
Trends and Exceptions in the Interaction of Hydroxamic Acid Derivatives of Common Di- and Tripeptides with Some 3d and 4d Metal Ions in Aqueous Solution


 and
and
Abstract
:1. Introduction
2. Results
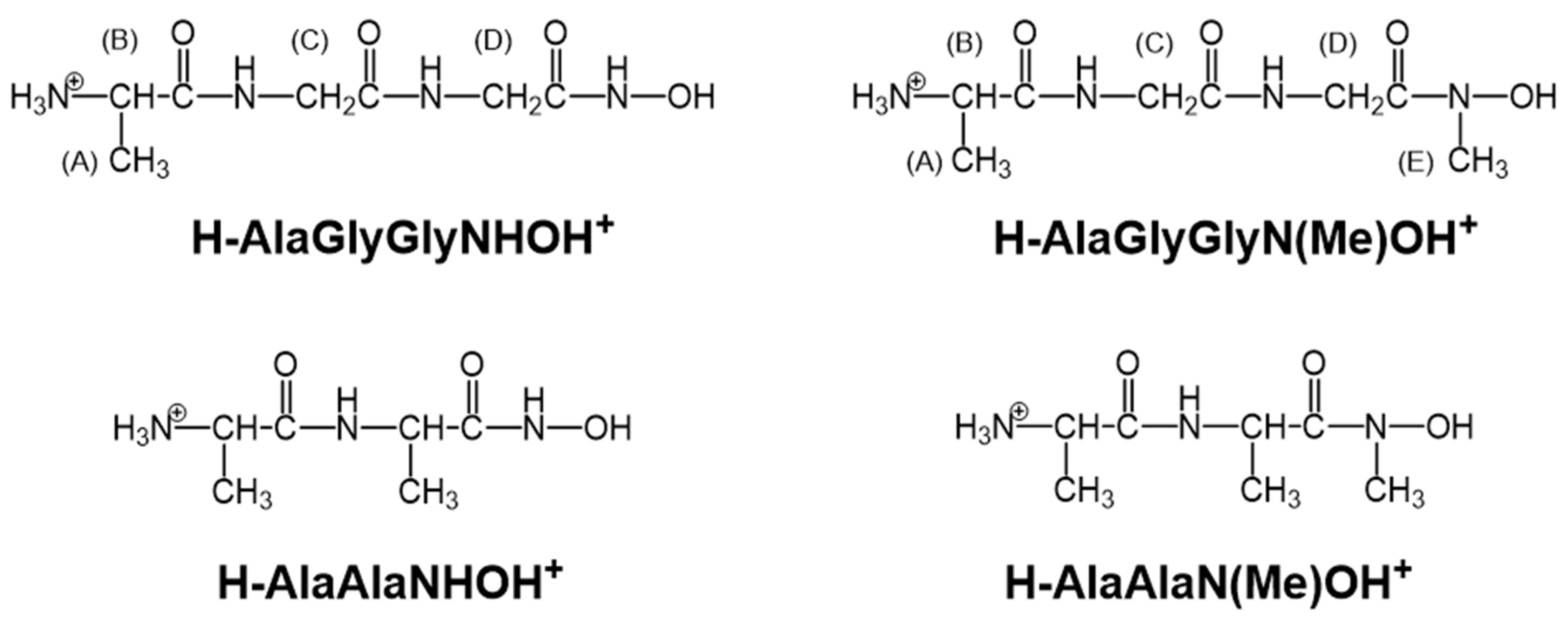
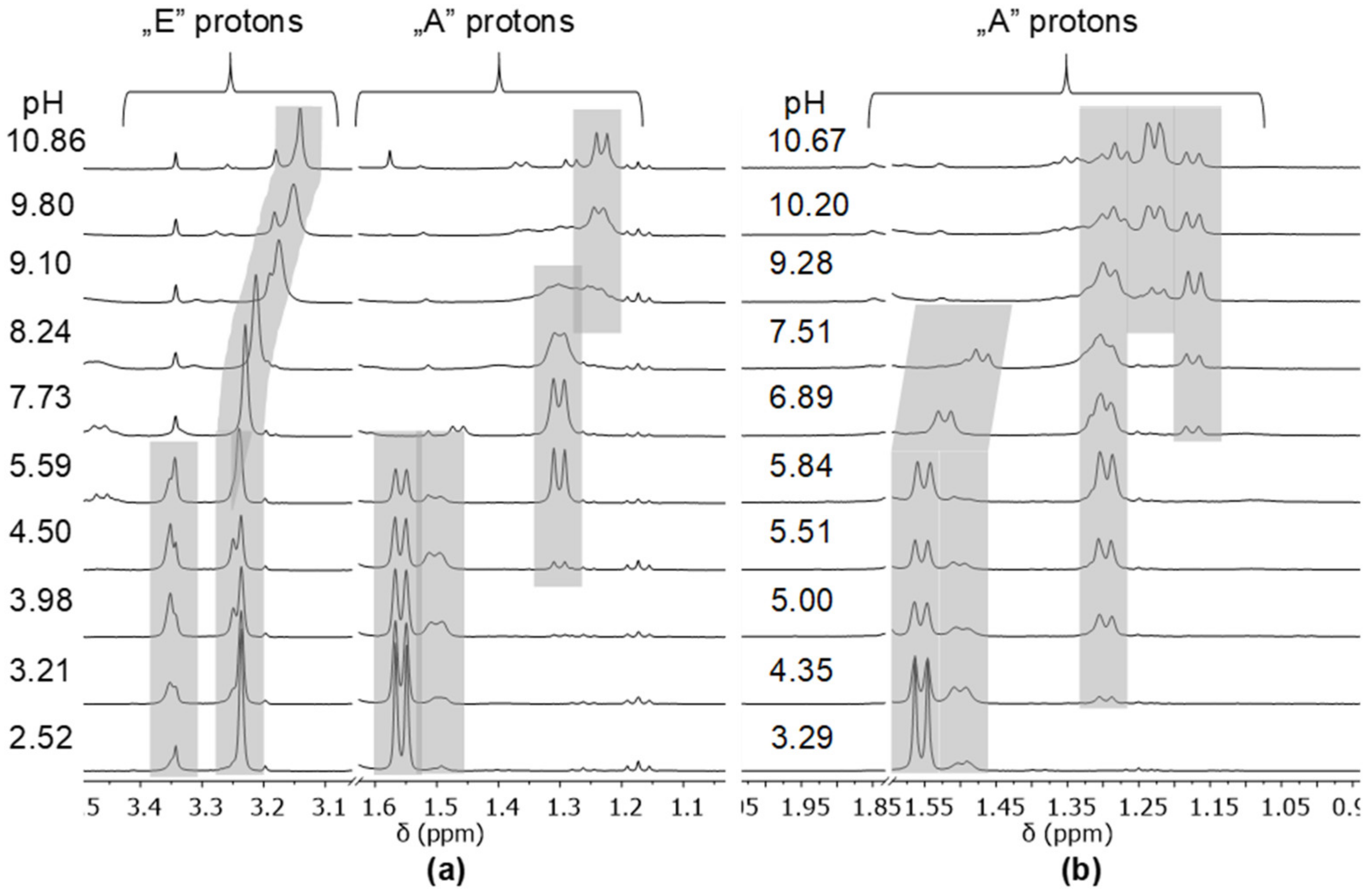
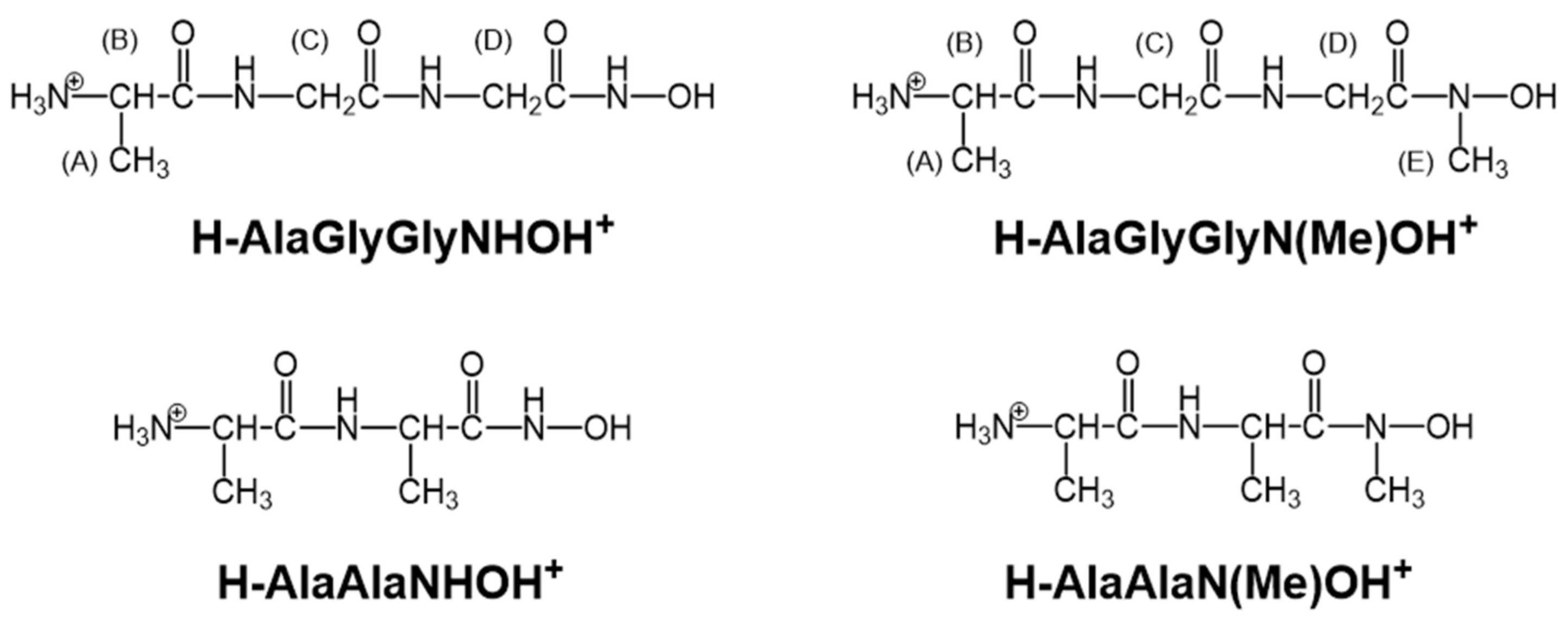
2.1. Protonation Equilibria of the Investigated Peptide Hydroxamic Acids
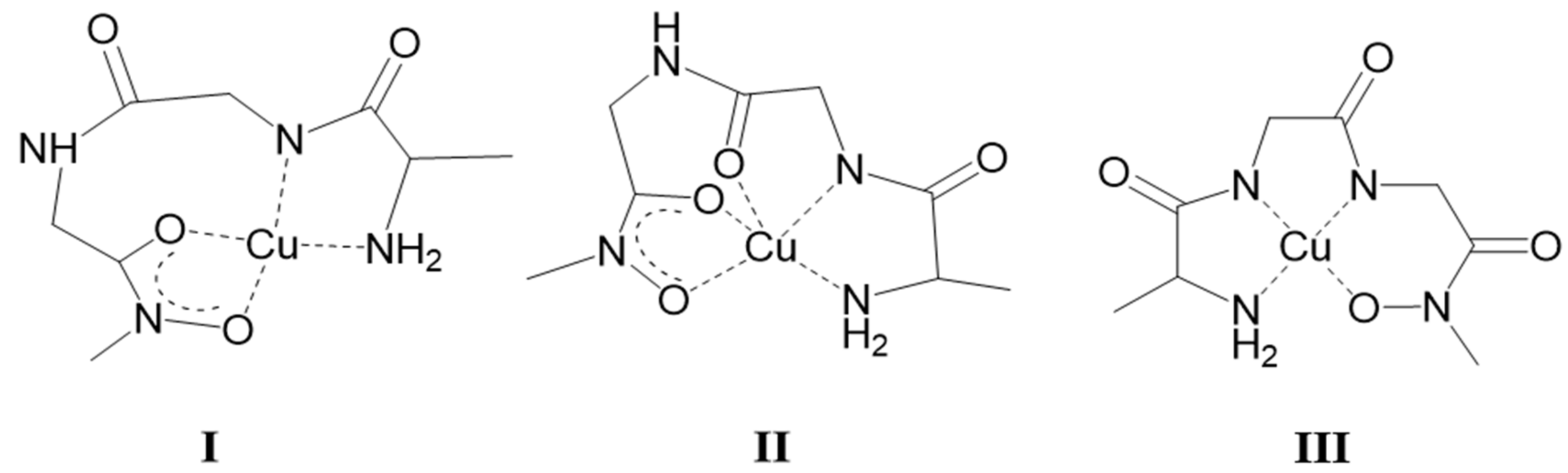
2.2. Complexation of AlaGlyGlyNHOH and AlaGlyGlyN(Me)OH with Selected 3d Ions—Fe(III), Ni(II), Cu(II), and Zn(II)
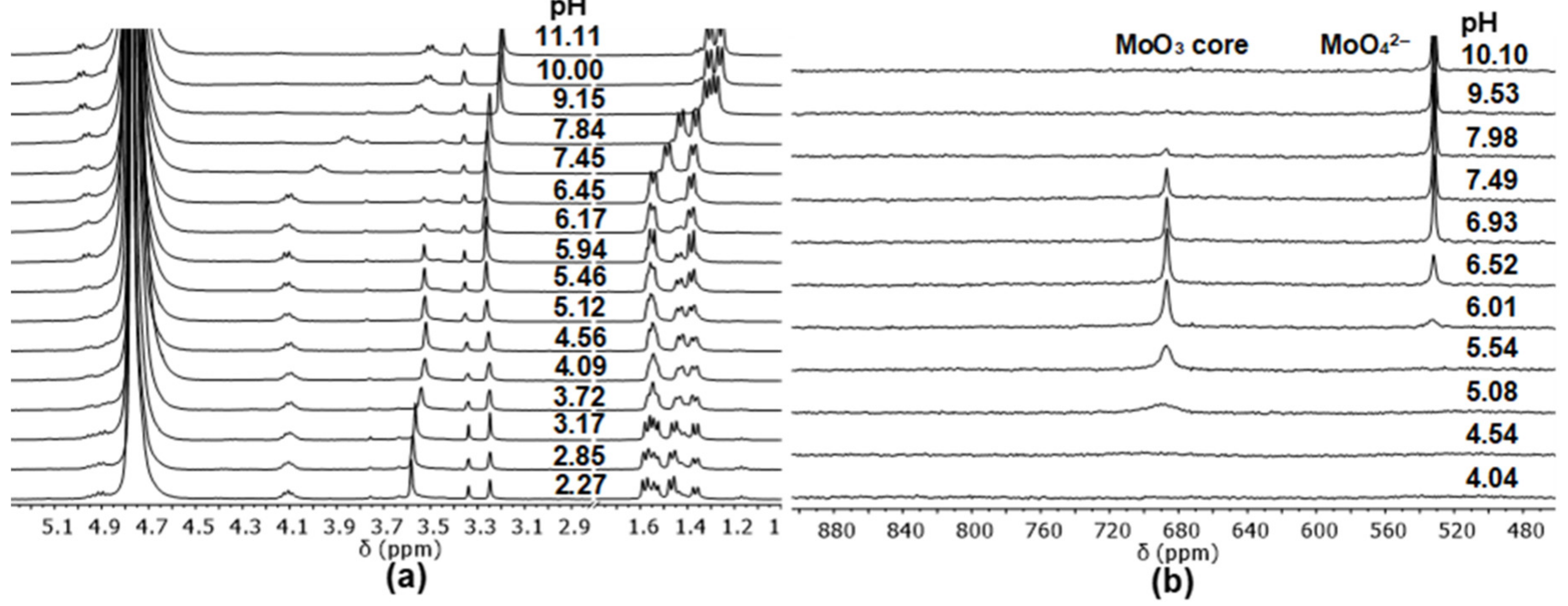
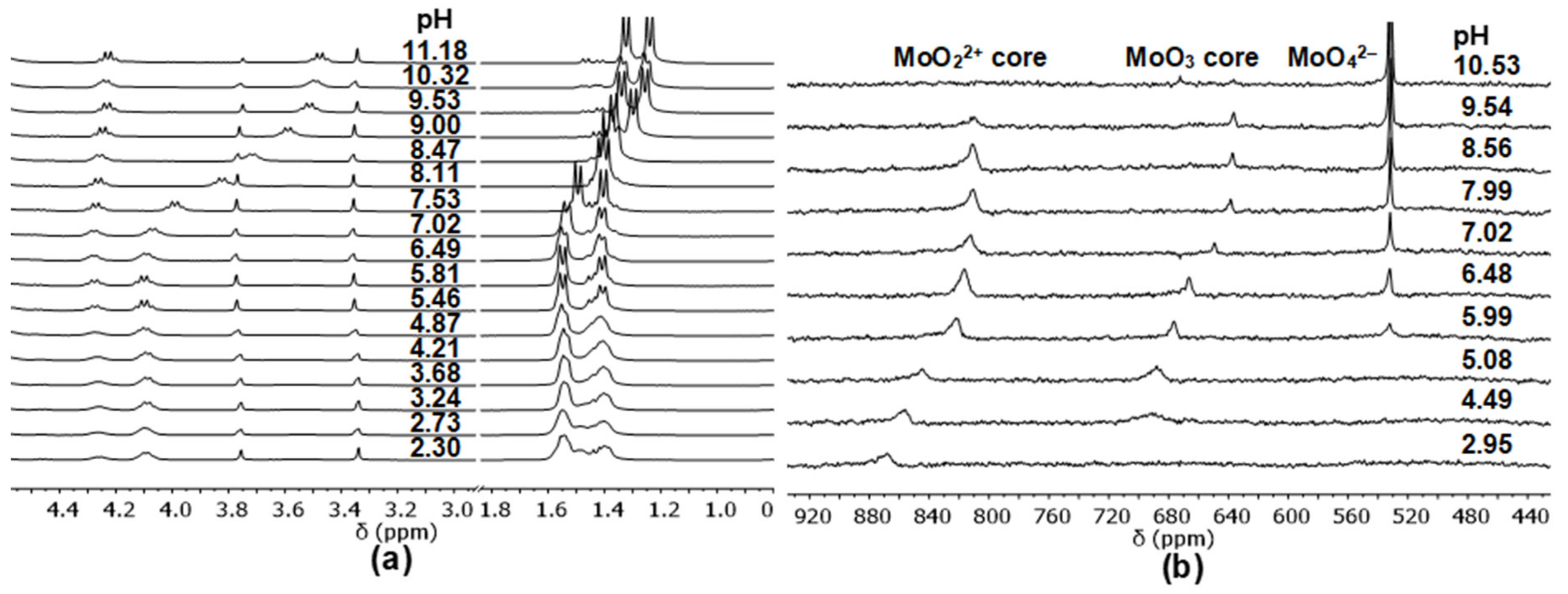
2.3. Complexation of AlaAlaNHOH, AlaAlaN(Me)OH, AlaGlyGlyNHOH, and AlaGlyGlyN(Me)OH with the Essential 4d Ion Mo(VI)
- 1.
- Although the characteristic signal of MoO42– appears at pH ca. 6 with this primary derivative (the same is seen in Figure 3b with the secondary counterpart), to some extent, the primary ligand remains in coordinative interaction with the MoO22+-core up to pH ca. 10. Moreover, the pH-dependence of the chemical shift of the quite broad signals of the complexes with MoO22+-core shows two inflexion points (two deprotonation processes). These experimental results support the formation of three different species with this core as a function of pH.
- 2.
- The very broad NMR signals of MoO3 oxygens are clearly observable at ca. 688 ppm at pH 4.49, and their chemical shifts show one inflexion point as a function of pH (indicating one deprotonation step and two types of species with MoO3 core). Furthermore, its intensity starts decreasing above pH 7 and almost disappears by pH 10.5.
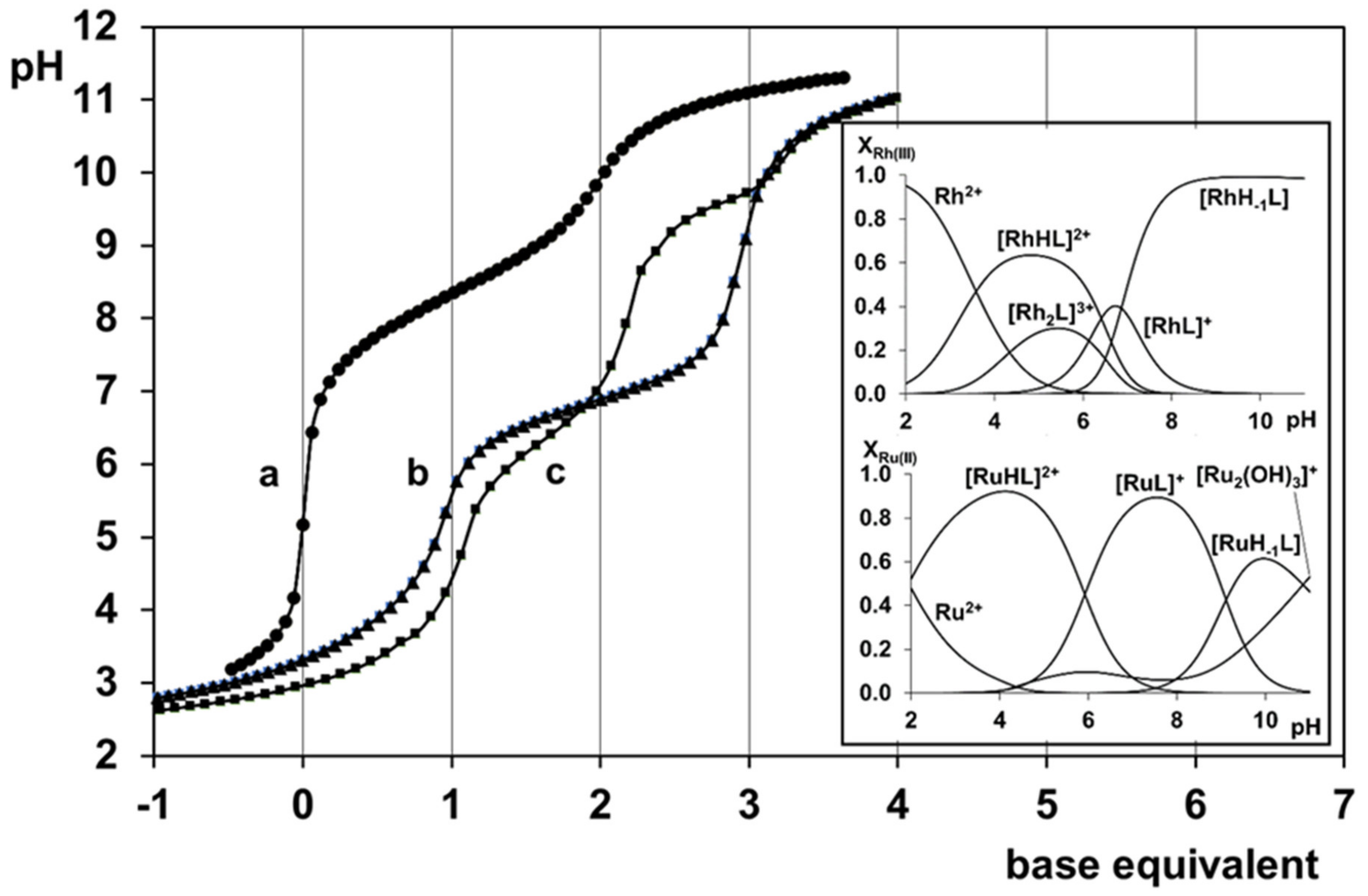
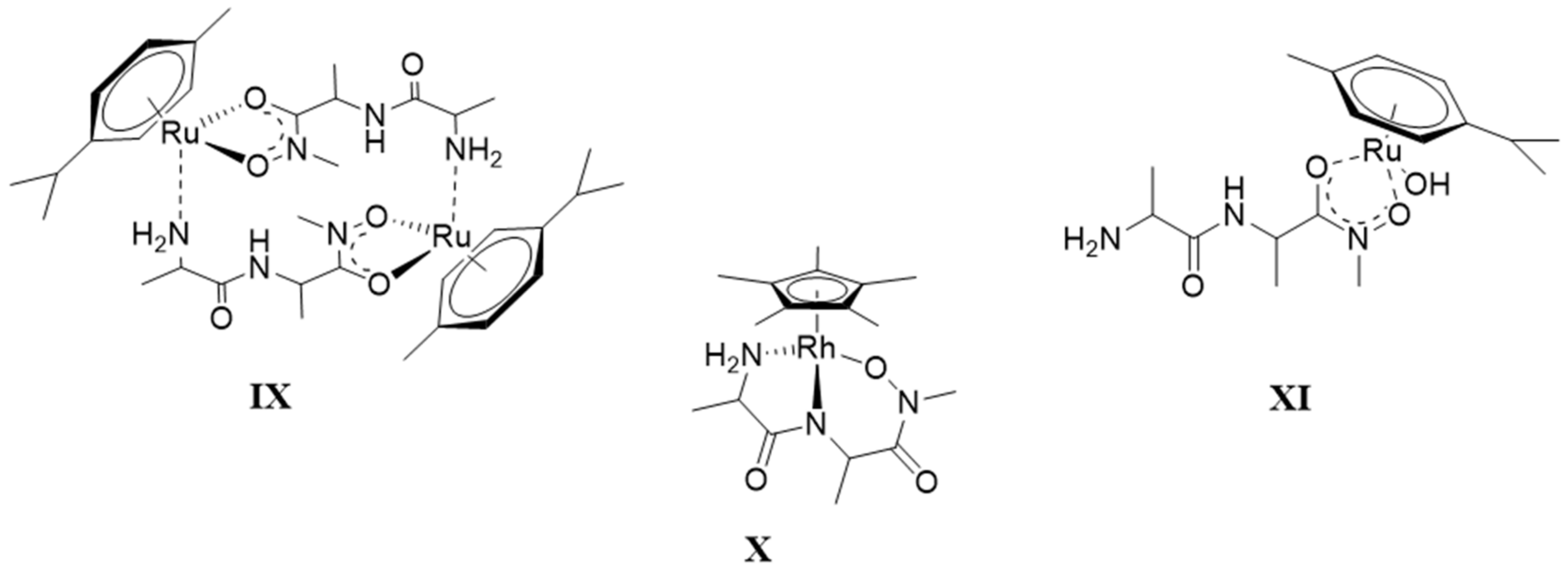
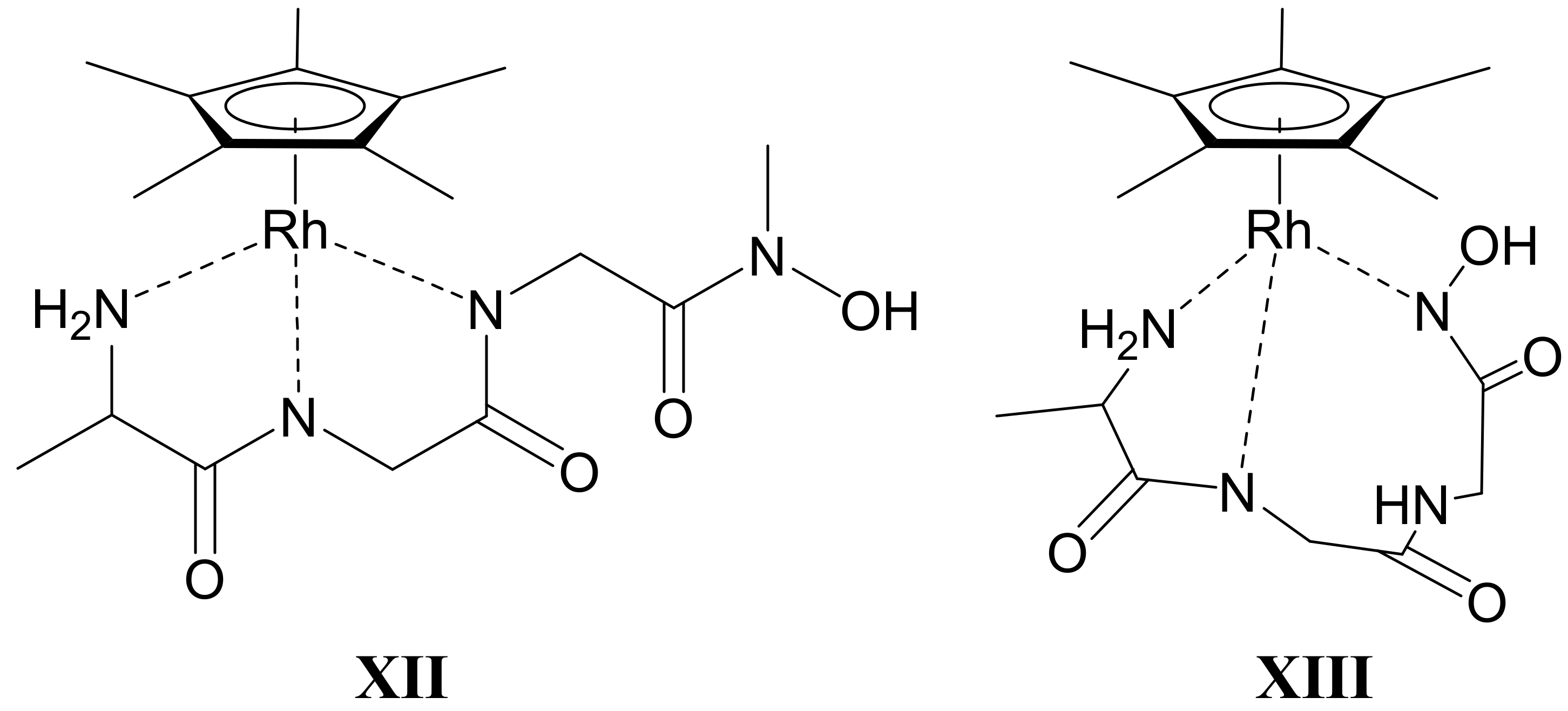
2.4. Complexation of AlaAlaNHOH or AlaAlaN(Me)OH with Two Half-Sandwich Type Cations—[(η6-p-cym)Ru(H2O)3]2+ or [(η5-Cp*)Rh(H2O)3]2+
2.5. Complexation of AlaGlyGlyNHOH or AlaGlyGlyN(Me)OH with the Two Half-Sandwich Type Cations—[(η6-p-cym)Ru(H2O)3]2+ or [(η5-Cp*)Rh(H2O)3]2+
3. Discussion
- 1.
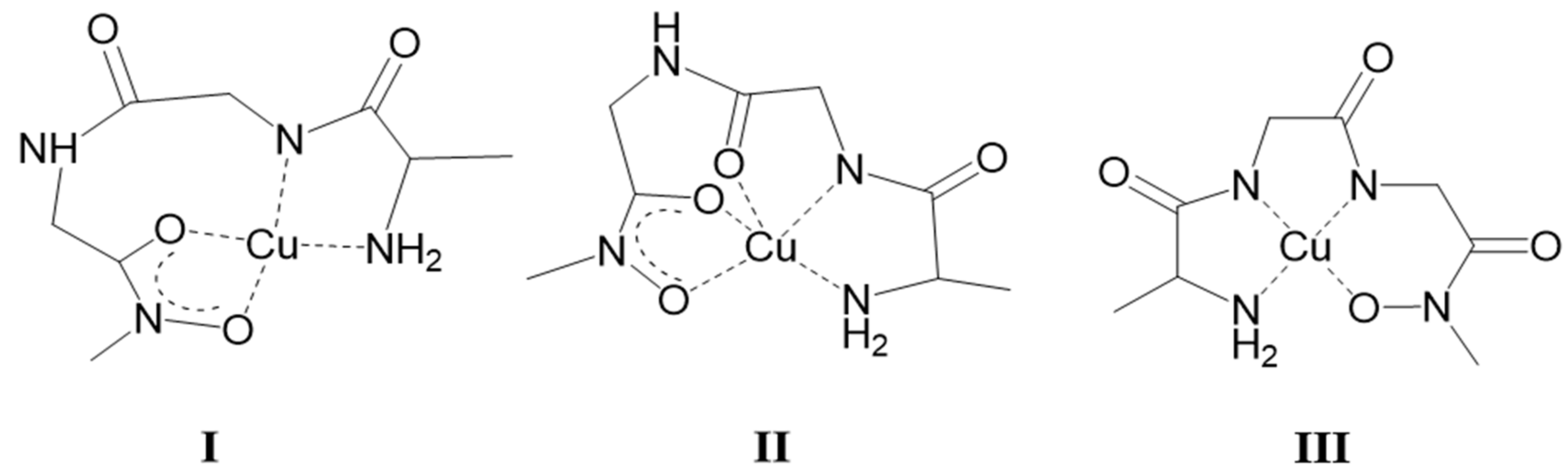
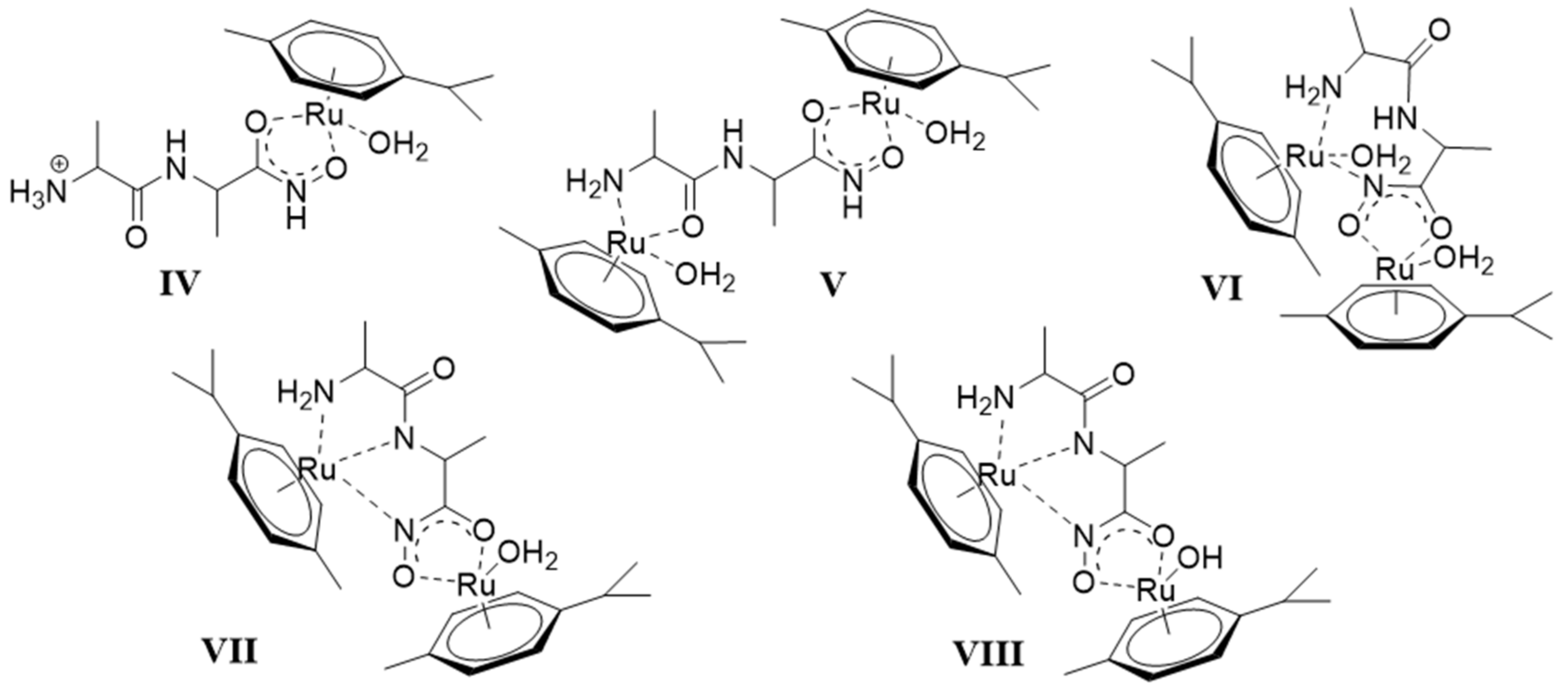
- Due to its highest conditional stability out of the possible ones under highly acidic conditions, the hydroxamate-type chelate, (O,O)hydr is the anchor with all the studied metal ions, Mo(VI), Fe(III), Cu(II), Ru(II), and Rh(III), with which the interaction starts at pH ca. 3 or below. The situation is somewhat different with Ni(II) and Zn(II), where the interaction starts at pH ca. 4 or somewhat above. In these latter systems, binding isomers can exist because the coordination starts either via the five-membered (O,O)hydr or five-membered (NH2,CO) chelate. For Zn(II), these initial interactions were followed by hydrolytic processes resulting in the formation of mixed hydroxido complexes.
- 2.
- Exclusive hydroxamate-type coordination could be detected in the whole pH-range with Fe(III) and Mo(VI) (or also hydroximate-type one in the case of Mo(VI) with primary derivatives at higher pH). No role of the amino-N or peptide amide-N(s) was found in the complexation with these two metals. Consequently, there is no any significant difference between their complexation with the studied dipeptide and tripeptide derivatives. However, significant difference was found between the Mo(VI)-binding ability of the primary and secondary derivatives, the former being much more effective Mo(VI)-chelators. This is due to the significantly higher stability of the doubly deprotonated hydroximato chelate (which was detected in the Mo(VI)–primary derivative ligand systems up to pH ca. 10). On the contrary, the mono-deprotonated (O,O)hydr chelate cannot compete with the hydrolysis of the Mo(VI) above the neutral pH.
- 3.
- Out of the investigated metals, Ni(II) showed the highest preference towards the N-donors over the hydroxamate oxygens. This allows only a minor role of the (O,O)hydr at the beginning of the interaction. After that, Ni(II) forms with the primary AlaGlyGlyNHOH in slow cooperative processes a planar, 4N-coordinated (NH2,Namide,Namide,Nhydr) 1:1 complex with very high stability, which exclusively exists above pH ca. 6. With the secondary counterpart, however, an (NH2,Namide,Namide,Ohydr)-coordinated planar complex is formed, with slightly lower stability and dominates only above pH ca. 7. The hydrolysis of the coordinated metal ion is hindered in both complexes, but, in the latter system, formation of bis-complexes in small extent can be observed in the presence of ligand excess. Nevertheless, even the secondary AlaGlyGlyN(Me)OH is a more effective ligand for Ni(II) (the situation is the same with Cu(II)) than the previously studied dipeptide derivatives [19,20].
- 4.
- Although the binding modes of the most stable Cu(II) complexes with AlaGlyGlyNHOH and with the secondary counterpart, the 4N-coordinated (NH2,Namide,Namide,Nhydr) and (NH2,Namide,Namide,Ohydr) planar 1:1 complexes, respectively, are the same as those with Ni(II); moreover their stability constants are significantly higher than those of the corresponding Ni(II) complexes (see Table 2), these species become predominant in the Cu(II)-containing systems in the higher pH-range only (above pH 8 and 9, respectively). This happens because the (O,O)hydr can play a crucial role in the interaction with Cu(II), and this is not the case for Ni(II). As a result of the measurable competition between the different types of potential donors even at neutral pH, the processes become very slow especially in the Cu(II)–primary tripeptide derivative system (too slow to allow conventional titrimetry). There is some dominance of the formation of oligonuclear (mostly dimeric or trimeric) complexes and, mainly with the secondary derivative, the formation of bis-hydroxamato intermediate complexes is also considerable.
- 5.
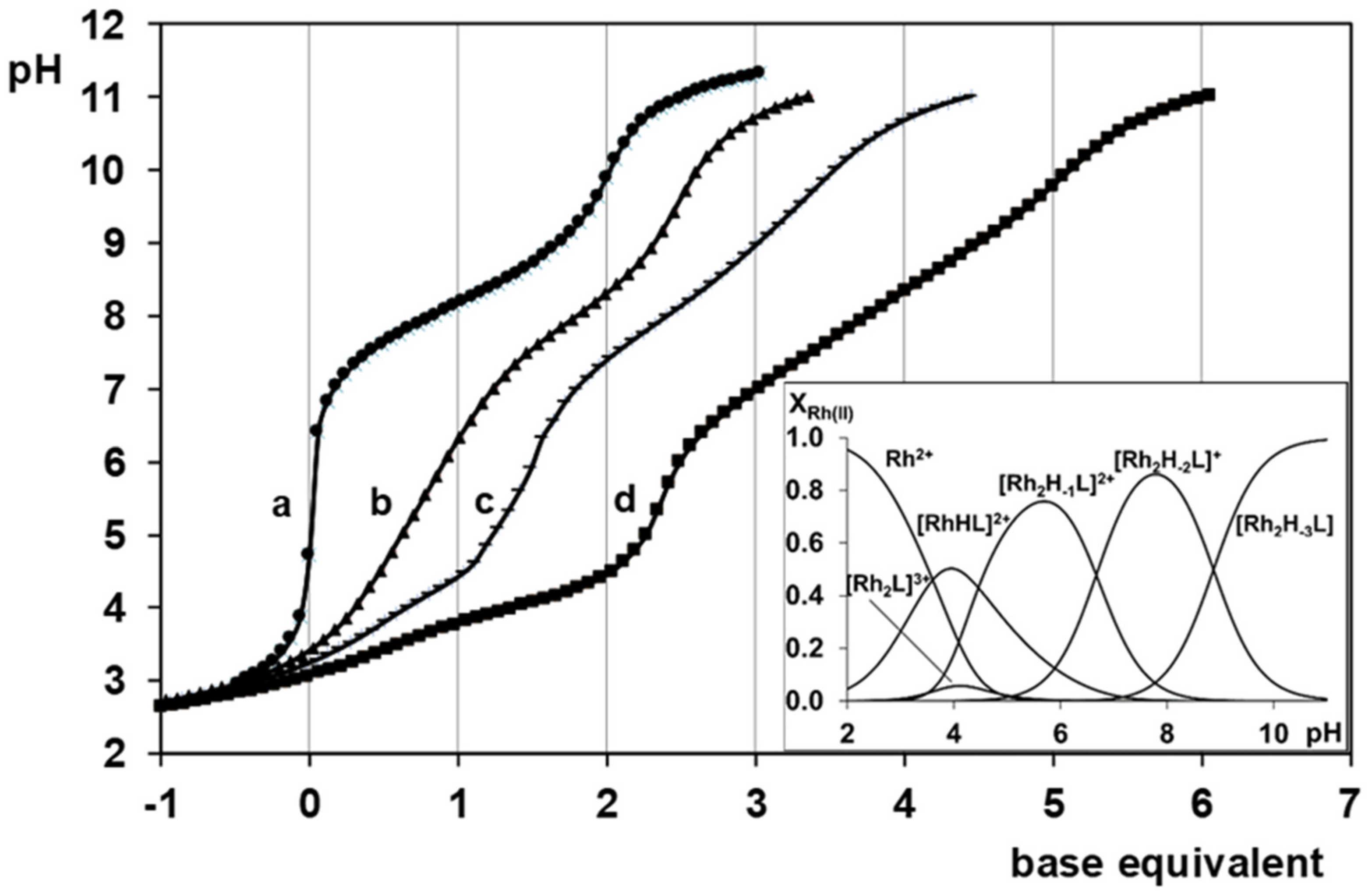
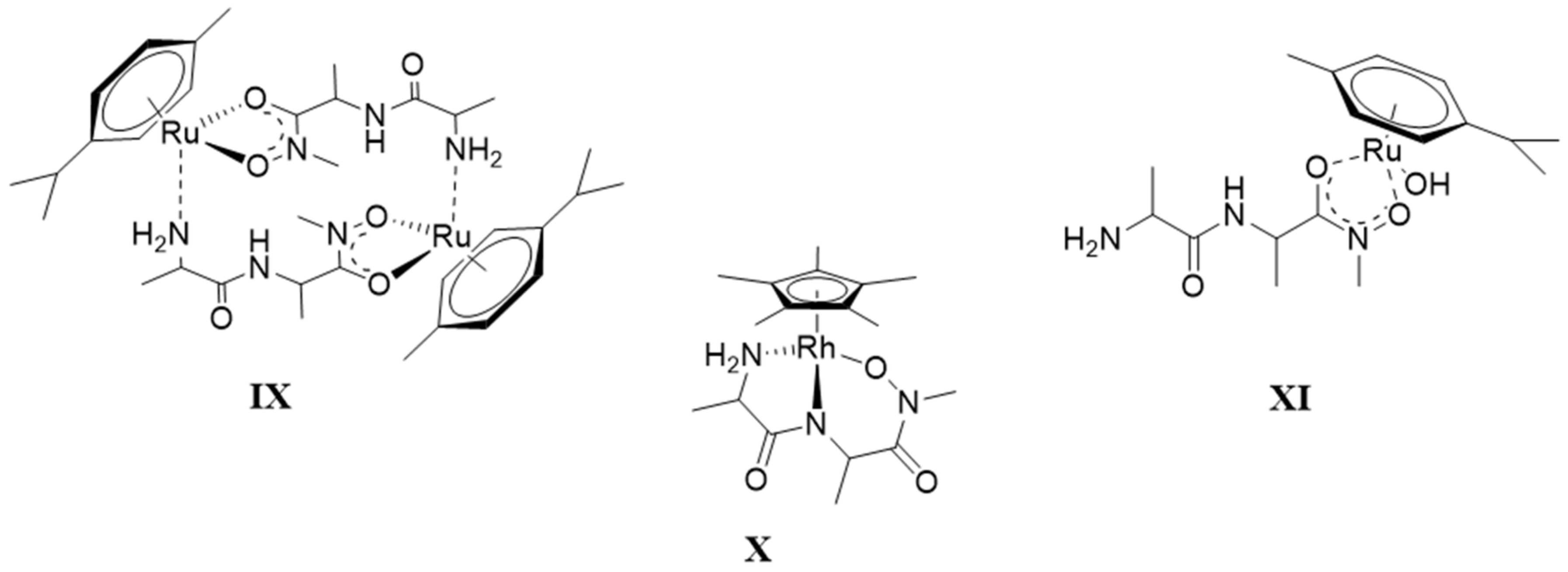
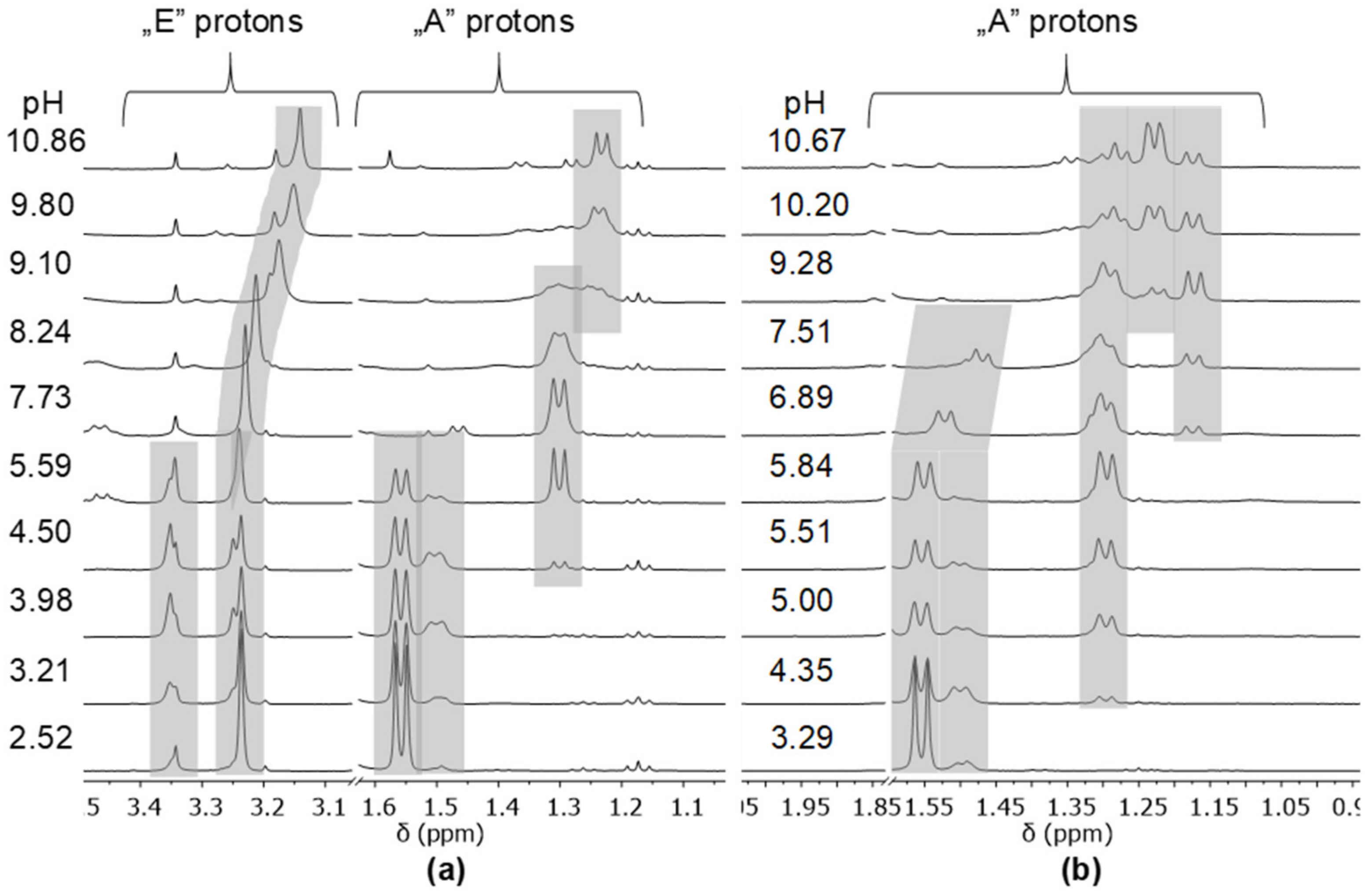
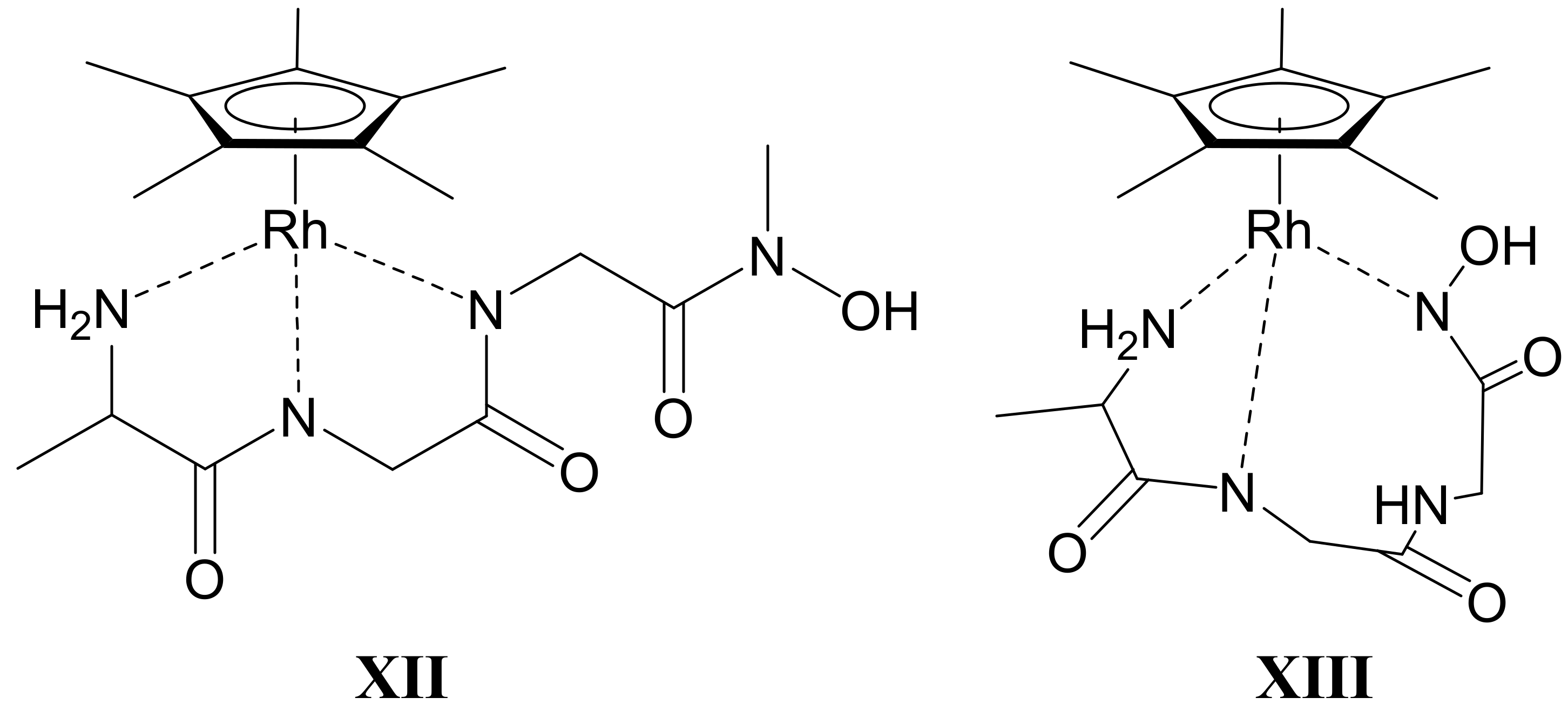
- There are only maximum three sites available for coordination for the studied half-sandwich type Ru(II) and Rh(III) cations. There is no doubt that with both cations and all four ligands the (O,O)hydr anchor-chelate can remain in the coordination sphere up to pH ca. 7 but is displaced by nitrogen donors, if there is no possibility for the formation of the hydroximate-(O,O) chelate, with the doubly deprotonated hydroxamic function. If, however, it can be formed, it remains in the coordination sphere, which is the case with the primary dipeptide derivative AlaAlaNHOH, and the N-donors can only coordinate to another metal ion. Consequently, dinuclear complexes predominate from slightly acidic conditions until the end of the investigated pH-range with both cations. In the most stable complex, the donor set (NH2,Namide,Nhydr) saturates the coordination sites of one metal, while the (O,O)hydr chelate and either a water, or above pH ca. 9–10, a hydroxide ion binds to another metal ion. On the contrary, mononuclear complexes dominate with the secondary counterpart, AlaAlaN(Me)OH, being the most stable an (NH2,Namide,Ohydr)-coordinated complex with the [(η5-Cp*)Rh(H2O)3]2+ cation. Due to the significantly higher affinity of [(η6-p-cym)Ru(H2O)3]2+ toward hydrolysis, it forms a mixed hydroxido complex involving an (NH2,Namide) chelate and a monodentate OH−.
- 6.
- The complex formation processes were too slow between [(η6-p-cym)Ru(H2O)3]2+ and the two tripeptide derivatives, AlaGlyGlyNHOH and AlaGlyGlyN(Me)OH, above pH ca. 6, where the interaction with the N-donors started. Consequently, solution equilibrium measurements were possible only on the [(η5-Cp*)Rh(H2O)3]2+-containing systems. With the secondary derivative containing three potential nitrogen donors, the initial (O,O)hydr chelate in the highly acidic pH range is partially replaced by the coordination of the NH2 group (most probably together with the neighboring CO) above pH ca. 4. This results in the formation of dinuclear species with mixed (O,O)hydr and (NH2, CO) coordination below pH ca. 7–7.5 as minor complexes, but mononuclear species with (NH2,Namide,Namide) binding mode (having the hydroxamate function is uncoordinated) dominate even in this intermediate pH-range.
- 7.
- The primary AlaGlyGlyNHOH contains four potential N-donors, which is more than enough to saturate the coordination sphere of a [(η5-Cp*)Rh(H2O)3]2+ cation. The results show a measurable competition between the C-terminal Namide and Nhydr between pH ca. 7–10, but above this pH, the complex with the (NH2,Namide,Namide) donor set, containing the hydroxamate function in uncoordinated form, has increasing dominance.
4. Materials and Methods
4.1. Materials and Stock Solutions
4.2. Solution Studies
[Fe(OH)]2+ logβ1,−1,0 = −3.21; [Fe(OH)2]+ logβ1,−2,0 = −6.73; [Fe2(OH)2]4+ logβ2,−2,0 = 4.09; [Fe3(OH)4]5+ logβ3,−4,0 = −7.58 [38].[HMoO4]– logβ1,1,0 = 4.03; [H2MoO4] logβ1,2,0 = 6.70; [H8(MoO4)7]6− logβ7,8,0 = 53.18; [H9(MoO4)7]5− logβ7,9,0 = 58.10; [H10(MoO4)7]4– logβ7,10,0 = 62.11; [H11(MoO4)7]3− logβ7,11,0 = 64.54 [30].[{(η5-Cp*)Rh}2(µ2-OH)2]2+ (logβ2,−2,0 = −8.53); [{(η5-Cp*)Rh}2(µ2-OH)3]+ (logβ2,−3,0 = −14.26) [39].
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Codd, R. Traversing the coordination chemistry and chemical biology of hydroxamic acids. Coord. Chem. Rev. 2008, 252, 1387–1408. [Google Scholar] [CrossRef]
- Gupta, S.P.; Sharma, A. The Chemistry of Hydroxamic Acids. In Hydroxamic acids: A unique Family of Chemicals with Multiple Biological Activities; Gupta S.P, Ed.; Springer: Berlin, Germany, 2013; pp. 1–17. [Google Scholar]
- Albrecht-Gary, A.M.; Crumbliss, A.L. Coordination chemistry of siderophores: thermodynamics and kinetics of iron chelation and release. In Metal Ions in Biological Systems; Sigel, A., Sigel, H., Eds.; Marcel Dekker: New York, NY, USA, 1998; Volume 35, pp. 239–327. [Google Scholar]
- Muri, E.M.F.; Nieto, M.J.; Sindelar, R.D.; Williamson, J.S. Hydroxamic acids as pharmacological agents. Curr. Med. Chem. 2002, 9, 1631–1653. [Google Scholar] [CrossRef] [PubMed]
- Marmion, C.J.; Parker, J.P.; Nolan, K.B. Hydroxamic acids: An Important Class of Metalloenzyme Inhibitors. In Comprehensive Inorganic Chemistry II: From Elements to Applications, 2nd ed.; Reedijk, J., Poeppelmeier, K.R., Eds.; Elsevier Ltd.: Amsterdam, The Netherlands, 2013; Volume 3, pp. 683–708. [Google Scholar] [CrossRef]
- Marmion, C.J.; Murphy, T.; Docherty, J.R.; Nolan, K.B. Hydroxamic acids are nitric oxide donors. Facile formation of ruthenium(II)-nitrosyls and NO-mediated activation of guanylate cyclase by hydroxamic acids. Chem. Commun. 2000, 1153–1154. [Google Scholar] [CrossRef]
- Kohli, R. Intra-and intermolecular hydrogen bonding in formohydroxamic acid. In Hydrogen bonding abilities of hydroxamic acid and its isosteres; Anchor Academic Publishing: Hamburg, Germany, 2016; pp. 58–112. [Google Scholar]
- Carrott, M.J.; Fox, O.D.; LeGurun, G.; Jones, C.J.; Mason, C.; Taylor, R.J.; Andrieux, F.P.L.; Boxall, C. Oxidation–reduction reactions of simple hydroxamic acids and plutonium(IV) ions in nitric acid. Radiochim. Acta 2008, 96, 333–343. [Google Scholar] [CrossRef]
- Farkas, E.; Enyedy, É.A. Interaction between iron(II) and hydroxamic acids: oxidation of iron(II) to iron(III) by desferrioxamine B under anaerobic conditions. J. Inorg. Biochem. 2001, 83, 107–114. [Google Scholar] [CrossRef]
- Kurzak, B.; Kozlowski, H.; Farkas, E. Hydroxamic and aminohydroxamic acids and their complexes with metal ions. Coord. Chem. Rev. 1992, 114, 169–2000. [Google Scholar] [CrossRef]
- Farkas, E.; Csóka, H.; Bell, G.; Brown, D.A.; Cuffe, L.P.; Fitzpatrick, N.J.; Glass, W.K.; Errington, W.; Kemp, T.J. Oxygen versus nitrogen co-ordination in complexes of MoVI and hydroxamate derivatives of α-amino acids: equilibrium, structural and theoretical studies. J. Chem. Soc. Dalton Trans. 1999, 16, 2789–2794. [Google Scholar] [CrossRef]
- Parajdi-Losonczi, P.L.; Bényei, A.C.; Kováts, É.; Timári, I.; Muchova Radosova, T.; Novohradsky, V.; Kasparkova, J.; Buglyó, P. [(η6-p-cymene)Ru(H2O)3]2+ binding capability of aminohydroxamates — A solution and solid state study. J. Inorg. Biochem. 2016, 160, 236–245. [Google Scholar] [CrossRef]
- Parajdi-Losonczi, P.L.; Buglyó, P.; Skakalova, H.; Kasparkova, J.; Lihi, N.; Farkas, E. Half-sandwich type rhodium(III)-aminohydroxamate complexes: the role of the position of the amino group in metal binding. New J. Chem. 2018, 42, 7659–7670. [Google Scholar] [CrossRef]
- Kurzak, B.; Farkas, E.; Glowiak, T.; Kozlowski, H. X-Ray and potentiometric studies on a pentanuclear copper(II) complex with β-alaninehydroxamic acid. J. Chem. Soc., Dalton Trans. 1991, 2, 163–167. [Google Scholar] [CrossRef]
- Tegoni, M.; Remelli, M. Metallacrowns of copper(II) and aminohydroxamates: Thermodynamics of self assembly and host–guest equilibria. Coord. Chem. Rev. 2012, 56, 289–315. [Google Scholar] [CrossRef]
- Ostrowska, M.; Fritsky, I.O.; Gumienna-Kontecka, E.; Pavlishchu, A.V. Metallacrown-based compounds: Applications in catalysis, luminescence, molecular magnetism, and adsorption. Coord. Chem. Rev. 2016, 327, 304–332. [Google Scholar] [CrossRef]
- Farkas, E.; Kiss, I. Complexes of peptidehydroxamates. Complex formation between transition metals and L-prolyl-L-leucylglycinehydroxamic acid [N-hydroxy-7-methyl-4-oxo-5-(pyrrolidine-2’-carboxamido)-3-azaoctanamide] and L-prolyl-L-leucinehydroxamic acid [N-hydroxy-4-methyl-2-(pyrrolidine-2’-carboxamido)pentanamide]. J. Chem. Soc. Dalton Trans. 1990, 3, 749–753. [Google Scholar] [CrossRef]
- Farkas, E.; Csapó, E.; Buglyó, P.; Damante, C.A.; Di Natale, G. Metal binding ability of histidine-containing peptidehydroxamic acids: Imidazole versus hydroxamate coordination. Inorg. Chim. Acta 2009, 362, 753–762. [Google Scholar] [CrossRef]
- Buglyó, P.; Nagy, E.M.; Farkas, E.; Sóvágó, I.; Sanna, D.; Micera, G. New insights into the metal ion-peptidehydroxamate interactions: Metal complexes of primary hydroxamic acid derivatives of common dipeptides in aqueous solution. Polyhedron 2007, 26, 1625–1633. [Google Scholar] [CrossRef]
- Buglyó, P.; Nagy, E.M.; Sóvágó, I.; Ozsváth, A.; Sanna, D.; Farkas, E. Metal ion binding capability of secondary (N-methyl) versus primary (N-H) dipeptide hydroxamic acids. Polyhedron 2016, 110, 172–181. [Google Scholar] [CrossRef]
- Ozsváth, A.; Farkas, E.; Diószegi, R.; Buglyó, P. Versatility and trends in the interaction between Pd(II) and peptide hydroxamic acids. New. J. Chem. 2019, 43, 8239–8249. [Google Scholar] [CrossRef]
- Moore, W.M.; Spilburg, C.A. Purification of human collagenases with a hydroxamic acid affinity column. Biochemistry 1986, 25, 5189–5195. [Google Scholar] [CrossRef]
- Melchart, M.; Sadler, P.J. Ruthenium Arene Anticancer Complexes. In Bioorganometallics: Biomolecules, Labeling, Medicine; Jaouen, G., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2005; pp. 39–64. [Google Scholar] [CrossRef]
- Mészáros, J.P.; Poljarevic, J.M.; Gál, G.T.; May, N.V.; Spengler, G.; Enyedy, É.A. Comparative solution and structural studies of half-sandwich rhodium and ruthenium complexes bearing curcumin and acetylacetone. J. Inorg. Biochem. 2019, 195, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Tejada-Jimenez, M.; Chamizo-Ampudia, A.; Calatrava, V.; Galvan, A.; Fernandez, E.; Llamas, A. From the Eukaryotic Molybdenum Cofactor Biosynthesis to the Moonlighting Enzyme mARC. Molecules 2018, 23, 3287. [Google Scholar] [CrossRef]
- Leimkühler, S.; Wuebbens, M.M.; Rajagopalan, K.V. The history of the discovery of the molybdenum cofactor and novel aspects of its biosythesis in bacteria. Coord. Chem. Rev. 2011, 255, 1129–1144. [Google Scholar] [CrossRef] [PubMed]
- Enyedy, É.A.; Csóka, H.; Lázár, I.; Micera, G.; Garibba, E.; Farkas, E. Effects of side-chain aminonitrogen donor atoms on metal complexation of aminohydroxamic acids: New diaminohydroxamates chelating Ni(II) more strongly than Fe(III). J. Chem. Soc. Dalton Trans. 2002, 13, 2632–2640. [Google Scholar] [CrossRef]
- Yokoi, H. ESP and optical absorption studies of various bis(N-salicylidenealkylaminato)copper(II) complexes with terahedrally-distoted coordination geometry. Bull. Chem. Soc. Japan 1974, 47, 3037–3040. [Google Scholar] [CrossRef]
- Yokoi, H.; Addison, A.W. Spectroscopic and redox properties of pseudotetrahedral copper(II) complexes. Their relation to copper proteins. Inorg. Chem. 1977, 16, 1341–1349. [Google Scholar] [CrossRef]
- Farkas, E.; Csóka, H.; Tóth, I. New insights into the solution equilibrium of molybdenum(VI)-hydroxamate systems: 1H and 17O-NMR spectroscopic study of Mo(VI)-desferrioxamine B and Mo(VI)-monohdroxamic acid systems. Dalton Trans. 2003, 8, 1645–1652. [Google Scholar] [CrossRef]
- Buglyo, P.; Parajdi-Losonczi, P.L.; Benyei, A.C.; Lihi, N.; Biro, L.; Farkas, E. Versatility of Coordination Modes in Complexes of Monohydroxamic Acids with Half-Sandwich Type Ruthenium, Rhodium, Osmium and Iridium Cations. Chem. Select 2017, 26, 8127–8136. [Google Scholar] [CrossRef]
- Bíró, L.; Farkas, E.; Buglyó, P. Hydrolytic behaviour and chloride ion binding capability of [Ru(η6-p-cym)(H2O)3]2+: A solution equilibrium study. Dalton Trans. 2012, 41, 285–299. [Google Scholar] [CrossRef]
- Gran, G. Determination of the Equivalent Point in Potentiometric Titrations. Acta Chem. Scand. 1950, 4, 559–577. [Google Scholar] [CrossRef]
- Bennett, M.A.; Smith, A.K. Arene ruthenium(II) complexes formed by dehydrogenation of cyclohexadienes with ruthenium(III) trichloride. J. Chem. Soc. Dalton Trans. 1974, 233–241. [Google Scholar] [CrossRef]
- Irving, H.M.; Miles, M.G.; Pettit, L.D. A study of some problems in determining the stoicheiometric proton dissociation constants of complexes by potentiometric titrations using a glass electrode. Anal. Chim. Acta 1967, 38, 475–488. [Google Scholar] [CrossRef]
- Zékány, I.; Nagypál, I. PSEQUAD: A Comprehensive Program for the Evaluation of Potentiometric and/or Spectrophotometric Equilibrium Data Using Analytical Derivatives. In Computational Methods for the Determination of Formation Constants; Leggett, D.J., Ed.; Plenum: New York, NY, USA, 1985; pp. 291–352. [Google Scholar]
- Gans, P.; Sabatini, A.; Vacca, A. SUPERQUAD: An improved general program for computation of formation constants from potentiometric data. J. Chem. Soc. Dalton Trans. 1985, 6, 1195–1200. [Google Scholar] [CrossRef]
- Baes, C.F.; Mesmer, R.E. Iron. In The Hydrolysis of Cations; John Wiley & Sons: Hoboken, NJ, USA, 1976; pp. 226–237. [Google Scholar]
- Dömötör, O.; Aicher, S.; Schmidlehner, M.; Novak, M.S.; Roller, A.; Jakupec, M.A.; Kandioller, W.; Hartinger, C.G.; Keppler, B.K.; Enyedy, É.A. Antitumor pentamethylcyclopentadienyl rhodium complexes of maltol and allomaltol: synthesis, solution speciation and bioactivity. J. Inorg. Biochem. 2014, 134, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Krezel, A.; Bal, W. A formula for correlating pKa values determined in D2O and H2O. J. Inorg. Biochem. 2004, 98, 161–166. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| I = 0.20 M KCl | I = 0.20 M KNO3 | |||
|---|---|---|---|---|
| Ligand | pK1 | pK2 | pK1 | pK2 |
| H-AlaAlaNHOH+ | 7.66 1 | 8.88 1 | 7.77(1) | 8.89(1) |
| H-AlaAlaN(Me)OH+ | 7.74 2 | 8.74 2 | 7.77(1) | 8.70(1) |
| H-AlaGlyGlyNHOH+ | 7.71(1) | 8.82(1) | 7.74(1) | 8.82(1) |
| H-AlaGlyGlyN(Me)OH+ | 7.66(1) | 8.63(1) | 7.69(1) | 8.57(1) |
| AlaAlaNHOH 1 | AlaAlaN(Me)OH 2 | AlaGlyGlyNHOH | AlaGlyGlyN(Me)OH | |
|---|---|---|---|---|
| [FeHL]3+ | 17.62 a | 18.08 a | 17.74(1) a | 17.64(1) a |
| [FeL]2+ | 15.09 | 14.67 | – | – |
| [FeH2L2]3+ | 34.57 | 33.8 | 34.02(5) | 33.70(7) |
| 34.00 a | 34.71 a | 34.00(2) a | 33.76(1) a | |
| [FeH3L3]3+ | 49.40 | 50.01 | 48.7(2) | 48.97(11) |
| 48.61 a | 49.51 a | 48.55(3) a | 48.16(2) a | |
| [NiHL]2+ | 12.50 | – | 12.79(3) | 11.6(2) |
| [NiL]+ | 6.30 | 6.01 | – | 5.04(7) |
| [NiH−1L] | −1.10 | – | – | −3.3(1) |
| [NiH−2L]− | −9.57 | – | −7.44(2) | −10.78(3) |
| [NiHL2]+ | – | 16.8 | – | 17.10(9) |
| [NiL2] | – | 9.13 | – | – |
| [CuHL]2+ | 14.70 | 15.02 | 14.45(7) | 14.66(2) |
| [Cu2L2]2+ | 23.20 | – | – | – |
| [CuH−1L] | 4.43 | 3.04 | – | 3.52(2) |
| [CuH−2L]− | −5.24 | −6.63 | −5.38(5) | −5.28(2) |
| [CuH2L2]2+ | – | 29.17 | – | 27.6(2) |
| [CuHL2]+ | – | 22.3 | – | 22.23(9) |
| [CuL2] | – | 14.8 | – | 14.8(1) |
| [CuH−1L2]− | – | 6.3 | – | – |
| [Cu3H−4L2] | 5.89 | – | – | – |
| [Cu3H−5L2]− | −3.56 | – | – | – |
| [Cu3H−6L2]2− | −14.28 | – | – | – |
| [ZnHL]2+ | 12.35 | 12.00 | 12.47(2) | 11.92(2) |
| [ZnL]+ | 5.64 | 5.11 | 5.07(6) | 4.55(2) |
| [ZnH−1L] | −2.58 | −3.23 | −3.18(2) | −3.46(3) |
| [ZnHL2]+ | 17.52 | 17.12 | – | – |
| Species | AlaAlaNHOH | AlaAlaN(Me)OH | AlaGlyGlyNHOH | AlaGlyGlyN(Me)OH |
|---|---|---|---|---|
| [MoO2(HL)2]2+ | 46.5(1) | 47.78(7) | 48.57(9) | 46.44(2) |
| [MoO3(HL)] | 23.89(7) | 24.52(5) | 24.37(7) | 23.92(2) |
| Fitting parameter (mL)a | 0.00317 | 0.00596 | 0.00662 | 0.00226 |
| Number of fitted data | 422 | 461 | 195 | 271 |
| Species | AlaAlaNHOH | AlaAlaN(Me)OH | ||
|---|---|---|---|---|
| [(η6-p-cym)Ru]2+ | [(η5-Cp*)Rh]2+ | [(η6-p-cym)Ru]2+ | [(η5-Cp*)Rh]2+ | |
| [MHL]2+ | 16.09(2) | 15.54(1) | 17.53(2) | 15.89(1) |
| [ML]+ | – | – | 11.61(3) | 9.48(3) |
| [MH−1L] | – | – | 2.50(5) | 2.62(2) |
| [M2L]3+ | 14.95(10) | 13.57(8) | – | 14.16(2) |
| [M2H−1L]2+ | 11.16(5) | 10.07(1) | – | – |
| [M2H−2L]+ | 4.63(6) | 3.40(3) | – | – |
| [M2H−3L] | –4.79(9) | –5.47(4) | – | – |
| Fitting parameter (mL)a | 0.00683 | 0.00417 | 0.00707 | 0.00575 |
| Number of fitted data | 302 | 183 | 201 | 244 |
| pKMHL | – | – | 5.92 | 6.41 |
| pKML | – | – | 9.11 | 6.86 |
| Species | AlaGlyGlyN(Me)OH | AlaGlyGlyNHOH | ||
|---|---|---|---|---|
| logβ | Coordinated Donor Atoms | logβ | Coordinated Donor Atoms | |
| [MHL]2+ | 15.53(4) | (O,O)hydr | 15.35(4) | (O,O)hydr |
| [ML]+ | 10.56(7) | (NH2, Namide) | 10.42(6) | (NH2, Namide) |
| [MH−1L] | 2.42(6) | (NH2, Namide) | 3.42(4) | (NH2, Namide, Nhydr) |
| [MH−2L]‒ | –7.02(7) | (NH2, Namide, Namide) | –6.91(3) | (NH2, Namide, Namide) |
| [M2L]3+ | 14.0(1) | (O,O)hydr + (NH2, CO) | 13.6(1) | (O,O)hydr + (NH2, CO) |
| [M2H−1L]2+ | 9.62(5) | (O,O)hydr + (NH2, Namide) | 9.53(3) | (O,O)hydr + (NH2, Namide) |
| [M2H−2L]+ | ‒ | 4.12(3) | (O,O)hydr + (NH2, Namide, Nhydr) | |
| Fitting parameter (mL)a | 0.0123 | 0.00599 | ||
| Number of fitted data | 265 | 256 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ozsváth, A.; Bíró, L.; Nagy, E.M.; Buglyó, P.; Sanna, D.; Farkas, E. Trends and Exceptions in the Interaction of Hydroxamic Acid Derivatives of Common Di- and Tripeptides with Some 3d and 4d Metal Ions in Aqueous Solution. Molecules 2019, 24, 3941. https://doi.org/10.3390/molecules24213941
Ozsváth A, Bíró L, Nagy EM, Buglyó P, Sanna D, Farkas E. Trends and Exceptions in the Interaction of Hydroxamic Acid Derivatives of Common Di- and Tripeptides with Some 3d and 4d Metal Ions in Aqueous Solution. Molecules. 2019; 24(21):3941. https://doi.org/10.3390/molecules24213941
Chicago/Turabian StyleOzsváth, András, Linda Bíró, Eszter Márta Nagy, Péter Buglyó, Daniele Sanna, and Etelka Farkas. 2019. "Trends and Exceptions in the Interaction of Hydroxamic Acid Derivatives of Common Di- and Tripeptides with Some 3d and 4d Metal Ions in Aqueous Solution" Molecules 24, no. 21: 3941. https://doi.org/10.3390/molecules24213941