluxed for 30 min under nitrogen. After cooling to room temperature, a solution of amine 39 and triethylamine (3 equiv) in dry dichloromethane (6 mL/mmol) was added. The resulting solution was refluxed until complete consumption of the amine as determined by TLC (2–5 h). The reaction was quenched with sodium hydrogen carbonate then extracted with dichloromethane (×3). The organic phase was washed with brine (×3) then concentrated by rotary evaporation to yield the amide, which was purified by flash chromatography if required.
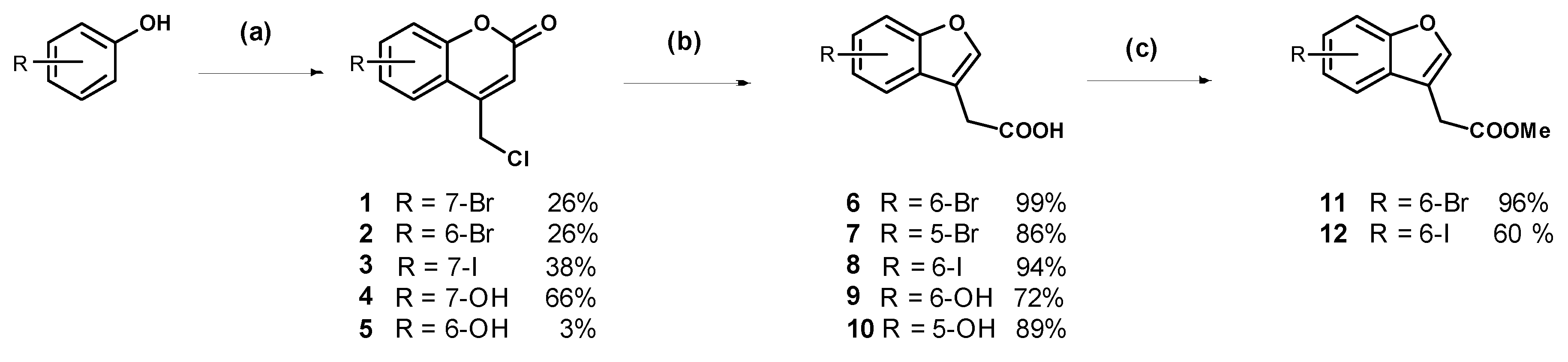
7-Bromo-4-chloromethylcoumarin (1): 3-Bromophenol (5.00 g, 28.9 mmol) was added to chilled 70% sulfuric acid (30 mL) and the stirring solution was cooled in an ice bath to less than 5 °C. Ethyl 4-chloroacetoacetate (3.90 mL) was added dropwise ensuring the internal temperature remained below 5 °C. The solution was stirred for two hours and was then warmed to room temperature and stirred for 22 h. The resulting suspension was poured onto ice water (500 mL) and the off-white precipitate was collected by vacuum filtration and recrystallised from ethanol:ethyl acetate (1:1) to afford the compound in question as white needles (2.04 g, 26%); mp 242–244 °C. δH (500 MHz, CDCl3) 7.56 (1H, d, J = 2.0 Hz, ArH), 7.54 (1H, d, J = 8.5 Hz, ArH), 7.47 (1H, dd, J = 7.0 Hz, 1.5, ArH), 6.58 (1H, s, CH), 4.64 (2H, s, CH2). δC (125 MHz, CDCl3) 159.4 (Cq), 154.1 (Cq), 149.0 (Cq), 127.9, 126.3 (Cq), 125.3, 120.7, 116.3, 116.1, 41.0 (CH2). LRMS (ESI): m/z 272.97 [M (79Br) + H]+, 274.96 [M (81Br) + H]+. HRMS (ESI): 294.9137 [M (79Br) + Na]+, 296.9117 [M (81Br) + H]+.
6-Bromo-4-chloromethylcoumarin (2): 4-Bromophenol (4.00 g, 23.1 mmol) was dissolved in methanesulfonic acid (21 mL) and cooled in an ice bath to 2 °C. Ethyl 4-chloroacetoacetate (3.12 mL) was added dropwise ensuring the temperature of the solution did not exceed 5 °C. The dark red solution stirred at 0 °C for 2 h then at room temperature for one week. The reaction mixture was poured onto ice then extracted with dichloromethane. The organic phase was washed with 1 M sodium hydroxide (×2) to remove the unreacted phenol; then the organic phase was dried (MgSO4) and concentrated to a light yellow semi-solid. Trituration with diethyl ether afforded the pure product as a light orange solid (1.66 g, 26%); mp 164–166 °C. δH (400 MHz, CDCl3) 7.79 (1H, s, ArH), 7.66 (1H, d, J = 8.8 Hz, ArH), 7.27 (1H, d, J = 8.8 Hz, ArH), 6.61 (1H, s, CH), 4.64 (2H, s, CH2). δC (100 MHz, CDCl3) 159.4 (Cq), 152.8 (Cq), 148.4 (Cq), 135.1, 126.8, 119.2, 118.9 (Cq), 117.3 (Cq), 116.9 (CH), 40.9 (CH2). LRMS (ESI): m/z 276.9 [M (79Br) + H]+, 278.9 [M (81Br) + H]+. HRMS (ESI): 296.9128 [M (79Br) + Na]+, 296.9099 [M (81Br) + Na]+.
4-(Chloromethyl)-7-iodo-2H-chromen-2-one (3): 3-Iodophenol (1.00 g, 4.55 mmol) was dissolved in methanesulfonic acid (5 mL) and cooled in an ice bath to 15 °C. Ethyl 4-chloroacetoacetate (619 µL, 4.55 mmol) was added drop-wise at a slow enough rate to keep the reaction mixture below 10 °C. The reaction stirred for one hour before being removed from the ice bath. After 40 h of stirring at room temperature the purple suspension was poured onto ice and the resulting precipitate was collected by vacuum filtration. The crude off-white solid was recrystallised from ethanol:ethyl acetate (1:1) to afford the compound in question as a white solid (554 mg, 38%); mp 215–217 °C. δH (400 MHz, CDCl3) 7.76 (1H, s, ArH), 7.67 (1H, d, J = 8.4 Hz, ArH), 7.37 (1H, d, J = 8.0 Hz, ArH), 6.59 (1H, s, CH), 4.63 (2H, s, CH2). δC (100 MHz, CDCl3) 159.3, 153.7, 149.1, 133.7, 126.6, 125.1, 116.8, 116.4, 97.9, 41.0. LRMS (ESI): m/z 320.90 [M + H]+.
7-Hydroxy-4-chloromethylcoumarin (
4): To a chilled solution of resorcinol (5.00 g, 45.4 mmol) in 70% H
2SO
4 (50 mL) was added, dropwise, ethyl 4-chloroacetoacetate (6.15 mL), while ensuring the temperature did not exceed 5 °C. The chilled solution stirred for two hours, then 20 h at room temperature. The reaction mixture was poured onto ice and the precipitate was collected by vacuum filtration. The light pink semi-solid was triturated with diethyl ether then recrystallised from ethanol to give the compound in question as a white solid (5.18 g, 57%); mp. 178–180 °C (lit. 185 °C) [
26]. The mother liquor was concentrated and purified by flash chromatography (20–40% ethyl acetate in hexanes) to afford the compound in question as a white solid (888 mg, 9%). δ
H (500 MHz, DMSO-
d6) 10.63 (1H, bs, s, OH), 7.67 (1H, d,
J = 8.5 Hz, ArH), 6.83 (1H, d,
J = 8.5 Hz, ArH), 6.74 (1H, s, ArH), 6.41 (1H, s, ArH), 4.94 (1H, s, CH
2). δ
C (100 MHz, DMSO-
d6) 161.9, 160.6, 155.8, 151.5, 127.0, 113.6, 111.5, 109.8, 103.0, 41.8.
6-Hydroxy-4-chloromethylcoumarin (
5): Hydroquinone (1.60 g, 14.7 mmol) was dissolved in 73% sulfuric acid (10 mL) by heating. The solution was cooled to 4 °C in an ice bath and ethyl 4-chloroacetoacetate (1.99 mL) was added dropwise ensuring the internal temperature remained below 5 °C. The dark red solution stirred in an ice bath for one hour then at room temperature for three days. The reaction was poured onto ice and the resulting light brown solid was collected by vacuum filtration. Recrystallisation from 1,4-dioxane afforded the compound in question as a light brown solid (100 mg, 3%); mp 220–222 °C (lit. 220–222 °C) [
27]. δ
H (500 MHz, DMSO-
d6) 9.83 (1H, br s, OH), 7.28 (1H, d,
J = 9.0 Hz, ArH), 7.13 (1H, s, ArH), 7.07 (1H, d,
J = 8.8 Hz, ArH), 6.64 (1H, s, CH), 5.00 (2H, s, CH
2). δ
C (100 MHz, DMSO-
d6) 160.3 (C
q), 154.2 (C
q), 150.8 (C
q), 147.1 (C
q), 120.7, 118.2, 118.1 (C
q), 116.1, 110.1 (CH), 41.8 (CH
2).
2-(6-Bromobenzofufran-3-yl)acetic acid (
6): Coumarin
1 (1.77 g, 6.48 mmol) was suspended in 2 M NaOH (37 mL) and stirred at 80 °C for 18 h. The resulting suspension was cooled to room temperature and acidified with concentrated hydrochloric acid to pH 1. The white precipitate was collected by vacuum filtration and dried under reduced pressure (1.63 g, 99%); mp 154–156 °C (lit. 156–157 °C) [
28]. δ
H (500 MHz, DMSO-
d6) 7.90 (1H, s, ArH), 7.86 (1H, s, ArH), 7.56 (1H, d,
J = 8.0 Hz, ArH), 7.42 (1H, d,
J = 8.25 Hz, ArH), 3.68 (2H, s, CH
2). δ
C (100 MHz, DMSO-
d6) 172.2 (C
q), 155.4 (C
q), 144.8, 127.7 (C
q), 126.2, 122.3, 117.3 (C
q), 114.9, 114.7 (C
q), 29.3 (CH
2).
2-(5-Bromobenzofuran-3-yl)acetic acid (
7): Coumarin
2 (252 mg, 0.920 mmol) was dissolved in 2 M NaOH (5.5 mL) and stirred at 80 °C for 16 h. TLC indicated complete consumption of the starting material. The solution was cooled to room temperature and acidified with concentrated hydrochloric acid. The product precipitated and was collected by vacuum filtration as a light grey solid (203 mg, 86%); mp 142–143 °C (lit. 142–143 °C) [
28]. δ
H (400 MHz, DMSO-
d6) 7.93 (1H, s, ArH), 7.81 (1H, s, ArH), 7.55 (1H, d,
J = 8.8 Hz, ArH), 7.44 (1H, d,
J = 8.6 Hz, ArH), 3.70 (2H, s, CH
2). δ
C (100 MHz, CDCl
3) 176.2 (C
q), 154.0 (C
q), 144.3, 129.4 (C
q), 127.6, 122.4, 116.0 (C
q), 113.1, 112.0 (C
q), 29.2 (CH
2). LRMS (ESI):
m/
z 253.5 [M (
79Br) + H]
+, 254.9 [M (
81Br) + H]
+. HRMS (ESI): 252.9521 [M (
79Br) + H]
+, 254.9509 [M (
81Br) + H]
+.
2-(6-Iodobenzofuran-3-yl)acetic acid (8): Coumarin 3 (480 mg, 1.50 mmol) was suspended in 2 M NaOH (10 mL) and stirred at 80 °C for 16 h. The resulting solution was cooled to room temperature and acidified with concentrated hydrochloric acid to pH 1. The white precipitate was collected by vacuum filtration and dried under reduced pressure (423 mg, 94%); mp 164–166 °C. δH (400 MHz, CDCl3) 7.86 (1H, s, ArH), 7.57–7.56 (2H, m, 2H), 7.31 (1H, d, J = 8.0 Hz, ArH), 3.73 (2H, s, CH2). δC (100 MHz, DMSO-d6) 172.2, 155.6, 144.3, 131,8, 128.0, 122.6, 120.5, 114.6, 89.0, 29.3. LRMS (ESI): m/z 300.80 [M − H]−. HRMS (ESI): 300.9397 [M − H]−.
2-(6-Hydroxybenzofuran-3-yl)acetic acid (
9): A suspension of coumarin
4 (200 mg, 0.950 mmol) in 2 M sodium hydroxide (10 mL) was stirred at 80 °C for 16 h. The resulting brown solution was cooled to room temperature and acidified with concentrated hydrochloric acid to pH 1. The compound in question precipitated as a brown solid which was collected by vacuum filtration (183 mg, 66%); mp 141–143 °C (lit. 142–143 °C) [
28]. δ
H (500 MHz, DMSO-
d6) 9.49 (1H, br s, OH), 7.65 (1H, s, ArH), 7.33 (1H, d,
J = 8.4 Hz, ArH), 6.87 (1H, s, ArH), 6.73 (1H, d,
J = 8.4 Hz, ArH), 3.59 (2H, s, CH2). δ
C (125 MHz, DMSO-
d6) 172.5 (C
q), 156.2 (C
q), 156.1 (C
q), 142.0, 120.6, 120.3 (C
q), 114.3 (C
q), 112.4, 98.1, 29.6 (CH
2). LRMS (ESI):
m/
z 191.0 [M − H]
−.
2-(5-Hydroxybenzofufran-3-yl)acetic acid (10): Coumarin 5 (93.3 mg, 0.4458 mmol) was suspended in 2 M sodium hydroxide (3 mL) and the resulting brown solution was stirred at 80 °C for 48 h. The solution was cooled to room temperature and acidified with concentrated hydrochloric acid to pH 1, then extracted with ethyl acetate (×3). The combined organic phases were dried over magnesium sulfate, then concentrated under reduced pressure to a light brown oil which crystallised to an off-white solid (76.5 mg, 89%); mp 128–130 °C. δH (400 MHz, DMSO-d6) 9.14 (1H, br s, OH), 7.75 (1H, s, ArH), 7.31 (1H, d, J = 8.8 Hz, ArH), 6.86 (1H, s, ArH), 6.72 (1H, d, J = 8.8 Hz, ArH), 3.58 (2H, s, CH2). LRMS (ESI): m/z 191. 0 [M − H]−.
Methyl 2-(6-bromobenzofuran-3-yl)acetate (11): Thionyl chloride (1.31 mL, 18.1 mmol) was added dropwise to chilled methanol (6 mL) and the solution was stirred in an ice bath. After one hour, 6 (1.54 g, 4.02 mmol) was added and the suspension was refluxed for 16 h. Upon cooling to room temperature, white crystals precipitated. The methanol was removed under reduced pressure and the resulting crude solid was purified by flash chromatography (30–70% ethyl acetate in hexanes) to afford the product as an oil, which crystallised to an off-white solid under reduced pressure (1.57 g, 97%); mp 70–71 °C. δH (400 MHz, CDCl3) 7.66 (1H, s, ArH), 7.61 (1H, s, ArH), 7.43 (1H, d, J = 8.0 Hz, ArH), 7.38 (1H, d, J = 8.0 Hz, ArH), 3.73 (3H, s, CH3), 3.69 (2H, s, CH2). δC (100 MHz, CDCl3) 170.8, 155.5, 143.4, 126.7, 126.1, 120.7, 117.9, 115.0, 113.1, 52.2, 29.4. HRMS (ESI): 304.9408, 306.9388 [M + K]+.
Methyl 2-(6-iodobenzofuran-3-yl)acetate (12): Thionyl chloride (932 µL) was added dropwise to chilled methanol (10 mL) and the solution was stirred for 50 min under nitrogen. The acid 8 (1.29 g, 4.28 mmol) was added and the suspension was heated to reflux for four hours. Upon cooling to room temperature, a precipitate formed. The suspension was chilled in an ice bath and water was added, causing more solid to precipitate. The precipitate was collected by vacuum filtration to give the compound in question as an off-white solid (945 mg, 70%); mp 80–82 °C. δH (400 MHz, CDCl3) 7.85 (1H, s, ArH), 7.57–7.54 (2H, m, ArH), 7.31 (1H, d, J = 8.2 Hz, ArH), 3.73 (3H, s, OCH3), 3.68 (2H, s, CH2). δC (100 MHz, CDCl3) 170.8 (Cq), 155.7 (Cq), 143.2, 131.7, 127.3 (Cq), 121.1, 120.8, 113.1 (Cq), 88.1 (Cq), 52.2 (OCH3), 29.4 (CH2). LRMS (ESI): m/z 339.0 [M + Na]+. HRMS (ESI): 296.9860 [M − H2O − H]−.
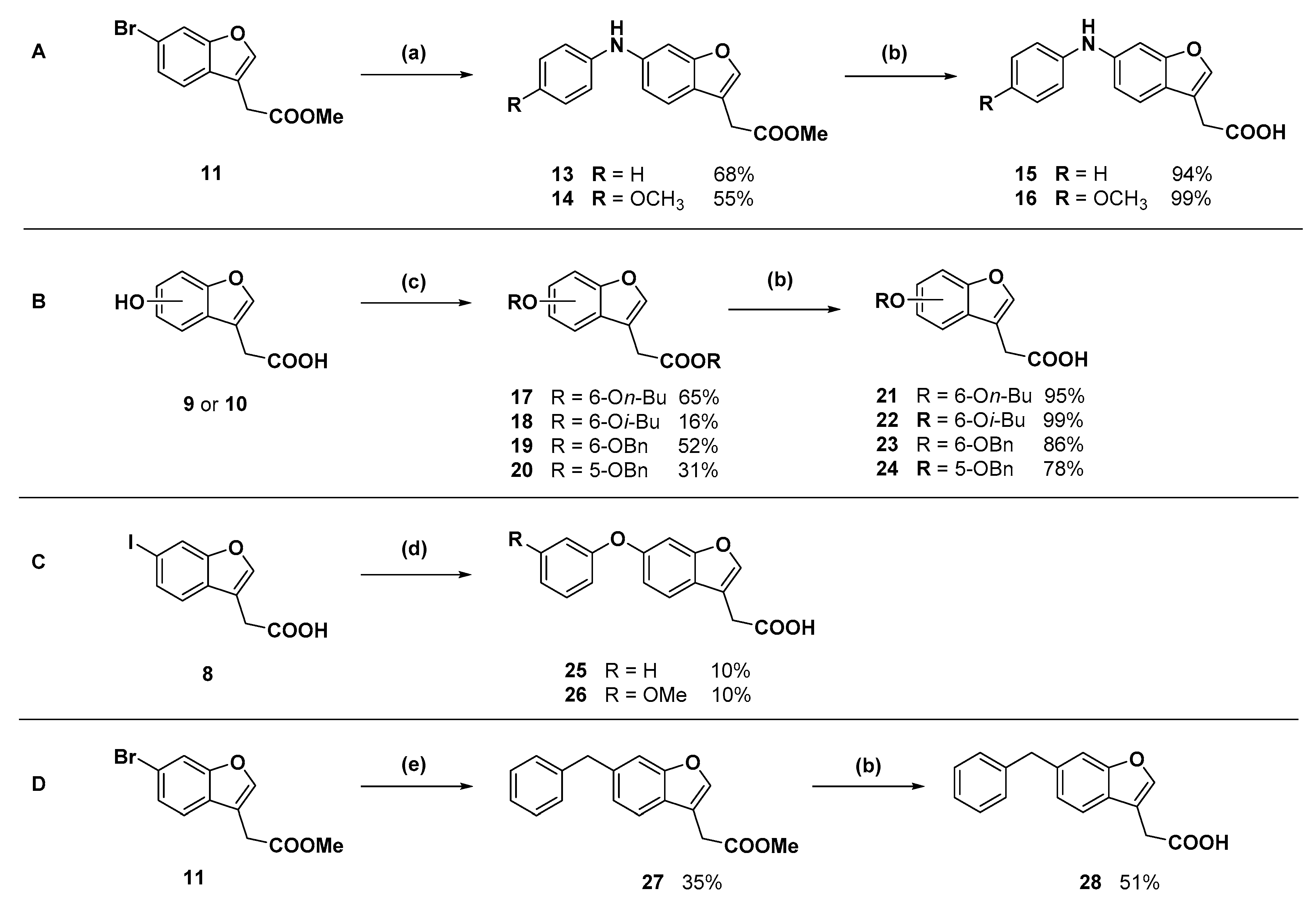
Methyl 2-(6-(phenylamino)benzofuran-3-yl)acetate (13): Compound 11 (250 mg, 0.929 mmol) was coupled to aniline (102.0 µL) according to General Procedure B. The crude orange oil was purified by flash chromatography (5–10% ethyl acetate in hexanes) to afford the compound in question as an orange oil (178 mg, 68%). δH (400 MHz, CDCl3) 7.52 (1H, s, ArH), 7.42 (1H, d, J = 8.4 Hz, ArH), 7.30–7.24 (3H, m, ArH), 7.08 (2H, d, J = 8.5 Hz, ArH), 6.99 (1H, d, J = 8.4 Hz, ArH), 6.93 (1H, t, J = 7.4 Hz, ArH), 3.74 (3H, s, OCH3), 3.68 (2H, s, CH2). δC (125 MHz, CDCl3) 171.2 (Cq), 156.3 (Cq), 143.5 (Cq), 141.9, 140.9 (Cq), 129.4, 121.8 (Cq), 120.9, 120.0, 117.5, 115.3, 113.0 (Cq), 100.9, 52.1 (CH3), 29.6 (CH2). LRMS (ESI): m/z 282.0 [M + H]+.
Methyl 2-(6-((4-methoxyphenyl)amino)benzofuran-3-yl)acetate (14): Compound 11 (100 mg, 0.372 mmol) was coupled to p-anisidine (54.9 mg) according to General Procedure B. The crude dark orange oil was purified by flash chromatography (dichloromethane) to afford the compound in question as a yellow oil (57.5 mg, 50%). δH (400 MHz, CDCl3) 7.47 (1H, s, ArH), 7.37 (1H, d, J = 8.4 Hz, ArH), 7.09 (2H, d, J = 8.9 Hz, ArH), 7.04 (1H, s, ArH), 6.89–6.83 (3H, m, ArH), 5.61 (1H, br s, NH), 3.81 (3H, s, OCH3), 3.73 (3H, s, OCH3), 3.66 (2H, s, CH2). δC (100 MHz, CDCl3) 171.2 (Cq), 156.6 (Cq), 155.2 (Cq), 143.2 (Cq), 141.3, 136.0 (Cq), 121.9, 120.5 (Cq), 119.9, 114.7, 113.3, 112.9 (Cq), 98.2, 55.5 (OCH3), 52.1 (OCH3), 29.6 (CH2). LRMS (ESI): m/z 312.1 [M + H]+.
2-(6-(Phenylamino)benzofuran-3-yl)acetic acid (15): Ester 13 (178 mg, 0.634 mmol) was hydrolysed using 2 M sodium hydroxide according to General Procedure A to afford the compound in question as a brown solid (160 mg, 94%); mp 127–130 °C. δH (400 MHz, CDCl3) 7.53 (1H, s, ArH), 7.43 (1H, d, J = 8.4 Hz, ArH), 7.30–7.25 (3H, m, ArH), 7.09 (2H, d, J = 8.4 Hz, ArH), 6.99 (1H, d, J = 8.4 Hz, ArH), 6.95 (1H, t, J = 7.4 Hz, ArH), 3.72 (2H, s, CH2). δC (125 MHz, CDCl3) 176.5 (Cq), 156.3 (Cq), 143.4 (Cq), 142.1, 141.0 (Cq), 129.4, 121.6 (Cq), 121.0, 120.0, 117.6, 115.3, 112.4 (Cq), 100.8, 29.5 (CH2). LRMS (ESI): m/z 265.96 [M − H]−. HRMS (ESI): 306.1621 [M + K]+.
2-(6-((4-Methoxyphenyl)amino)benzofuran-3-yl)acetic acid (16): Ester 14 (50.0 mg, 0.161 mmol) was hydrolysed using 2 M methanolic sodium hydroxide (5 equiv, 402 µL) according to General Procedure A to afford the compound in question as a light brown solid (47.9 mg, 100%); mp 120–122 °C. δH (400 MHz, CDCl3) 7.49 (1H, s, ArH), 7.38 (1H, d, J = 8.4 Hz, ArH), 7.13 (3H, m, ArH), 6.93 (1H, d, J = 8.4 Hz, ArH), 6.89–6.83 (3H, m, ArH), 3.81 (3H, s, OCH3), 3.70 (2H, s, CH2). δC (100 MHz, CDCl3) 176.2 (Cq), 156.6 (Cq), 155.4 (Cq), 143.4 (Cq), 141.6, 135.9 (Cq), 122.1, 120.4 (Cq), 120.0, 114.8, 113.4, 112.3 (Cq), 98.3, 55.6 (OCH3), 29.5 (CH2). LRMS (ESI): m/z 296.1 [M − H]−. HRMS (ESI): 296.0959 [M − H]−.
Butyl 2-(6-butoxybenzofuran-3-yl)acetate (17): Compound 9 (200 mg. 1.041 mmol) was reacted with n-butyl bromide according to General Procedure C using acetonitrile as solvent, to give the compound in question as a yellow oil (205 mg, 65%) which did not require purification. δH (500 MHz, CDCl3) 7.52 (1H, s, ArH), 7.41 (1H, d, J = 9.0 Hz, ArH), 6.99 (1H, s, ArH), 6.88 (1H, d, J = 8.5 Hz, ArH), 4.13 (2H, t, J = 6.8 Hz, OCH2), 4.00 (2H, t, J = 6.5 Hz), OCH2), 3.65 (2H, s, CH2), 1.79 (2H, m, CH2), 1.61 (2H, m, CH2), 1.53 (2H, m, CH2), 1.35 (2H, m, CH2), 0.97 (3H, t, J = 7.5 Hz, CH3), 0.91 (3H, t, J = 7.3 Hz, CH3). δC (100 MHz, CDCl3) 170.8 (Cq), 157.7 (Cq), 156.2 (Cq), 141.8, 120.9 (Cq), 119.7, 113.1 (Cq), 112.3, 96.8, 68.3 (OCH2), 64.9 (OCH2), 31.3 (CH2), 30.6 (CH2), 29.9 (CH2), 19.3 (CH2), 19.1 (CH2), 13.8 (CH3), 13.6 (CH3). LRMS (ESI): m/z 305.1 [M + H]+.
Isobutyl 2-(6-isobutoxybenzofuran-3-yl)acetate (18): Compound 9 (200 mg, 1.04 mmol) was reacted with i-butyl bromide according to General Procedure C using acetonitrile as solvent, to give the compound in question as a yellow oil (50.0 mg, 16%) which did not require further purification. δH (400 MHz, CDCl3) 7.52 (1H, s, ArH), 7.41 (1H, d, J = 8.4 Hz, ArH), 6.99 (1H, s, ArH), 6.89 (1H, d, J = 8.4 Hz, ArH), 3.91 (2H, d, J = 6.8 Hz, CH2), 3.75 (2H, d, J = 6.4 Hz, OCH2), 3.67 (2H, s, CH2), 2.11 (1H, sept, J = 6.8 Hz, CH), 1.92 (1H, sept, J = 6.8 Hz, CH), 1.04 (6H, d, J = 6.4 Hz, 2CH3), 0.90 (6H, d, J = 6.8 Hz, 2CH3). δC (100 MHz, CDCl3) 170.8 (Cq), 157.8 (Cq), 156.2 (Cq), 141.8, 120.9 (Cq), 119.7, 113.1 (Cq), 112.3, 96.8, 75.1 (OCH2), 71.2 (OCH2), 29.9 (CH2), 28.3 (CH), 29.7 (CH), 19.3 (CH3), 19.0 (CH3). LRMS (ESI): m/z 305.1 [M + H]+.
Benzyl 2-(6-(benzyloxyl)benzofuran-3-yl)acetate (
19): Compound
9 (315 mg, 1.64 mmol) was reacted with benzyl bromide according to General Procedure C using acetone as solvent. Purification by flash chromatography (1–10% ethyl acetate in hexanes) afforded the compound in question as a white solid (315 mg, 52%); mp 77–79 °C (lit. 77–78.5 °C) [
29]. δ
H (400 MHz, CDCl
3) 7.53 (1H, s, ArH), 7.46–745 (2H, m, ArH), 7.41–7.33 (9H, m, ArH), 7.07, (1H, s, ArH), 6.94 (1H, d,
J = 8.4 Hz, ArH), 5.16 (2H, s, OCH
2), 5.11 (2H, s, OCH
2), 3.71 (2H, s, CH
2). δ
C (100 MHz, CDCl
3) 170.5, 157.2, 156.1, 142.0, 136.9, 135.6, 128.6, 128.5, 128.3, 128.3, 128.0, 127.5, 121.2, 119.9, 112.8, 112.5, 97.3, 70.6, 66.8, 29.8. LRMS (ESI):
m/
z 373.1 [M + H]
+.
Benzyl 2-(5-benzyloxy)benzofuran-3-yl)acetate (20): Compound 10 (75.0 mg, 0.390 mmol) was reacted with benzyl bromide (21.3 µL) according to General Procedure C using acetone as solvent. Purification by flash chromatography (1–30% ethyl acetate in hexanes) gave the compound in question as a white solid (44.7 mg, 31%); mp 70–72 °C. δH (400 MHz, CDCl3) 7.60 (1H, s, ArH), 7.46–7.32 (11H, m, ArH), 7.06 (1H, d, J = 2.4 Hz, ArH), 7.00 (1H, dd, J = 9.0 Hz, 2.6, ArH), 5.16 (2H, s, OCH2), 5.01 (2H, s, OCH2), 3.72 (2H, s, CH2). δC (100 MHz, CDCl3) 170.4 (Cq), 155.1 (Cq), 150.3 (Cq), 143.7, 137.1 (Cq), 135.6 (Cq), 128.6, 128.5, 128.4, 128.3, 128.1 (Cq), 127.9, 127.6, 114.1, 113.0 (Cq), 112.0, 103.5, 70.8 (CH2), 66.9 (CH2), 29.9 (CH2). LRMS (ESI): m/z 373.1 [M + H]+.
2-(6-Butoxybenzofuran-3-yl)acetic acid (21): Ester 17 (205 mg, 0.673 mmol) was hydrolysed using 2 M methanolic sodium hydroxide (5 equiv, 1.68 mL) according to General Procedure A to give the compound in question as a white solid (143 mg, 86%); mp 108–109 °C. δH (500 MHz, CDCl3) 7.53 (1H, s, ArH), 7.40 (1H, d, J = 8.8 Hz, ArH), 7.00 (1H, s, ArH), 6.89 (1H, d, J = 8.4 Hz, ArH), 3.99 (2H, t, J = 6.4 Hz, OCH2), 3.71 (2H, s, CH2), 1.79 (2H, p, J = 6.8 Hz, CH2), 1.51 (2H, sextet, J = 7.6 Hz, CH2), 0.99 (3H, t, J = 7.4 Hz, CH3). δC (100 MHz, CDCl3) 176 (Cq), 157.8 (Cq), 156.3 (Cq), 142.0, 120.6 (Cq), 119.7, 112.4, 112.3 (Cq), 96.8, 68.3 (OCH2), 31.3 (CH2), 29.4 (CH2), 19.3 (CH2), 13.8 (CH3). LRMS (ESI): m/z 247.2 [M − H]−.
2-(6-Isobutoxybenzofuran-3-yl)acetic acid (22): Ester 18 (50.0 mg, 0.164 mmol) was hydrolysed using 2 M methanolic sodium hydroxide (5 equiv, 411 µL) according to General Procedure A. The crude compound required purification by RP-HPLC. The fractions containing the product were lyophilised to give the compound in question as a white solid (6 mg, 15%); mp 130–132 °C. δH (400 MHz, CDCl3) 7.53 (1H, s, ArH), 7.40 (1H, s, J = 8.4 Hz, ArH), 6.99 (1H, s, ArH), 6.90 (1H, d, J = 8.6 Hz, ArH), 3.75 (2H, d, J = 6.4 Hz, OCH2), 3.71 (2H, s, CH2), 2.11 (1H, sept, J = 6.4 Hz, CH), 1.05 (6H, d, J = 6.8 Hz, CH3). δC (100 MHz, CDCl3) 157.9 (Cq), 156.3 (Cq), 142.0, 120.6 (Cq), 119.6, 112.4, 112.3 (Cq), 96.9, 75.1 (OCH2), 29.7 (CH2), 19.3 (CH3). One quaternary carbon not observed. LRMS (ESI): m/z not found.
2-(6-((Benzyloxy)benzofuran-3-yl)acetic acid (23): Ester 19 (248 mg, 0.665 mmol) was hydrolysed using 2 M sodium hydroxide (5 equiv, 1.66 mL) according to General Procedure A. The compound in question was afforded after recrystallisation from chloroform as a crystalline white solid (124 mg, 61%); mp 153–155 °C. δH (500 MHz, CDCl3) 7.54 (1H, s, ArH), 7.46–7.32 (7H, m, ArH), 7.08 (1H, s, ArH), 6.98 (1H, d, J = 8.5 Hz, ArH), 5.11 (2H, s, OCH2), 3.71 (2H, s, CH2). δC (100 MHz, CDCl3) 176.3, 157.4, 156.1, 142.2, 136.9, 128.6, 128.0, 127.5, 121.1, 119.8, 112.6, 112.3, 97.5, 70.6, 29.5. LRMS (ESI): m/z 281.0 [M − H]−. HRMS (ESI): 281.0848 [M − H]−.
2-(5-(Benzyloxy)benzofuran-3-yl)acetic acid (24): Ester 20 (28.7 mg, 0.0771 mmol) was hydrolysed using 2 M methanolic sodium hydroxide (5 equiv, 193 µL) according to General Method B to give the compound in question as a white solid (16.9 mg, 78%); mp 147–149 °C. δH (400 MHz, CDCl3) 7.61 (1H, s, ArH), 7.47–7.30 (6H, m, ArH), 7.10 (1H, s, ArH), 7.00 (1H, dd, J = 8.8 Hz, 2.0, ArH), 5.10 (2H, s, OCH2), 3.71 (2H, s, CH2). δC (100 MHz, CDCl3) 176.2 (Cq), 155.2 (Cq), 150.3 (Cq), 143.9, 137.1 (Cq), 128.6, 127.9, 127.6, 114.1, 112.4 (Cq), 112.1, 103.6, 70.9 (OCH2), 29.4 (CH2). One quaternary carbon not observed. LRMS (ESI): m/z 281.0 [M − H]−. HRMS (ESI): 281.0848 [M − H]−.
2-(6-Phenoxybenzofuran-3-yl)acetic acid (25): Compound 8 (302 mg, 1.00 mmol) was coupled to phenol according to General Procedure D. The crude compound was purified by flash chromatography (2.5% methanol in dichloromethane) to give a red solid (61.3 mg) that was recrystallised from acetonitrile to give the compound in question as a white solid (7.1 mg, 3%). The mother liquor was concentrated and purified by semi-preparative RP-HPLC. The fractions containing the product were lyophilised to give the compound in question as a white solid (19.7 mg, 7%); mp 107–109 °C. δH (400 MHz, CDCl3) 7.60 (1H, s, ArH). 7.50 (1H, d, J = 8.4 Hz, ArH), 7.36–7.32 (2H, m, ArH), 7.19–7.09 (2H, m, ArH), 7.03–7.00 (3H, m, ArH), 7.74 (2H, s, CH2). δC (100 MHz, CDCl3) 176.2 (Cq), 157.6 (Cq), 155.8 (Cq), 155.3 (Cq), 143.2 (Cq), 129.8, 123.2, 120.1, 118.6, 115.3, 112.4 (Cq), 102.6, 29.7 (CH2). One carbon signal obscured. LRMS (ESI): m/z 267.2 [M − H]−. HRMS (ESI): 267.0689 [M − H]−.
2-(6-(3-Methoxyphenoxy)benzofuran-3-yl)acetic acid (26): Compound 8 (302 mg, 1.00 mmol) was coupled to 3-methoxyphenol (149 mg) according to General Procedure D. The crude brown oil was purified by flash chromatography (2.5% methanol in dichloromethane) and subsequent recrystallisation from aqueous ethanol afforded the compound in question as an off-white solid (30.0 mg, 10%); mp. 91–93 °C. δH (400 MHz, CDCl3) 7.62 (1H, s, ArH), 7.50 (1H, d, J = 8.5 Hz, ArH), 7.22 (1H, t, J = 8.5 Hz, ArH), 7.15 (1H, s, ArH), 7.02 (1H, d, J = 8.5 Hz, ArH), 7.66 (1H, d, J = 8.5 Hz, ArH), 6.61–6.58 (2H, m, ArH), 3.78 (3H, s, OCH3), 3.75 (2H, s, CH2). LRMS (ESI): m/z 299.11 [M + H]+. HRMS (ESI): 297.0800 [M − H]−.
Methyl 2-(6-benzylbenzofuran-3-yl)acetate (27): A three-necked flask charged with zinc powder (535 mg), lithium chloride (433 mg), and a stir bar was flame dried under reduced pressure. The flask was cooled to room temperature and filled with nitrogen. Dry tetrahydrofuran (5 mL) was added via cannula and the suspension was cooled to 0 °C. Benzyl bromide (486 µL) was added dropwise via syringe and the suspension stirred for 1.5 h. The suspension was then transferred via cannula to an evacuated, flame dried flask containing compound 11 (250 mg, 0.929 mmol), Pd(OAc)2 (17.9 mg), and XPhos (35.9 mg). The flask was filled with nitrogen then heated to 50 °C. The resulting black reaction mixture was stirred for 18 h, cooled to room temperature, and then treated with 1 M HCl and allowed to stir for 1 min. The organic phase was collected and the aqueous phase was extracted with ethyl acetate (×3). The combined organic phases were dried over magnesium sulfate then concentrated under reduced pressure to a dark yellow oil. The crude oil was purified by flash chromatography (2.5% ethyl acetate in hexanes) to give the compound in question as a yellow oil (60.5 mg, 35%). δH (400 MHz, CDCl3) 7.57 (1H, s, ArH), 7.46 (1H, d, J = 8.1 Hz, ArH), 7.31–7.29 (2H, m, ArH), 7.22–718 (3H, m, ArH), 7.12 (1H, d, J = 8.1 Hz, ArH), 4.09 (2H, s, CH2), 3.72 (3H, s, OCH3), 3.68 (2H, s, CH2). δC (100 MHz, CDCl3) 171.1 (Cq), 155.6 (Cq), 142.6, 141.1 (Cq), 138.1 (Cq), 128.9, 128.4, 126.1, 125.7 (Cq), 123.9, 119.4, 112.9 (Cq), 111.7, 52.1 (OCH3), 42.0 (CH2), 29.5 (CH2). LRMS (ESI): m/z 303.1 [M + Na]+.
2-(6-Benzylbenzofuran-3-yl)acetic acid (28): Ester 27 (60.5 mg, 0.216 mmol) was hydrolysed using 2 M methanolic sodium hydroxide (5 equiv, 540 µL) according to General Procedure A to give the compound in question as a while solid (29.2 mg, 51%); mp 124–127 °C. δH (400 MHz, CDCl3) 7.58 (1H, s, ArH), 7.47 (1H, s, ArH), 7.32–7.28 (3H, m, ArH), 7.23–7.21 (3H, m, ArH), 7.13 (1H, d, J = 8.0 Hz, ArH), 4.10 (2H, s, CH2), 3.73 (2H, s, CH2). δC (100 MHz, CDCl3) 176.9, 155.6 (Cq), 142.8, 141.1 (Cq), 138.3 (Cq), 128.9, 128.5, 126.1, 125.5 (Cq), 124.0, 199.4, 112.2 (Cq), 111.8, 42.0 (CH2), 29.5 (CH2). LRMS (ESI): m/z 265.0 [M − H]−. HRMS (ESI): 265.0895 [M − H]−.
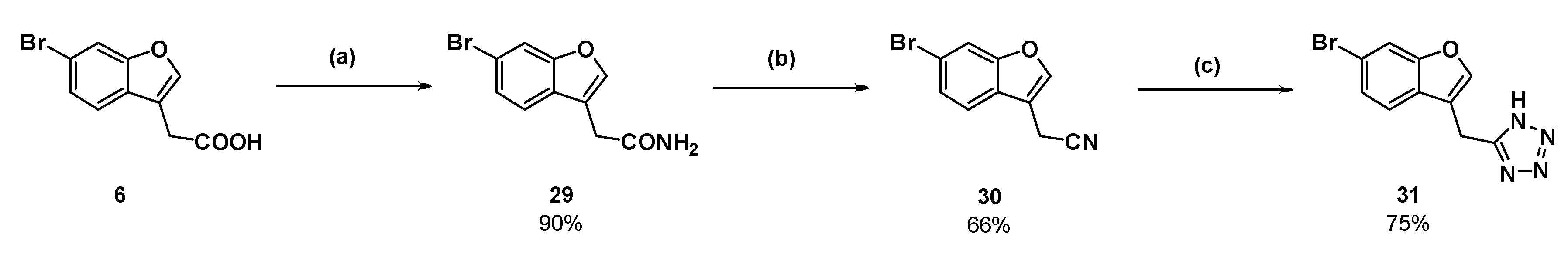
2-(6-Bromobenzofuran-3-yl)acetamide (29): Compound 6 (150 mg, 0.588 mmol) was dissolved in dichloromethane (6 mL) in an atmosphere of nitrogen. Phosphorous pentachloride (122 mg, 0.588 mmol) was added and the solution was heated to reflux for 30 min. The reaction cooled to room temperature and ammonia gas (produced by heating an aqueous solution of ammonium hydroxide to 50 °C) was bubbled through the pale-yellow solution. Within 10 s, a white solid precipitated out of solution. After several minutes, the ammonia gas was removed from the reaction then the dichloromethane was removed under reduced pressure. The white residue was partitioned between ethyl acetate and water. The aqueous phase was washed with ethyl acetate (×2) then the combined organic phases were dried over magnesium sulfate and concentrated under reduced pressure to afford the amide as a white solid (135 mg, 90%); mp 140–143 °C. δH (400 MHz, CDCl3) 7.85–7.84 (2H, m, ArH), 7.57 (1H, d, J = 8.4 Hz, ArH), 7.53 (1H, br s, NH), 7.42 (1H, d, J = 8.4 Hz, ArH), 6.98 (1H, br s, NH), 3.47 (2H, s, CH2). δC (100 MHz, DMSO-d6) 171.6 (Cq), 155.4 (Cq), 144.5, 127.8 (Cq), 126.1, 122.3, 117.3 (Cq), 115.7 (Cq), 115.0, 30.6 (CH2). LRMS (ESI): m/z 253.9 [M + H]+. HRMS (ESI): 275.9617 [M + Na]+.
2-(6-Bromobenzofuran-3-yl)acetonitrile (30): Amide 29 (110 mg, 0.431 mmol) was suspended in dry toluene (4.5 mL) in an atmosphere of nitrogen. Distilled thionyl chloride (47.0 µL) was added and the reaction mixture was heated to reflux. The starting material dissolved upon heating. After 1.5 h, TLC showed a new product had formed and only a small amount of starting material remained. The reaction was cooled to room temperature, and then the toluene and excess thionyl chloride were removed under reduced pressure. The resulting dark orange oil was dissolved in ethyl acetate and washed with a saturated solution of sodium hydrogen carbonate and brine (×2). The organic phase was dried (MgSO4) and concentrated to a dark orange oil under reduced pressure. The crude oil was purified by flash chromatography (10% ethyl acetate in hexanes) to afford the nitrile as a dark pink solid (66.9 mg, 66%); mp 88–90 °C. δH (400 MHz, CDCl3) 7.70 (1H, s, ArH), 7.65 (1H, s, ArH), 7.46 (1H, d, J = 8.4 Hz, ArH), 7.43 (1H, d, J = 8.4 Hz, ArH), 3.75 (2H, s, CH2). δC (100 MHz, CDCl3) 155.7(Cq), 143.2, 126.7, 125.1 (Cq), 119.9, 118.8 (Cq), 116.3 (Cq), 115.4, 110.2 (Cq), 13.0 (CH2). LRMS (ESI): m/z not found. I.R (KBr) 2254 cm−1 (-C≡N).
5-((6-Bromobenzofuran-3-yl)methyl)-1H-tetrazole (31): Nitrile 30 (57 mg, 0.241 mmol), sodium azide (47.1 mg), and triethylammonium chloride (99.7 mg) were suspended in dry toluene (1 mL) in an atmosphere of nitrogen. The suspension was stirred at reflux for 3 days. The reaction was cooled to room temperature and water was added. The aqueous phase was collected and acidified with concentrated hydrochloric acid. Upon acidification, the compound in question precipitated as an off-white solid which was collected by vacuum filtration (50.2 mg, 75%); mp 184–185 °C. δH (400 MHz, DMSO-d6) 7.98 (1H, s, ArH), 7.90 (1H, s, ArH), 7.67 (1H, d, J = 8.4 Hz, ArH), 7.42 (1H, d, J = 8.0 Hz, ArH), 4.39 (2H, s, CH2). δC (100 MHz, DMSO-d6) 155.5 (Cq), 144.9, 126.9 (Cq), 126.4, 121.8, 117.7 (Cq), 115.3 (Cq), 115.2, 18.3 (CH2). One quaternary carbon was not observed. LRMS (ESI): m/z 279.0 [M (79Br) + H]+, 280.9 [M (81Br) + H]+. HRMS (ESI): 276.9758 [M (79Br) − H]−, 278.9738 [M (81Br) − H]−.
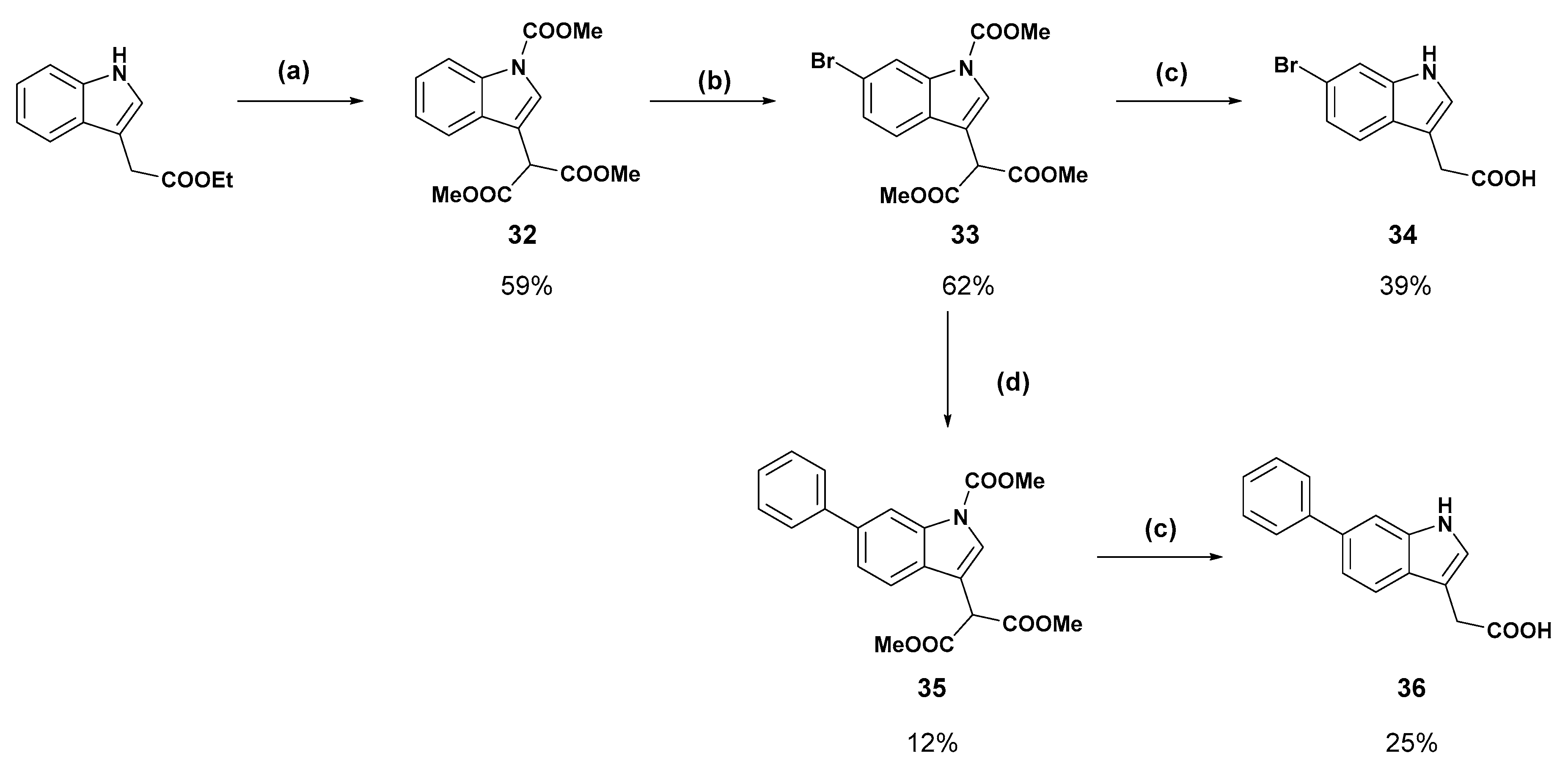
Dimethyl 2-(1-(methoxycarbonyl)-1H-indole-3-yl)malonate (
32): Based on the method of Suarez–Castillo [
19]. A solution of ethyl-3-indole acetate (1.5 g, 7.38 mmol) in dry tetrahydrofuran (10 mL) was added to a suspension of sodium hydride (1.18 g as 60% dispersion in mineral oil, 29.5 mmol) in dry tetrahydrofuran (40 mL) under an atmosphere of nitrogen. The suspension was stirred until the formation of gas was no longer observed (5–10 min). To the resulting green suspension was added dimethyl carbonate (12.0 mL, 142 mmol) and the reaction mixture was stirred at reflux for 16 h, after which time the colour became maroon. The reaction was cooled to room temperature and the tetrahydrofuran was removed under reduced pressure. The resulting residue was partitioned between a saturated solution of sodium hydrogen carbonate and diethyl ether. The aqueous phase was washed with diethyl ether (×2) and the combined organic phases were washed with brine (×3), dried over magnesium sulfate, and concentrated to a red oil by rotary evaporation. A small portion of ether was added and heated to boil. Upon cooling, a solid precipitated which was recrystallised from diethyl ether and the mother liquor was purified by flash chromatography (dichloromethane) to afford the titled product as an off-white solid (1.33 g, 59%); mp 108–110 °C (lit. 108–110 °C) [
30]. δ
H (500 MHz, CDCl
3) 8.20 (1H, d,
J = 7.5 Hz, ArH), 7.82 (1H, s, ArH), 7.57 (1H, d,
J = 7.5 Hz, ArH), 7.36 (1H, t,
J = 7.5 Hz, ArH), 7.28 (1H, t,
J = 7.5, ArH), 4.90 (1H, s, CH), 4.04 (3H, s, OCH
3), 3.78 (6H, s, (OCH
3)
2). δ
C (100 MHz, CDCl
3) 168.0 (C
q), 151.2 (C
q), 135.3 (C
q), 129.1 (C
q), 125.1, 125.0, 123.2, 119.2, 115.3, 112.8 (C
q), 53.9 (OCH
3), 53.0 (OCH
3), 49.0 (CH).
Dimethyl 2-(6-bromo-1-(methoxycarbonyl)-1H-indole-3-yl)malonate (
33): According to the method of Suarez–Castillo [
19]. Malonate
32 (1.25 g, 4.09 mmol) was dissolved in carbon tetrachloride (50.0 mL), treated with bromine (1.68 mL), and stirred at room temperature for 24 h. A saturated solution of sodium sulfite (50 mL) was added and stirred vigorously for 24 h. The aqueous phase was extracted with dichloromethane (×2) and the combined organic phases were washed with brine (×3), dried over magnesium sulfate, and concentrated under reduced pressure to an orange oil. Diethyl ether (15 mL) was added and the solution was cooled until a precipitate formed which was collected and recrystallised from diethyl ether to afford the titled product as an off-white solid (826 mg, 52%). The mother liquor was purified by flash chromatography (5–20% ethyl acetate in hexanes) to afford an off-white solid after trituration with diethyl ether (157 mg, 10%); mp 110–112 °C (lit. 110–112 °C) [
19]. δ
H (400 MHz, CDCl
3) 8.38 (1H, br s, ArH), 7.79 (1H, s, ArH), 7.45 (1H, d,
J = 8.4 Hz, ArH), 7.40 (1H, d,
J = 8.4 Hz, ArH), 4.85 (1H, s, CH), 4.04 (3H, s, NCOOCH
3), 3.78 (6H, s, (COOCH
3)
2). δ
C (100 MHz, CDCl
3) 167.8 (C
q), 150.8 (C
q), 135.9 (C
q), 127.9, 126.5, 125.6 (C
q), 120.6, 118.8 (C
q), 118.4, 112.7 (C
q), 54.1 (CH
3), 53.1 (CH
3), 49.0 (CH).
2-(6-Bromo-1H-indol-3-yl)acetic acid (34): Malonate 33 (200 mg, 0.640 mmol) was dissolved in methanol:water (9:1) and treated with potassium hydroxide (180 mg, 3.21 mmol) and refluxed for 18 h. The reaction was cooled to room temperature and concentrated under reduced pressure. The resulting residue was dissolved in water and the aqueous solution was washed with ethyl acetate. The aqueous phase was then acidified with 1 M hydrochloric acid to pH 2 and the cloudy suspension was extracted with ethyl acetate (×3). The combined organic layers were dried over magnesium sulfate, then concentrated under reduced pressure to a light yellow solid. Recrystallisation from chloroform afforded the titled product as a white solid (64.0 mg, 39%); mp 178–180 °C δH (400 MHz, DMSO-d6) 11.04 (1H, br s, NH/COOH), 7.52 (1H, s, ArH), 7.44 (1H, d, J = 8.4 Hz, ArH), 7.25 (1H, s, ArH), 7.10 (1H, d, J = 8.4 Hz, ArH), 3.62 (2H, s, CH2). δC (100 MHz, DMSO-d6) 173.4, 137.4, 126.8, 125.6, 121.8, 120.9, 114.4, 114.3, 108.6, 31.3. LRMS (ESI): m/z 252.3 [M (79Br) − H)−, 253.9 [M (81Br) − H]−.
Dimethyl 2-(1-(methoxycarbonyl)-6-phenyl-1H-indol-3-yl)malonate (35): Bromide 33 (250 mg, 0.651 mmol), phenylboronic acid (476 mg), caesium fluoride (297 mg), and Pd(dppf)Cl2 (24.0 mg) were dissolved in degassed acetonitrile:water (4:1) in an atmosphere of nitrogen. The suspension was heated to 90 °C and stirred for 18 h. The reaction was partitioned between ethyl acetate and brine and the organic layer was separated, washed with brine (×2), and dried over magnesium sulfate. The ethyl acetate was removed under reduced pressure then the crude brown solid was purified by flash chromatography (20% ethyl acetate in hexane, then 2.5–5% methanol in dichloromethane). The fractions containing the product were concentrated under reduced pressure and triturated with diethyl ether to afford the compound in question as a white solid (30.0 mg, 12%); mp 124–126 °C. δH (400 MHz, CDCl3) 8.47 (1H, bs s, ArH), 7.85 (1H, s, ArH), 7.68 (2H, d, J = 7.2 Hz, ArH), 7.65 (1H, d, J = 8.3 Hz, ArH) 7.55 (1H, d, J = 8.2 Hz, ArH), 7.46 (2H, t, J = 7.6 Hz, ArH), 7.36 (1H, t, J = 7.4 Hz, ArH), 4.94 (1H, s, CH), 4.05 (3H, s, OCH3), 3.81 (6H, s, OCH3) δC (100 MHz, CDCl3) 168.0 (Cq), 151.1 (Cq), 141.5 (Cq), 138.6 (Cq), 135.9 (Cq), 128.7, 128.2 (Cq), 127.5, 127.1, 125.5, 122.8, 119.5, 113.9, 112.8 (Cq), 53.9 (CH3), 53.0 (OCH3), 49.1 (CH).
2-(6-Phenyl-1H-indol-3-yl)acetic acid (36): Malonate 35 (30 mg, 78.7 µmol) was dissolved in methanol:water (9:1) and treated with potassium hydroxide (22.0 mg). The reaction refluxed for 16 h before additional potassium hydroxide (8.80 mg) was added. After one hour at reflux the reaction was cooled to room temperature and concentrated under reduced pressure. The resulting residue was dissolved in water and the aqueous solution was washed with ethyl acetate. The aqueous phase was then acidified with 1 M hydrochloric acid to pH 2 and the cloudy suspension was extracted with ethyl acetate (×3). The combined organic layers were dried over magnesium sulfate, then concentrated under reduced pressure to a light yellow solid. Purification by flash chromatography (10% methanol in dichloromethane) and subsequent recrystallisation from acetonitrile afforded the compound in question as a white solid (5.0 mg, 25%); mp 214–216 °C. δH (500 MHz, Acetone-d6) 7.69 (4H, m, ArH), 7.43 (2H, t, J = 7.8, ArH), 7.36 (1H, d, 8.3, ArH), 7.34 (1H, s, ArH), 7.30 (1H, t, J = 7.4 Hz, ArH), 3.78 (2H, s, CH2). δC (100 MHz, Acetone-d6) 172.4 (Cq), 142.5 (Cq), 134.7 (Cq), 127.7, 127.0, 126.4, 124.6, 124.4 (Cq), 119.1, 118.5, 109.6, 108.3 (Cq), 30.6 (CH2). LRMS (ESI): m/z 252.1 [M + H]+. HRMS (ESI): m/z 250.0895 [M − H]−.
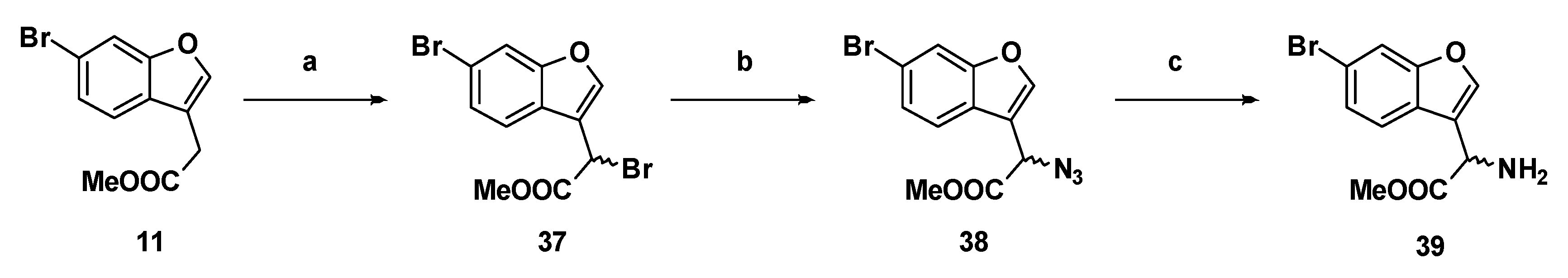
Methyl 2-bromo-2-(6-bromobenzofuran-3-yl)acetate (37): Compound 11 (500 mg, 1.86 mmol), N-bromosuccinimide (363.8 mg, 2.04 mmol) and azobisisobutyronitrile (61.0 mg, 0.370 mmol) were suspended in carbon tetrachloride (25 mL) and the mixture was refluxed for two hours. The reaction mixture was cooled to room temperature, filtered through a bed of Celite®, and the carbon tetrachloride was removed by distillation. The crude compound was purified by flash chromatography (0.5% ethyl acetate in hexanes) to afford the compound in question as an off-white solid (523 mg, 81%); mp 73–75 °C. δH (400 MHz, CDCl3) 7.88 (1H, s, ArH), 7.69 (1H, s, ArH), 7.66 (1H, d, J =8.4 Hz, ArH), 7.45 (1H, d, J = 8.4 Hz, ArH), 5.57 (1H, s, CH), 3.84 (3H, s, OCH3). δC (100 MHz, CDCl3) 168.0, 155.9, 144.5, 126.7, 124.6, 121.9, 118.7, 116.2, 115.3, 53.5, 36.5, 29.6. LRMS (ESI): m/z 368.52 [M (79Br, 79Br) + Na]+, 370.93 [M (79Br, 81Br) + Na]+, 372.92 [M (81Br, 81Br) + Na]+. HRMS (ESI): m/z 344.9786 [M (79Br, 79Br) − H]−.
Methyl-2-azido-2-(6-bromobenzofuran-3-yl)acetate, (38): Bromide 37 (760 mg, 2.18 mmol) was dissolved in dry N,N-dimethylformamide (20 mL) in an atmosphere of nitrogen. The stirring solution was chilled to −40 °C in a bath of acetonitrile/dry ice. Sodium azide (426 mg, 6.55 mmol) was added and the colourless solution gradually became yellow. The reaction was monitored by TLC, and after 50 min the solution was removed from the bath. Ethyl acetate and 1 M hydrochloric acid were added and the aqueous phase was extracted with ethyl acetate (×2). The combined organic phases were washed with brine (×5), dried over magnesium sulfate, then concentrated under reduced pressure to yield the product as a yellow oil (594 mg, 88%). δH (400 MHz, CDCl3) 7.72 (1H, s, ArH), 7.70 (1H, d, 1.6, ArH), 7.55 (1H, d, J = 8.4 Hz, ArH), 7.43 (1H, dd, J = 8.4 Hz, 1.6, ArH), 5.16 (1H, s, CH), 3.83 (3H, s, OCH3). δC (100 MHz, CDCl3) 168.7 (Cq), 155.8 (Cq), 144.2, 126.9 (Cq), 124.4 (Cq), 121.2, 118.8 (Cq), 115.4, 114.4 (Cq), 57.2 (CH), 53.2 (OCH3). LRMS (ESI): m/z not found.
Methyl-2-amino-2-(6-bromobenzofuran-3-yl)acetate (39): Azide 38 (114 mg, 0.369 mmol) was dissolved in dry methanol in an atmosphere of nitrogen. Adams catalyst (2.5% w/w) was added and the flask was purged three times with hydrogen. The reaction stirred at room temperature under hydrogen for 5 h, at which time TLC indicated negligible starting material. The reaction was filtered through Celite®, then concentrated to afford the amine as a yellow oil (97.1 mg, 0.342 mmol, 92%). δH (500 MHz, CDCl3) 7.66 (1H, d, J 1.5, ArH), 7.63 (1H, s, ArH), 7.56 (1H, d, J = 8.0 Hz, ArH), 7.38 (1H, dd, J = 8.3 Hz, 1.5, ArH), 4.84 (1H, s, CH), 3.74 (1H, s, CH3). δC (100 MHz, CDCl3) 173.4 (Cq), 155.9 (Cq), 143.0, 126.3, 125.0 (Cq), 121.3, 119.8 (Cq), 118.1 (Cq), 115.2, 52.6 (OCH3), 50.5 (CH). LRMS (ESI): m/z 283.9 [M (79Br) + H]+, 285.9 [M (81Br) + H]+.
Methyl 2-Acetamido-2-(6-bromobenzofuran-3-yl)acetate (40): Amine 39 (100 mg, 0.326 mmol) was coupled to acetyl chloride (25.6 µL) according to General Procedure F to form the compound in question as an off-white solid (62.0 mg, 58%); mp 166–169 °C. δH (400 MHz, CDCl3) 7.65 (1H, s, ArH), 7.62 (1H, s, ArH), 7.49 (1H, d, J = 8.4 Hz, ArH), 7.37 (1H, d, J = 8.4 Hz, ArH), 6.58 (1H, d, J = 7.2 Hz, NH), 5.84 (1H, d, J = 7.5 Hz, CH), 3.76 (3H, s, OCH3), 2.02 (3H, s, CH3). δC (100 MHz, CDCl3) 170.6, 169.6, 155.7, 143.9, 126.6, 124.6, 120.9, 118.4, 116.5, 115.2, 53.0, 47.7, 22.9. LRMS (ESI): m/z 325.9 [M (79Br) + H]+, 327.9 [M (81Br) + H]+.
Methyl 2-benzamido-2-(6-bromobenzofuran-3-yl) acetate (41): Benzoic acid was coupled to amine 39 (100 mg, 0.626 mmol) according to general procedure G. The crude amide was purified by flash chromatography (dichloromethane) to give the compound in question as a white solid (69.3 mg, 55%); mp 161–162 °C. δH (500 MHz, CDCl3) 7.80–7.78 (2H, m, ArH), 7.69 (1H, s, ArH), 7.66 (1H, s, ArH), 7.56 (1H, d, J = 8.4 Hz, ArH), 7.51 (1H, t, J = 7.4 Hz, ArH), 7.43–7.39 (2H, m, ArH), 7.38 (1H, d, J = 8.4 Hz, ArH), 7.16 (1H, d, J = 7.1 Hz, NH), 6.04 (1H, d, J = 7.2 Hz, CH), 3.80 (3H, s, OCH3). δC (100 MHz, CDCl3) 170.7, 166.8, 155.8, 144.1, 133.2, 132.1, 128.6, 127.1, 126.6, 124.7, 120.9, 118.4, 116.5, 115.3, 53.1, 48.2. LRMS (ESI): m/z 388.0 [M (79Br) + H]+, 390.1 [M (81Br) + H]+.
Methyl 2-(6-bromobenzofuran-3-yl)-2-(2-(4-methoxyphenyl)acetamido)acetate (42): Amine 39 (46.3 mg, 0.163 mmol) was coupled to 4-methoxyphenylacetic acid (27.1 mg) according to General Procedure G to give the compound in question as a light yellow solid (37.0 mg, 52%); mp 114–116 °C. δH (400 MHz, CDCl3) 7.64 (1H, s, ArH), 7.53 (1H, s, ArH), 7.31 (2H, s, ArH), 7.15 (2H, d, J = 8.4 Hz, ArH), 6.87 (2H, d, J = 8.4 Hz, ArH), 6.39 (1H, br d, J = 7.2 Hz, NH), 5.80 (1H, d, J = 7.6 Hz, CH), 3.80 (3H, s, OCH3), 3.73 (1H, s, OCH3), 3.55 (2H, s, CH2). δC (100 MHz, CDCl3) 170.9 (Cq), 170.4 (Cq), 159.0 (Cq), 155.7 (Cq), 143.8, 130.4, 126.5, 126.0 (Cq), 124.5 (Cq), 120.9, 118.3 (Cq), 116.3 (Cq), 115.2, 114.5, 55.3 (OCH3), 53.0 (CH), 48.0 (OCH3), 42.5 (CH2). LRMS (ESI): m/z 432.0 [M (79Br) + H]+, 434.1 [M (81Br) + H]+.
Methyl 2-(6-bromobenzofuran-3-yl)-2-(2-(2-fluorophenyl)acetamido)acetate (43): Amine 39 (87.0 mg, 0.284 mmol) was coupled to 2-fluorophenylacetic acid (43.8 mg) according to General Procedure G to give the compound in question as a light yellow solid (105 mg, 71%); mp 154–156 °C. δH (400 MHz, CDCl3) 7.64 (1H, s, ArH), 7.57 (1H, s, ArH), 7.37 (1H, d, J = 8.4 Hz, ArH), 7.33 (1H, d, J = 8.4 Hz, ArH), 7.31–7.26 (2H, m, ArH), 7.14–7.05 (2H, m, ArH), 5.58 (1H, d, J = 7.2 Hz, NH), 5.81 (1H, d, J = 7.2 Hz, CH), 3.74 (3H, s, OCH3), 3.64 (2H, s, CH2). δC (100 MHz, CDCl3) 170.3, 169.4, 162.1, 159.6, 155.7, 143.9, 131.6, 131.5, 129.6, 129.5, 126.5, 124.6, 124.6, 124.5, 121.5, 121.3, 120.9, 118.4, 116.2, 115.7, 115.5, 115.2, 53.0, 48.1, 36.6. LRMS (ESI): m/z 420.2 [M (79Br) + H]+, 422.0 [M (81Br) + H]+.
Methyl 2-(6-bromobenzofuran-3-yl)-2-(methylsulfonamido)acetate (44): Amine 39 (150 mg, 0.528 mmol) was coupled to mesyl chloride (45 µL) according to General Procedure F. The crude yellow oil was purified by flash chromatography (dichloromethane) to afford the compound in question as a light-yellow oil (115 mg, 60%). δH (400 MHz, CDCl3) 7.69 (1H, s, ArH), 7.68 (1H, s, ArH), 7.52 (1H, d, J = 8.4 Hz, ArH), 7.39 (1H, d, J = 8.4 Hz, ArH), 5.74 (1H, d, J = 6.7 Hz, NH), 5.46 (1H, d, J = 6.7 Hz, CH), 3.77 (3H, s, OCH3), 2.84 (3H, s, CH3). δC (100 MHz, CDCl3) 170.0 (Cq), 155.8 (Cq), 144.2, 126.9, 124.0 (Cq), 121.0, 118.7 (Cq), 116.0 (Cq), 115.4, 53.4 (CH), 51.4 (OCH3), 42.1 (CH3). LRMS (ESI): m/z 361.9 [M (79Br) + H]+, 363.9 [M (81Br) + H]+.
Methyl 2-(6-bromobenzofuran-3-yl)-2-(phenylsulfonamido)acetate (45): Amine 39 (75 mg, 245 µmol) was coupled to benzenesulfonyl chloride according to General Procedure F. The crude yellow solid was purified by flash chromatography (15% ethyl acetate in hexanes) to afford the compound in question as a pale yellow solid (16.0 mg, 15%); mp 133–135 °C. δH (400 MHz, CDCl3) 7.74–7.72 (2H, m, ArH), 7.57 (1H, s, ArH), 4.48–7.45 (2H, m, ArH), 7.38–7.33 (3H, m, ArH), 7.30 (1H, d, J = 8.4 Hz, ArH), 5.84 (1H, d, J = 7.6 Hz, NH), 5.30 (1H, d, J = 7.6 Hz, CH), 3.59 (3H, s, OCH3). δC (100 MHz, CDCl3) 169.5, 155.64, 144.0, 139.5, 132.8, 128.8, 127.0, 126.6, 124.1, 121.0, 118.4, 115.5, 115.1, 53.2, 51.4. LRMS (ESI): m/z 446.4 [M (79Br) + H]+, 448.2 [M (81Br) + H]+.
2-Acetamido-2-(6-bromobenzofuran-3-yl)acetic acid (46): Ester 40 (61.0 mg, 0.187 mmol) was hydrolysed using 2 M methanolic sodium hydroxide (5 equiv, 468 µL) according to General Procedure A to give the compound in question as a light yellow solid (57.0 mg, 98%); mp 194–196 °C. δH (500 MHz, Acetone-d6) 7.96 (1H, s, ArH), 7.78 (1H, s, ArH), 7.70 (1H, d, J = 8.4 Hz, ArH), 7.45 (1H, d, J = 8.4 Hz, ArH), 5.83–5.81 (1H, m, CH), 1.99 (3H, s, CH3). δC (100 MHz, Acetone-d6) 171.4 (Cq), 170.5 (Cq), 156.7 (Cq), 145.6, 127.1, 126.4 (Cq), 123.0, 118.6 (Cq), 118.3 (Cq), 115.7, 48.6 (CH), 22.6 (CH3). LRMS (ESI): m/z 309.94 [M (79Br) + H]+, 311.82 [M (81Br) + H]+.
2-Benzamido-2-(6-bromobenzofuran-3-yl)-2-acetic acid (47): Ester 41 (69.3 mg, 0.179 mmol) was hydrolysed using 2 M methanolic sodium hydroxide (5 equiv, 448 µL) according to General Procedure A to give the compound in question as a white solid (64.2 mg, 96%); mp 209–212 °C. δH (500 MHz, DMSO-d6) 9.18 (1H, d, J = 7.1 Hz, NH), 8.13 (1H, s, ArH), 7.93–7.89 (3H, m, ArH), 7.70 (1H, d, J = 8.4 Hz, ArH), 7.54 (1H, t, J = 8.4 Hz, ArH), 7.50–7.44 (3H, m, ArH), 5.83 (1H, d, J = 7.2 Hz, CH). δC (100 MHz, DMSO-d6) 171.0 (Cq), 166.5 (Cq), 155.0 (Cq), 145.1, 133.59 (Cq), 131.6, 128.3, 127.7, 126.1 (Cq), 125.8, 122.3, 117.2 (Cq), 116.8 (Cq), 114.7, 48.2 (CH). LRMS (ESI): m/z 747.5 [2 M]+. HRMS (ESI): m/z 371.9920 [M (79Br) − H]−, 373,9904 [M (81Br) − H]−.
2-(6-Bromobenzofuran-3-yl)-2-(2-(4-methoxyphenyl)acetamido)acetic acid (48): Ester 42 (15 mg, 34.7 µmol) was hydrolysed using 2 M methanolic sodium hydroxide (5 equiv, 87.0 µL) according to General Procedure A to afford the compound in question as an off-white solid (8.4 mg, 58%); mp 159–161 °C. δH (400 MHz, CD3OD) 7.80 (1H, s, ArH), 7.71 (1H, s, ArH), 7.48 (1H, d, J = 8.4 Hz, ArH), 7.35 (1H, d, J = 8.4 Hz, ArH), 7.19 (2H, d, 8.6, ArH), 6.84 (2H, d, J = 8.6 Hz, ArH), 5.70 (1H, s, CH), 3.77 (3H, s, OCH3), 3.52 (2H, s, CH2). LRMS (ESI): m/z 415.89 [M (79Br) + H]+, 417.85 [M (81Br) + H]+. HRMS (ESI): m/z 416.0196 [M (79Br) − H]−, 418.0181 [M (81Br) − H]−.
2-(6-Bromobenzofuran-3-yl)-2-(2-(2-fluorophenyl)acetamido)acetic acid (49): Ester 43 (43.5 mg, 0.104 mmol) was hydrolysed using 2 M methanolic sodium hydroxide (5 equiv, 259 µL) according to General Procedure A to afford the compound in question as a light yellow solid (20.0 mg, 50%); mp 209–211 °C. δH (500 MHz, DMSO-d6) 13.09 (1H, br s, COOH), 8.97 (1H, d, J = 7.0 Hz, NH), 8.07 (1H, s, ArH), 7.93 (1H, s, ArH), 7.64 (1H, d, J = 8.5 Hz, ArH), 7.48 (1H, d, J = 8.5 Hz, ArH), 7.34–7.25 (2H, m, ArH), 7.15–7.10 (2H, m, ArH), 5.59 (1H, d, J = 7.0 Hz, CH), 3.60 (2H, s, CH2). δC (125 MHz, DMSO-d6) 171.4, 169.7, 161.1 (d, J = 243 Hz), 155.6, 145.3, 132.2 (d, J = 3.75 Hz), 129.13, 129.07, 126.6, 125.8, 124.6 (d, J = 3.73 Hz), 122.7, 117.8, 117.4, 115.4 (d, J = 21 Hz), 115.2, 48.3, 35.1. LRMS (ESI): m/z 404.4 [M − H]−. HRMS (ESI): m/z 403.9985 [M (79Br) − H]−, 405.997 [M (81Br) − H]−.
2-(6-Bromobenzofuran-3-yl)-2-(methylsulfonamido)acetic acid (50): Ester 44 (115 mg, 318 µmol) was hydrolysed using 2 M methanolic sodium hydroxide (5 equiv, 794 µL) according to General Method A to afford the compound in question as a light yellow solid (75 mg, 68%); mp 140–142 °C. δH (400 MHz, DMSO-d6) 8.16 (1H, d, J = 8.3 Hz, ArH), 8.06 (1H, s, ArH), 7.91 (1H, s, ArH), 7.73 (1H, d, J = 8.4 Hz, ArH), 7.48 (1H, d, J = 8.4 Hz, ArH), 5.34 (1H, d, J = 8.3 Hz, CH), 2.92 (2H, s, CH2). δC (100 MHz, DMSO-d6) 171.3 (Cq), 155.6 (Cq), 145.3, 126.5, 125.5 (Cq), 122.8, 117.8 (Cq), 117.7 (Cq), 155.2, 51.4 (CH), 41.6 (CH3). LRMS (ESI): m/z 345.95 [M (79Br) + H]+, 347.84 [M (81Br) + H]+. HRMS (ESI): m/z 345.9425 [M (79Br) − H]−, 347.9403 [M (81Br) − H]−.
2-(6-Bromobenzofuran-3-yl)-2-(phenylsulfonamido)acetic acid (51): Ester 45 (15.0 mg, 35.4 µmol) was hydrolysed using 2 M methanolic sodium hydroxide (5 equiv, 88.5 µL) according to General Method A to afford the compound in question as an off-white solid (13.3 mg, 92%); mp 179–182 °C. δH (500 MHz, Acetone-d6) 7.83 (1H, s, ArH), 7.82–7.80 (2H, m, ArH), 7.69 (1H, s, ArH), 7.62 (1H, d, J = 8.4 Hz, ArH), 7.52 (1H, t, J = 7.4 Hz, ArH), 7.43 (2H, t, J = 7.7 Hz, ArH), 7.38 (1H, d, J = 8.4 Hz, ArH), 5.37 (1H, s, CH). δC (125 MHz, Acetone-d6) 170.5, 156.6, 145.8, 142.0, 133.3, 129.7, 127.8, 127.0, 125.8, 123.0, 118.6, 117.8, 115.6, 52.2. HRMS (ESI): m/z 407.9707 [M (79Br) − H]−, 409.9614 [M (81Br) − H]−.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






















