
Synthesis of Resins Using Epoxies and Humins as Building Blocks: A Mechanistic Study Based on In-Situ FT-IR and NMR Spectroscopies



Abstract
:1. Introduction
2. Results
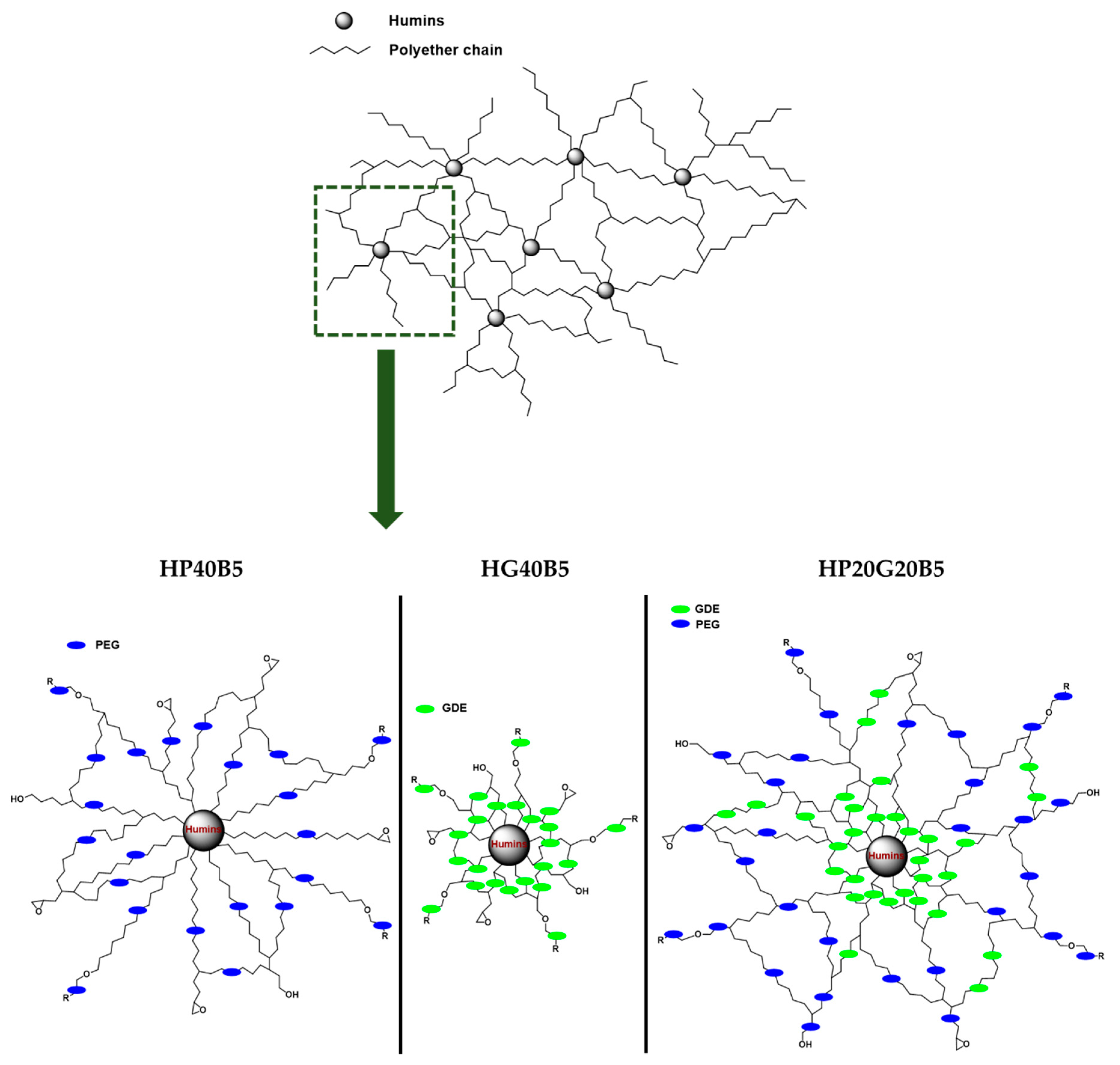
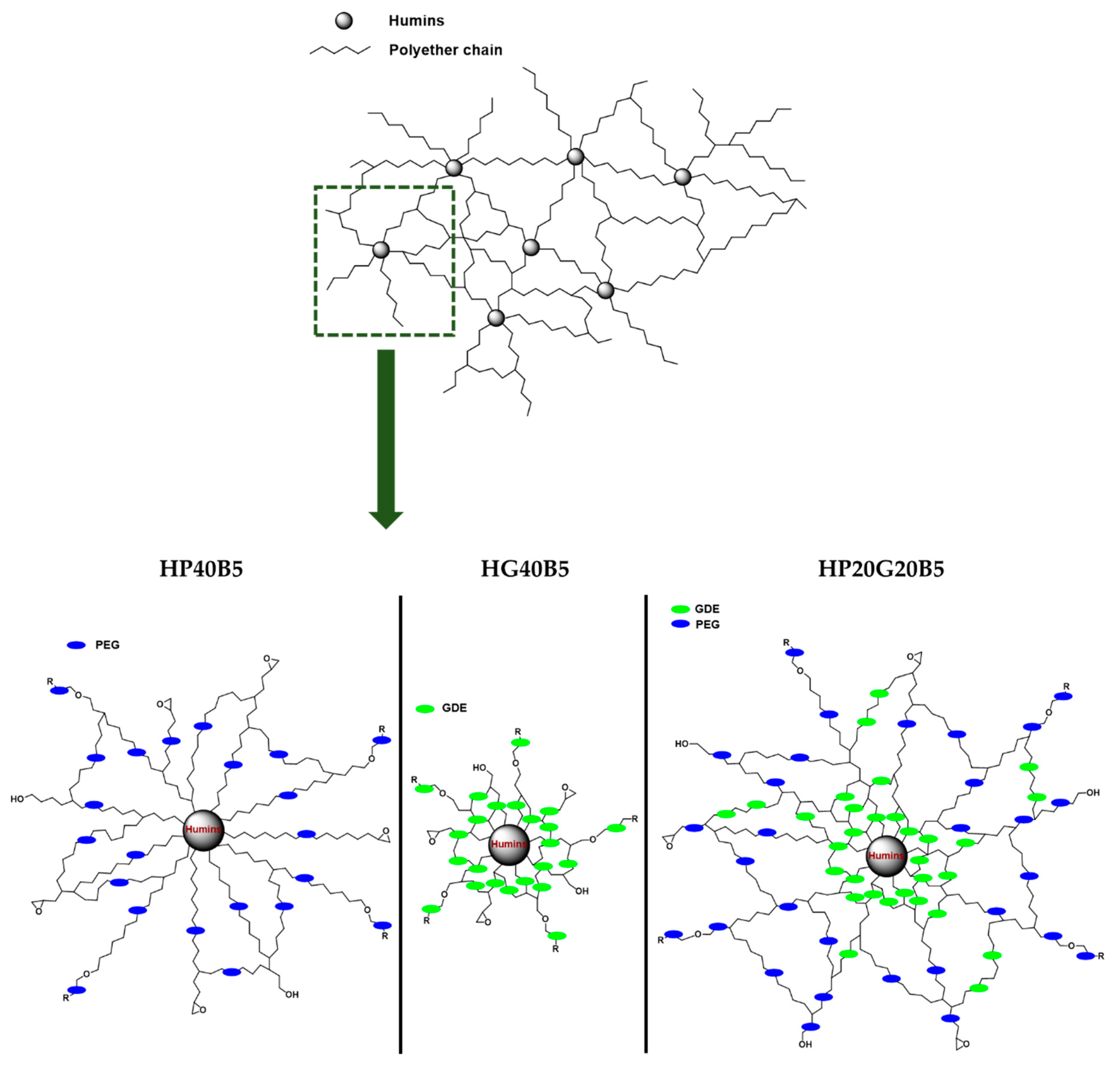
Synthesis of Copolymers
3. Discussion
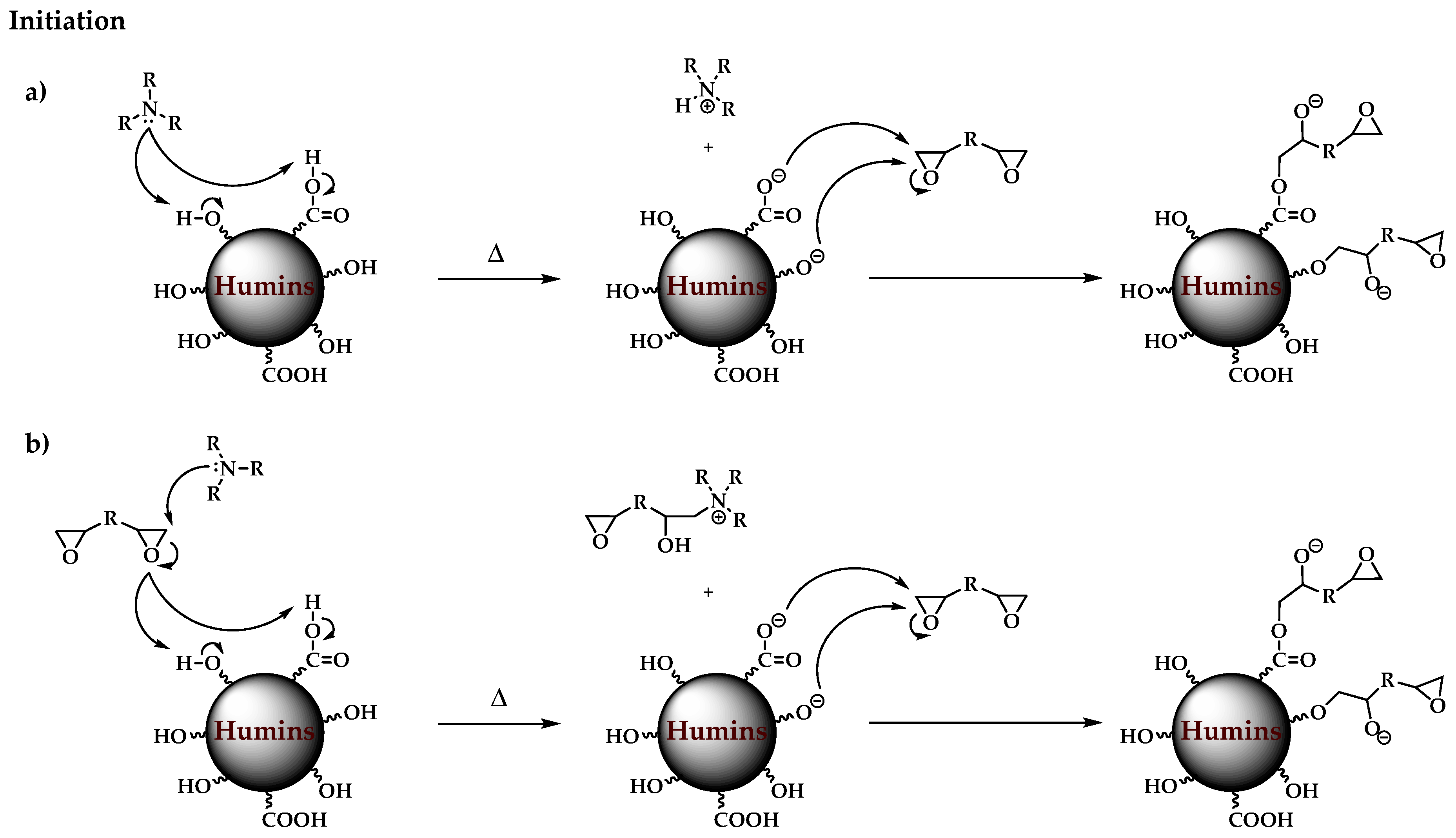
3.1. Plausible Polymerization Mechanism
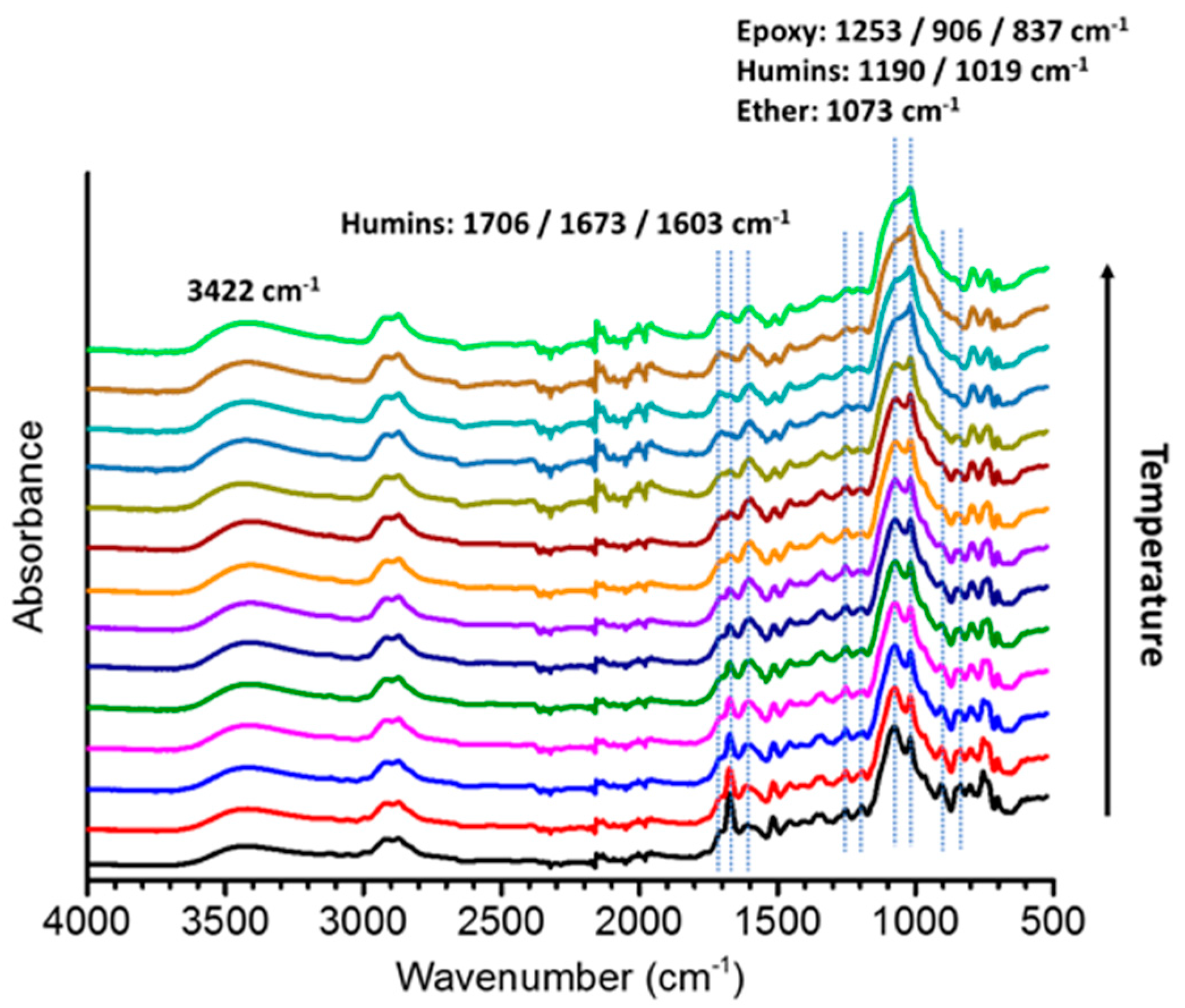
3.2. FT-IR Spectroscopy
3.2.1. Raw Materials
3.2.2. Copolymers Based on Humins–Epoxides Thermosets
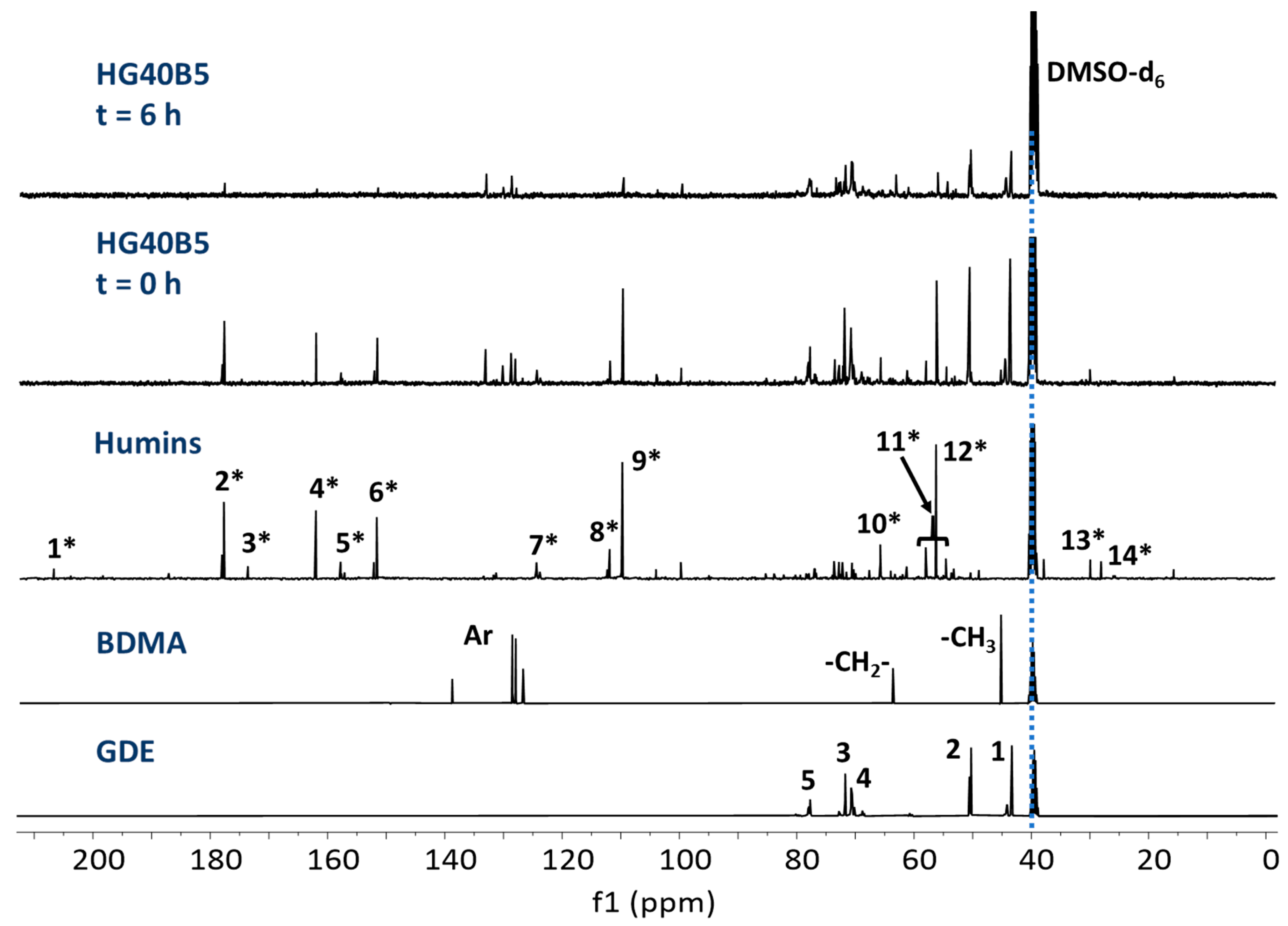
3.3. NMR Spectroscopy
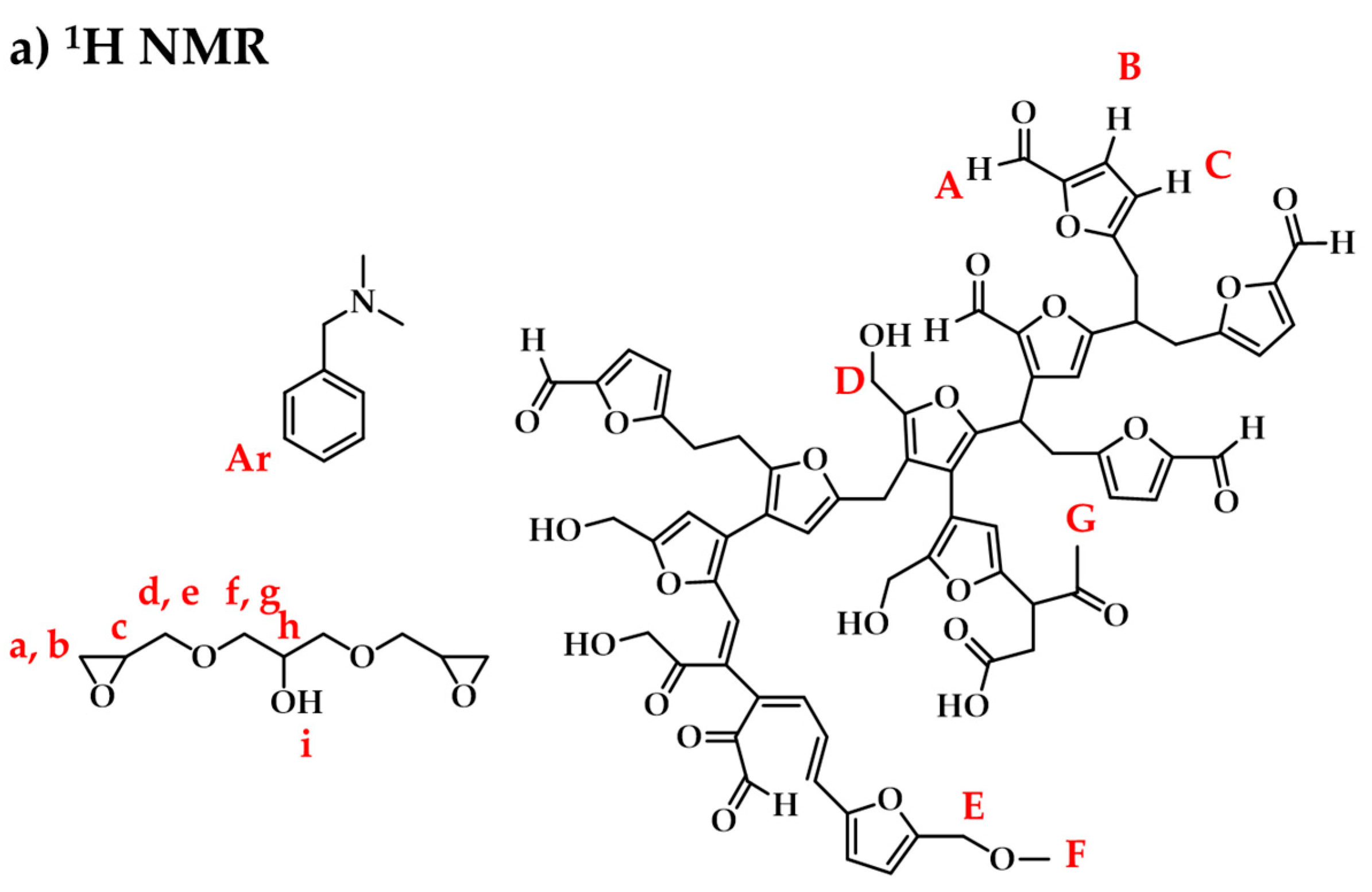
3.3.1. Raw Materials and Initial Mixtures
3.3.2. Humins–Epoxy Thermosets
4. Materials and Methods
4.1. Materials
4.2. Bulk Preparation of Resins
4.3. FT-IR Spectroscopy
4.4. NMR Spectroscopy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gandini, A.; Lacerda, T.M. From monomers to polymers from renewable resources: Recent advances. Prog. Polym. Sci. 2015, 48, 1–39. [Google Scholar] [CrossRef]
- Sousa, A.F.; Vilela, C.; Fonseca, A.C.; Matos, M.; Freire, C.S.R.; Gruter, G.J.M.; Coelho, J.F.J.; Silvestre, A.J.D. Biobased Polyesters and Other Polymers from 2,5-Furandicarboxylic Acid: A Tribute to Furan Excellency. Polym. Chem. 2015, 6, 5961–5983. [Google Scholar] [CrossRef]
- Llevot, A.; Grau, E.; Carlotti, S.; Grelier, S.; Cramail, H. From Lignin-Derived Aromatic Compounds to Novel Biobased Polymers. Macromol. Rapid Commun. 2016, 37, 9–28. [Google Scholar] [CrossRef] [PubMed]
- Imre, B.; García, L.; Puglia, D.; Vilaplana, F. Reactive Compatibilization of Plant Polysaccharides and Biobased Polymers: Review on Current Strategies, Expectations and Reality. Carbohydr. Polym. 2019, 209, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Romain, C.; Williams, C.K. Sustainable Polymers from Renewable Resources. Nature 2016, 540, 354–362. [Google Scholar] [CrossRef]
- Gandini, A.; Lacerda, T.M.; Carvalho, A.J.F.; Trovatti, E. Progress of Polymers from Renewable Resources: Furans, Vegetable Oils, and Polysaccharides. Chem. Rev. 2016, 116, 1637–1669. [Google Scholar] [CrossRef]
- Mashouf Roudsari, G.; Mohanty, A.K.; Misra, M. Green Approaches to Engineer Tough Biobased Epoxies: A Review. ACS Sustain. Chem. Eng. 2017, 5, 9528–9541. [Google Scholar] [CrossRef]
- Kumar, S.; Krishnan, S.; Mohanty, S.; Nayak, S.K. Synthesis and Characterization of Petroleum and Biobased Epoxy Resins: A Review. Polym. Int. 2018, 67, 815–839. [Google Scholar] [CrossRef]
- Xu, J.; Li, Z.; Wang, B.; Liu, F.; Liu, Y.; Liu, F. Recyclable Biobased Materials Based on Diels-Alder Cycloaddition. J. Appl. Polym. Sci. 2019, 136, 1–10. [Google Scholar] [CrossRef]
- Haas, M.J.; McAloon, A.J.; Yee, W.C.; Foglia, T.A. A Process Model to Estimate Biodiesel Production Costs. Bioresour. Technol. 2006, 97, 671–678. [Google Scholar] [CrossRef]
- Schweizer, A. Caramel and Humin. A Contribution to the Knowledge of the Decomposition Products of Sugars. Recl. Des Trav. Chim. Des Pays Bas 1938, 57, 345–382. [Google Scholar] [CrossRef]
- Schweizer, A. The Composition of the Humins Produced by the Action of Sulphuric Acid on Some Organic Substances. Recl. Des Trav. Chim. Des Pays Bas 1940, 59, 781–784. [Google Scholar] [CrossRef]
- Cheng, Z.; Everhart, J.L.; Tsilomelekis, G.; Nikolakis, V.; Saha, B.; Vlachos, D.G. Structural Analysis of Humins Formed in the Brönsted Acid Catalyzed Dehydration of Fructose. Green Chem. 2018, 20, 997–1006. [Google Scholar] [CrossRef]
- Van Zandvoort, I.; Koers, E.J.; Weingarth, M.; Bruijnincx, P.C.A.; Baldus, M.; Weckhuysen, B.M. Structural Characterization of 13C-Enriched Humins and Alkali-Treated 13C Humins by 2D Solid-State NMR. Green Chem. 2015, 17, 4383–4392. [Google Scholar] [CrossRef]
- Herzfeld, J.; Rand, D.; Matsuki, Y.; Daviso, E.; Mak-Jurkauskas, M.; Mamajanov, I. Molecular Structure of Humin and Melanoidin via Solid State NMR. J. Phys. Chem. B 2011, 115, 5741–5745. [Google Scholar] [CrossRef] [PubMed]
- Sumerskii, I.V.; Krutov, S.M.; Zarubin, M.Y. Humin-like Substances Formed under the Conditions of Industrial Hydrolysis of Wood. Russ. J. Appl. Chem. 2010, 83, 320–327. [Google Scholar] [CrossRef]
- Patil, S.K.R.; Heltzel, J.; Lund, C.R.F. Comparison of Structural Features of Humins Formed Catalytically from Glucose, Fructose, and 5-Hydroxylmethylfurfuraldehyde. Energy Fuels 2012, 26, 5281–5293. [Google Scholar] [CrossRef]
- Van Zandvoort, I.; Wang, Y.; Rasrendra, C.B.; Van Eck, E.R.H.; Bruijnincx, P.C.A.; Heeres, H.J.; Weckhuysen, B.M. Formation, Molecular Structure, and Morphology of Humins in Biomass Conversion: Influence of Feedstock and Processsing Conditions. ChemSusChem 2013, 6, 1745–1758. [Google Scholar] [CrossRef]
- Tsilomelekis, G.; Orella, M.J.; Lin, Z.; Cheng, Z.; Zheng, W.; Nikolakis, V.; Vlachos, D.G. Molecular Structure, Morphology and Growth Mechanisms and Rates of 5-Hydroxymethyl Furfural (HMF) Derived Humins. Green Chem. 2016, 18, 1983–1993. [Google Scholar] [CrossRef]
- Shi, N.; Liu, Q.; Ju, R.; He, X.; Zhang, Y.; Tang, S.; Ma, L. Condensation of α-Carbonyl Aldehydes Leads to the Formation of Solid Humins during the Hydrothermal Degradation of Carbohydrates. ACS Omega 2019, 4, 7330–7343. [Google Scholar] [CrossRef]
- Hoang, T.M.C.; van Eck, E.R.H.; Bula, W.P.; Gardeniers, J.G.E.; Lefferts, L.; Seshan, K. Humin Based By-Products from Biomass Processing as a Potential Carbonaceous Source for Synthesis Gas Production. Green Chem. 2015, 17, 959–972. [Google Scholar] [CrossRef]
- Filiciotto, L.; Balu, A.M.; Romero, A.A.; Rodríguez-Castellón, E.; Van der Waal, J.C.; Luque, R. Bening-by-Design Preparation of Humin-Based Iron Oxide Initiatoric Nanocomposites. Green Chem. 2017, 17, 4423–4434. [Google Scholar] [CrossRef]
- Kang, S.; Fu, J.; Deng, Z.; Jiang, S.; Zhong, G.; Xu, Y.; Guo, J.; Zhou, J. Valorization of Biomass Hydrolysis Waste: Activated Carbon from Humins as Exceptional Sorbent for Wastewater Treatment. Sustainability 2018, 10, 1795. [Google Scholar] [CrossRef]
- Chernysheva, D.V.; Chus, Y.A.; Klushin, V.A.; Lastovina, T.A.; Pudova, L.S.; Smirnova, N.V.; Kravchenko, O.A.; Chernyshev, V.M.; Ananikov, V.P. Sustainable Utilization of Biomass Refinery Wastes for Accessing Activated Carbons and Supercapacitor Electrode Materials. ChemSusChem 2018, 11, 3599–3608. [Google Scholar] [CrossRef] [PubMed]
- Tosi, P.; Van Klink, G.P.M.; Celzard, A.; Fierro, V.; Vincent, L.; de Jong, E.; Mija, A. Auto-Crosslinked Rigid Foams Derived from Biorefinery Byproducts. ChemSusChem 2018, 11, 2797–2809. [Google Scholar] [CrossRef] [PubMed]
- Mija, A.; de Jong, E.; van der Waal, J.C.; van Klink, G. Humins Containing Foam. WO 2017074183 A1 20170504, 4 May 2017. [Google Scholar]
- Blank, W.J.; He, Z.A.; Picci, M. Catalyssis of the Epoxy-Carboxyl Reaction. J. Coat. Technol. 2002, 74, 33–41. [Google Scholar] [CrossRef]
- Vidil, T.; Tournilhac, F.; Musso, S.; Robisson, A.; Leibler, L. Control of Reactions and Network Structures of Epoxy Thermosets. Prog. Polym. Sci. 2016, 62, 126–179. [Google Scholar] [CrossRef]
- Ellis, B. Introduction to the Chemistry, Synthesis, Manufacture and Characterization of Epoxy Resins. In Chemistry and Technology of Epoxy Resins; Ellis, B., Ed.; Springer Science Business Media: Berlin, Germany, 1993; pp. 1–36. [Google Scholar]
- Pin, J.-M.; Guigo, N.; Vincent, L.; Sbirrazzuoli, N.; Mija, A. Copolymerization as a Strategy to Combine Epoxidized Linseed Oil and Furfuryl Alcohol: The Design of a Fully Bio-based Thermoset. ChemSusChem 2015, 8, 4149–4161. [Google Scholar] [CrossRef]
- Dinu, R.; Mija, A. Polyfuranic Frame Networks with Elastomeric Behaviour Based on Humins Biorefinery By-Products. Green Chem. 2019. [Google Scholar] [CrossRef]
- Rozenberg, B.A. Thermodynamics and Mechanism of Reactions of Epoxy Oligomers with Amines. Adv. Polym. Sci. 1986, 75, 113–165. [Google Scholar]
- Ashcroft, W.R. Curing Agents for Epoxy Resins. In Chemistry and Technology of Epoxy Oligomers with Amines; Ellis, B., Ed.; Springer Science + Business Media: Berlin, Germany, 1993; pp. 37–71. [Google Scholar]
- Braun, M. Fundamentals and Transition-State Models. Aldol Additons of Group 1 and 2 Enolates. In Modern Aldol Reactions; Mahrwald, R., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Baden-Württemberg, Germany, 2008; Volume 1, pp. 1–61. [Google Scholar]
- Patil, S.K.R.; Lund, C.R.F. Formation and Growht of Humins via Aldol Addition and Condensation during Acid-Catalyzed Conversion of 5-Hydroxymethylfurfural. Energy Fuels 2011, 25, 4745–4755. [Google Scholar] [CrossRef]
- List, B. Amine-Catalyzed Aldol Reactions. In Modern Aldol Reactions; Mahrwald, R., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Baden-Württemberg, Germany, 2008; Volume 1, pp. 161–200. [Google Scholar]
- Markert, M.; Mulzer, M.; Schetter, B.; Mahrwald, R. Amine-Catalyzed Direct Aldol Addition. J. Am. Chem. Soc. 2007, 129, 7258–7259. [Google Scholar] [CrossRef] [PubMed]
- Muralidhara, A.; Tosi, P.; Mija, A.; Sbirrazzuoli, N.; Len, C.; Engelen, V.; De Jong, E.; Marlair, G. Insights on Thermal and Fire Hazards of Humins in Support of Their Sustainable Use in Advanced Biorefineries. ACS Sustain. Chem. Eng. 2018, 6, 16692–16701. [Google Scholar] [CrossRef]
- Heltzel, J.; Patil, S.K.R.; Lund, C.R.F. Humin Formation Pathways. In Reactions and Mechanisms in Thermocatalytic Biomass Conversion II; Schlaf, M.Z., Zhang, Z.C., Eds.; Springer Science + Business: Singapore, 2016; pp. 105–118. [Google Scholar]
- Guzmán, D.; Ramis, X.; Fernández-Francos, X.; De la Flor, S.; Serra, A. Preparation of New Biobased Coatings from a Triglycidyl Eugenol Derivative through Thiol-Epoxy Click Reaction. Prog. Org. Coat. 2018, 114, 259–267. [Google Scholar] [CrossRef]
- Pin, J.-M.; Guigo, N.; Mija, A.; Vincent, L.; Sbirrazzuoli, N.; Van der Waal, J.C.; De Jong, E. Valorization of Biorefinery Side-Stream Products: Combination of Humins with Polyfurfuryl Alcohol for Composite Elaboration. ACS Sustain. Chem. Eng. 2014, 2, 2182–2190. [Google Scholar] [CrossRef]
- Munteanu, S.B.; Vasile, C. Spectral and Thermal Characterization of Styrene-Butadiene Copolymers with Different Architectures. J. Optoelectron. Adv. Mater. 2005, 7, 3135–3148. [Google Scholar]
- Montero, A.L.; Montero, L.A.; Martínez, R.; Spange, S. Ab Initio Modelling of Crosslinking in Polymers. J. Mol. Struct. THEOCHEM 2006, 770, 99–106. [Google Scholar] [CrossRef]
- Vandenberg, E.J. Polymerization of Glycidol and Its Derivatives: A New Rearrangement Polymerization. J. Polym. Sci. Pol. Chem. 1985, 23, 915–949. [Google Scholar] [CrossRef]
- Sunder, A.; Hanselmann, R.; Frey, H.; Mülhaupt, R. Controlled Synthesis of Hyperbranched Polyglycerols by Ring-Opening Multibranching Polymerization. Macromolecules 1999, 32, 4240–4246. [Google Scholar] [CrossRef]
- Gao, C.; Yan, D. Hyperbranched Polymers: From Synthesis to Applications. Prog. Polym. Sci. 2004, 29, 183–275. [Google Scholar] [CrossRef]
- Voit, B.I.; Lederer, A. Hyperbranched and Highly Branched Polymer Architectures—Synthetic Strategies and Major Characterization Aspects. Chem. Rev. 2009, 109, 5924–5973. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Xu, Z.; Zhang, D. Synthesis and Application of Epoxy-Ended Hyperbranched Polymers. Chem. Eng. J. 2018, 343, 283–302. [Google Scholar] [CrossRef]
- Huang, Y. Concerning the Solvent Effect in the Aldol Condensation. Monatsh. Chem. 2000, 131, 521–523. [Google Scholar] [CrossRef]
- Kapoor, M.; Majumber, A.B.; Nath Gupta, M. Promiscuous Lipase-Catalyzed C-C Bond Formation Reactions Between 4 Nitrobenzaldehyde and 2-Cyclohexen-1-One in Biphasic Medium: Aldol and Morita-Baylis-Hillman Adduct Formations. Catal. Lett. 2015, 145, 527–532. [Google Scholar] [CrossRef]
- Emrick, T.; Chang, H.; Fréchet, J.M.J. The Preparation of Hyperbranched Aromatic and Alipathic Polyether Epoxies by Chloride-catalyzed Proton Transfer Polymerization from ABn and A2 + B3 Monomers. J. Polym. Sci. Pol. Chem. 2000, 38, 4850–4869. [Google Scholar] [CrossRef]
- Ahn, K.D.; Kim, M.H. Enhanced Cationic Photocuring of Epoxides with Styrene Oxide as a Reactive Diluent. Prog. Org. Coat. 2012, 73, 194–201. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds HG40B5, HP40B5 and HP20G20B5 are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Initial w/w Ratios: Humins/Diepoxy Monomer/Initiator |
|---|---|
| HG40B5 | Humins (55)/GDE (40)/BDMA (5) |
| HP40B5 | Humins (55)/PEGDE (40)/BDMA (5) |
| HP20G20B5 | Humins (55)/PEGDE (20)/GDE (20)/BDMA (5) |
| Signals | 1H NMR Chemical Shift (ppm) | 13C NMR Chemical Shift (ppm) |
|---|---|---|
| a, 1 | 4.40 | 65.3 |
| b, 2 | 3.40–3.60 | 70.3 |
| c, 3 | 3.45 | 79.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montané, X.; Dinu, R.; Mija, A. Synthesis of Resins Using Epoxies and Humins as Building Blocks: A Mechanistic Study Based on In-Situ FT-IR and NMR Spectroscopies. Molecules 2019, 24, 4110. https://doi.org/10.3390/molecules24224110
Montané X, Dinu R, Mija A. Synthesis of Resins Using Epoxies and Humins as Building Blocks: A Mechanistic Study Based on In-Situ FT-IR and NMR Spectroscopies. Molecules. 2019; 24(22):4110. https://doi.org/10.3390/molecules24224110
Chicago/Turabian StyleMontané, Xavier, Roxana Dinu, and Alice Mija. 2019. "Synthesis of Resins Using Epoxies and Humins as Building Blocks: A Mechanistic Study Based on In-Situ FT-IR and NMR Spectroscopies" Molecules 24, no. 22: 4110. https://doi.org/10.3390/molecules24224110
APA StyleMontané, X., Dinu, R., & Mija, A. (2019). Synthesis of Resins Using Epoxies and Humins as Building Blocks: A Mechanistic Study Based on In-Situ FT-IR and NMR Spectroscopies. Molecules, 24(22), 4110. https://doi.org/10.3390/molecules24224110