1. Introduction
Selective transformation of one specific functional group in a molecule, containing multiple groups of similar reactivity is of the utmost relevance for targeted synthesis of molecules with pharmaceutical activities, as well as materials with well-defined properties for technical applications. Particularly, ester hydrolysis is one of the most fundamental chemical transformations. Successful strategies for the preferential monohydrolysis of a number of different esters have been reported under often strong basic conditions [
1,
2,
3], however general applicability is limited by competing elimination reactions under these conditions and chemoselectivity regarding a wide spectrum of multiester compounds. No study has yet been reported for the selective transformation of biphenyl-based esters using lipase biocatalysis. Biphenyl-based building blocks with ester functions represent an important class of precursors that give rise to compounds with anti-infective, anti-inflammatory, and anti-cancer potencies [
4,
5,
6,
7,
8,
9,
10], as well as in technical applications, such as in solar cells, where they are used as linkers for dyes [
11] and hole transport materials [
12], in metal organic frameworks for gas storage [
13], and as catalysts [
13,
14] in lithium ion batteries [
15] or rectification devices [
16], in nanocomposite thin films for use in organic light emitting diodes (OLED) [
17], or even as material in HPLC columns for the isolation of aromatic compounds [
18]. Chemoselective hydrolysis and subsequent functionalization of biphenyl ester-based compounds therefore allow the generation of tailored molecules with increased potency and selectivity for their biological targets, improved bioavailability (absorption-distribution-metabolism-excretion-toxicity, ADMET) profiles, or unique magnetic or electronic properties.
Biocatalysis strategies using lipases have become an attractive tool for modern organic synthesis in industry and academic research, due to the broad accepted substrate spectra and the usually mild conditions that can be applied, such as low temperatures and neutral pH values [
19,
20,
21]. Their synthetic use reduces the risk of side reactions considering the high chemo-, regio-, and stereoselectivity of lipases [
22,
23]. Given their tolerance for organic solvents, varying the ratios of water and organic solvent mixtures provides a means to control enzymatic reaction and selectivities. In addition, it offers the advantage to even convert small molecules with low solubilities in aqueous medium, in order to biocatalytically obtain the desired target products. Lipases catalyze a plethora of different reactions, including hydrolysis, esterification, transesterification, and alcoholysis [
24], the major physiological function of lipases however is the hydrolytic cleavage of triacylglycerides for energy production, and their ubiquitous expression and functions in diverse cellular compartments, which facilitate enzyme activity under a variety of conditions [
19,
20,
21]. Industrial applications of lipases include the generation of important pharmaceuticals through transesterifications and ester cleavage [
19,
25,
26,
27]. They are additionally used for biodiesel production in food and flavor industry as well as waste water treatment and generation of agrochemicals [
24]. In organic synthesis, lipases are often used for desymmetrization and dynamic kinetic resolution towards enantiopure compounds [
28,
29]. Furthermore, lipases have been shown to biocatalytically convert benzylic esters [
30,
31]. However, the conversion of symmetric biphenyl-based ester building blocks to unsymmetric esters has not yet been investigated.
Here, we present the combination of eco- and cost-friendly biomimetic oxidative phenol coupling with lipase-catalyzed monohydrolysis of biphenyl esters for the targeted synthesis of unsymmetric biphenyl-based ester building blocks. We systematically analyzed the catalytic activities of a series of lipases from different organisms for their potency to hydrolyze biphenyl esters. We are investigating particularly the effect of the reaction conditions, in terms of water–organic solvent ratios, pH, type of solvents, and reaction time, on the efficacy and chemoselectivity of the lipases, as well as their accepted substrate spectrum. Accompanying molecular docking experiments helped to rationalize the observed catalytic activities and chemoselectivities. Our study provides a biomimetic strategy using lipase catalysis for the controlled synthesis of unsymmetric biphenyl ester building blocks under mild conditions.
3. Discussion
Lipases are versatile biocatalysts for numerous targeted chemical transformations and widely used tools in organic synthesis, however their efficiency and selectivity to hydrolyze esters with biphenyl core moieties has not been examined so far. Here we established a biomimetic strategy for the chemoselective synthesis of unsymmetric biphenyl-based ester building blocks by combining iron(III)-catalyzed oxidative coupling reactions to construct the biphenyl core moiety, with selective monohydrolysis of the biphenyl diesters using lipases. This synthetic route offers a simple, environmentally friendly, and inexpensive access to unsymmetric biphenyl ester building blocks under mild conditions and can be used for the synthesis of a wide range of biphenyl esters with broad technical and pharmaceutical applications. We systematically addressed the impact of the reaction conditions on the catalytic activity and chemoselectivity of the lipases, particularly the effect of the reaction time and temperature, as well as the pH, water–organic solvent ratio, and type of organic solvent.
The bioinspired oxidative phenol coupling using the cheap and non-toxic iron(III) catalyst FeCl
3·6 H
2O offered us an efficient solid-phase homocoupling of phenol esters in one step to produce symmetric biphenyl-based methylesters in high yields. In the second step, we used lipase biocatalysis to chemoselectively hydrolyze one of the two equivalent ester groups. For that purpose, we profiled a series of nine lipases from different organisms, covering bacteria, fungi, and vertebrates, for their efficacy to catalyze the hydrolytic ester cleavage of the biphenyl esters. Five of the lipases were found to be able to cleave the biphenyl esters under different reaction conditions with a marked dependency of the enzymatic activity and particularly the chemoselectivity on the reaction time, temperature, pH value, and organic solvent content. Our results are in good agreement with previous studies, which reported that the activity of some of the lipases was critically dependent on the temperature and DMSO concentration [
37].
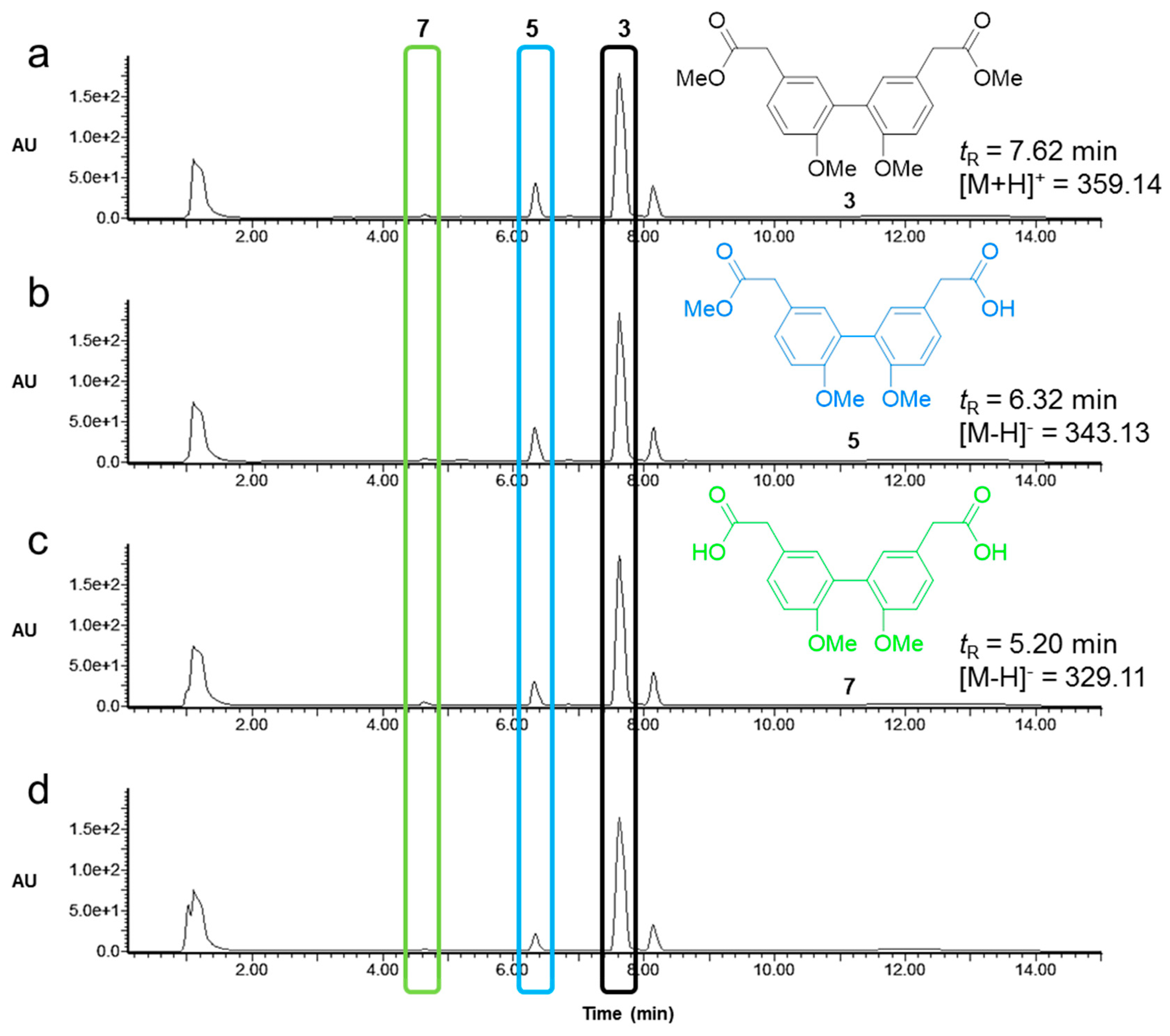
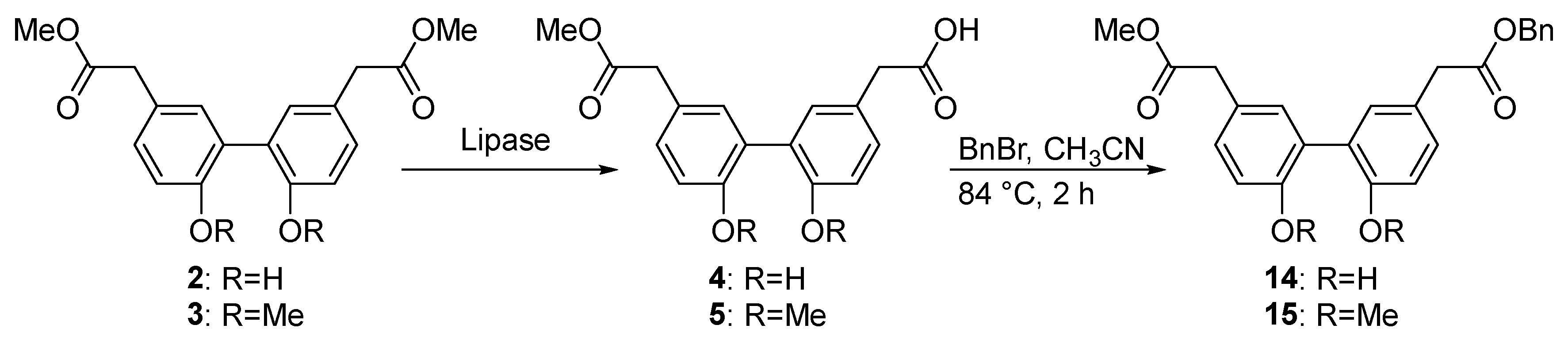
The identified active lipases CALB, CRL, RNL, MML, and PFL, capable of hydrolyzing biphenyl esters, showed a general acceptance of ortho substitutions at the biphenyl core moiety and hence, an advantageous level of promiscuity, allowing therefore their manifold use for biocatalytic conversions of a broad spectrum of biphenyl ester building blocks. For the biphenyl ester 2 with free hydroxyl groups in the ortho position of the core moiety, we found optimal reaction conditions that favor the formation of the unsymmetric product 4 by a factor of two over the symmetric product 6 using CALB at a moderate pH of 8, 25 °C, and 10% DMSO (v/v) after a reaction time of 2 h. Biphenyl 3 with methoxy groups at the biphenyl moiety was quickly converted to the symmetric product 7 under the same conditions. However, increasing the solvent content to 40% DMSO (v/v) and elevating the reaction temperature critically affected the activity and particularly the chemoselectivity of the four lipases CRL, MML, RNL, and PFL to efficiently and almost exclusively produce the unsymmetric biphenyl ester 5 by monohydrolysis of solely one ester group in the biphenyl diester. The chemoselective hydrolysis of only one ester group in the molecule was independent of the reaction time, and the overall yields increased steadily up to 48 h. The highest yields of unsymmetric biphenyl ester were obtained with lipases MML and PFL. The hydrolysis efficacies of the four lipases correlated well with predicted binding affinities by complementing computational docking experiments. Lipases such as PFL and MML featured favorable binding poses in proximity to the catalytic triad in the active site, and hence higher binding affinities of the biphenyl esters.
We were able to successfully transfer the optimized reaction conditions to a preparative scale and isolate the unsymmetric esters 4 and 5. Interestingly, the yields of the purified unsymmetric products were significantly higher than the yields obtained at an analytical scale, underpinning the potential of using lipases as biocatalysts for the selective monohydrolysis of biphenyl esters.
Biphenyl esters have been shown to represent an interesting class of building blocks for the development of pharmaceuticals and functionalized materials for use in a wide range of technical applications. This approach has great potential for the chemoselective conversion of a single functional group out of multiple groups with similar reactivities in a molecule, which can be challenging through traditional synthetic approaches, and the selective synthesis of unsymmetric biphenyls. Our biomimetic two-step strategy offers a series of advantages compared to other synthetic routes, such as the Suzuki–Miyaura coupling, the most frequently used synthetic approach for the synthesis of biphenyl core moieties, including no need for aryl halides, low cost, and mild reaction conditions, whereas the usually basic conditions of the Suzuki–Miyaura coupling include degradation of reactants, environmentally friendly, and no transesterifications by the solvent. In addition, the present study opens a novel facet for the effective and selective total synthesis of natural products, such as the pharmaceutically relevant ellagitannins, which possess biphenyl ester motifs connected to a glucopyranose, and which exhibit anti-infective, anti-inflammatory, and anti-cancer activities.
Summarizing our studies, we were able to establish a selective and efficient biomimetic strategy towards unsymmetric biphenyl esters by combining iron-catalyzed phenol coupling and lipase biocatalysis for the selective ester monohydrolysis under mild conditions. Varying critical reaction parameters, including time, pH, temperature, and solvent, control the activity and chemoselectivity of the enzymatic ester hydrolysis reaction.
4. Materials and Methods
4.1. Chemical Methods—General Information
Reactions with air- or moisture-sensitive reactants were performed under oxygen-free and dry inert atmosphere (nitrogen or argon). Chemicals were purchased from ABCR, Acros, AppliChem, AlfaAesar, Carl Roth, Fluka, Fluorochem, Sigma Aldrich, Merck and TCI and used without further purification. Dry solvents were freshly distilled or directly purchased from commercial suppliers and if not otherwise stated directly used without further purification. Silica gel column chromatography was performed under pressure with silica (60 Å, size 35–70 µM) purchased from Merck. TLC on silica gel was performed with silica coated glass slides from Merck (spot detection with UV or staining potassium-permanganate, ninhydrine or bromocresol green). Nuclear magnetic resonance spectra were recorded with a Bruker DPX-400 spectrometer. 13C NMR spectra were measured without 1H coupling. Chemical shifts were reported in ppm and calibrated to residual proton signal of deuterated solvents (CDCl3: δ = 7.26 ppm, CD3OD: δ = 3.31 ppm). Multiplicities are described using the following abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet. 13C NMR spectra were reported as values relative to the residual solvent signal (CDCl3: δ = 77.16 ppm, CD3OD: δ = 49.00 ppm) as internal standards. High resolution mass spectra were obtained with Micromass LCT via loop-mode injection from a Waters (Alliance 2695) HPLC system with achieved ionization by electrospray ionization (ESI). Calculated and found masses are given in the synthetic procedures.
4.2. Analytical LC-MS
Data from analytical LC–MS were obtained using a Waters 2767 sample manager connected to Waters 2545 pumps and system fluidics organizer (SFO), a Phenomenex Kinetex column (2.6 µ, C18, 100 Å, 4.6 × 100 mm) equipped with a Phenomenex Security Guard precolumn (Luna C5 300 Å) eluted at 1.0 mL·min−1 with a Waters 1998 Diode Array detector (200–600 nm), Waters 2424 ELSD, and Waters SQD-2 mass detector operating simultaneously in ES+ and ES- modes between 100 m/z and 650 m/z. The solvent gradient (A: HPLC grade ddH2O containing 0.05% formic acid; B: HPLC grade CH3CN containing 0.045% formic acid) was as follows: start: 10% B, 10 min 10%→90% B, 2 min 90% B, 1 min 90%→10% B, 2 min 10% B (total time: 15 min).
4.3. Preparative LC-MS
Purification of compounds with preparative LC-MS was done using a Waters mass-directed autopurification system comprising a Waters 2767 autosampler, a Waters 2545 pump system and SFO, and a Phenomenex Kinetex Axia column (5 µ, C18, 100 Å, 21.2 × 250 mm) equipped with a Phenomenex Security Guard precolumn (Luna C5 300 Å), eluting at 20 mL·min-1 at ambient temperature (solvents as mentioned in analytical LC-MS). The post-column flow was split in a ratio 100:1 and the minority flow was made up of HPLC grade MeOH (0.045% formic acid) to 1 mL·min−1 for simultaneous analysis by diode array (Waters 2998), evaporative light scattering (Waters 2424), and ESI mass spectrometry in positive and negative modes (Waters SQD-2). Detected peaks were collected into glass test tubes. The solvent was evaporated, and the residue analyzed by NMR.
4.4. Dimethyl-2,2‘-(6,6‘-dihydroxy-[1,1‘-biphenyl]-3,3‘-diyl)diacetate (2)
Silica gel (3.62 g) was added to a solution of FeCl3 ∙ 6 H2O (1.76 g, 6.51 mmol) in diethyl ether (95 mL) and methanol (5 mL) at room temperature and stirred for 5 min. The reaction mixture was concentrated under reduced pressure and the obtained yellow solid was stirred for 18 h in vacuo (1.5 mbar) at 55 °C. A solution of 4-hydroxyphenyl acetic acid (1, 61.7 mg, 0.41 mmol) in dichloromethane (15 mL) was added at room temperature. After thorough mixing the solvent was evaporated. The remaining dark solid was stirred in vacuo (1.5 mbar) at 60 °C. After 60 h, the solid was dissolved in methanol (25 mL) and filtered through Celite. The solvent was removed in vacuo and the crude product was purified by silica gel chromatography (first column: petrol ether—EtOAc, 1:1; second column: petrol ether—EtOAc, 2:1) to provide a highly viscous, colorless oil of 2 (40.26 mg, 0.122 mmol, 60%): 1H-NMR (400 MHz, CDCl3): δ (ppm) = 7.17 (dd, J = 2.39 Hz, J = 8.19 Hz, 2H), 7.14 (d, J = 2.39 Hz, 2H), 6.91 (d, J = 8.19 Hz, 2H), 6.21 (bs, 2H), 3.70 (s, 6H), 3.59 (s, 4H). 13C-NMR (100 MHz, CDCl3): δ (ppm) = 172.73, 152.16, 132.18, 130.59, 126.71, 124.26, 117.09, 52.21, 40.18. HRMS m/z calculated for C18H18O6 Na [M + Na]+: 353.1003; found 353.1001.
4.5. Dimethyl-2,2′-(6,6′-dimethoxy-[1,1′-biphenyl]-3,3′-diyl)-diacetate (3)
Potassium carbonate (0.11 g, 0.78 mmol) was added to a solution of biphenyl acetate 2 (0.12 g, 0.36 mmol) in dry DMF (1 mL) at room temperature. The reaction mixture was heated to 50 °C and stirred for 45 min, before it was cooled to room temperature and a solution of methyl iodide (0.05 mL, 0.85 mmol) in dry DMF (0.1 mL) was added slowly. The mixture was stirred for 14 h at 50 °C, after which it was cooled to room temperature and quenched by the addition of H2O (13 mL). The organic layer was separated and extracted with CH2Cl2. NaOH (10%, 2.6 mL) was added to the combined organic layers and stirred for 2 h. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2. The combined organic layers were washed with H2O, dried over Na2SO4, and concentrated. The crude product was purified by silica gel chromatography (petrol ether—EtOAc, 2:1) to yield a colorless oil of 3 (0.12 g, 90 %): 1H-NMR (400 MHz, CDCl3): δ (ppm) = 7.24 (dd, J = 2.05 Hz, J = 8.53 Hz, 2H), 7.15 (d, J = 2.05 Hz, 2H), 6.92 (d, J = 8.53 Hz, 2H), 3.76 (s, 6H), 3.69 (s, 6H), 3.59 (s, 4H). 13C-NMR (100 MHz, CDCl3): δ (ppm) = 172.37, 156.20, 132.36, 129.41, 127.57, 125.68, 111.19, 55.80, 52.01, 40.35. HRMS m/z calculated for C20H22O6 Na [M + Na]+: 381.1416; found 381.1314.
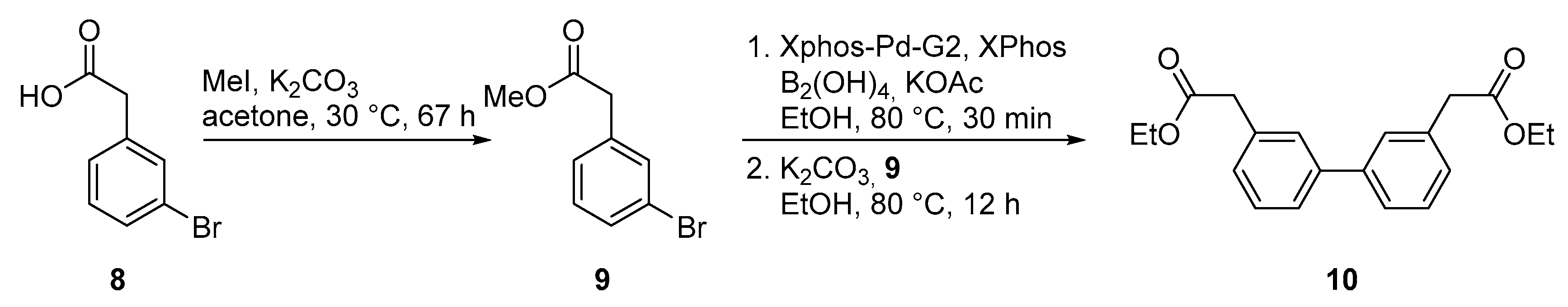
4.6. Methyl-2-(3-bromophenyl)acetate (9)
3-Bromo phenyl acetic acid (8, 4 g, 18.7 mmol, 1.0 eq) and dried K2CO3 (8.84 g, 56 mmol, 3.0 eq) were dissolved in acetone (40 mL) under inert conditions. Methyl iodide (10.5 mL, 56 mmol, 3.0 eq) was added dropwise and the reaction mixture was stirred for 67 h at 30 °C. After cooling the reaction to room temperature, the suspension was filtered with Celite and the solvent was removed in vacuo. The crude product was purified by silica gel chromatography (PE:EtOAc/4:1) yielding the product methyl-2-(3-bromophenyl)acetate (9) after evaporation as colorless oil in 100% yield (4.25 g, 18.7 mmol, quant.). Rf (PE:EtOAc/3:1): 0.85; 1H-NMR (400 MHz, CD3Cl): δ = 7.44 (d, J = 1.5 Hz, 1H), 7.41 (dt, J = 6.5, 2.2 Hz, 1H), 7.23–7.17 (m, 2H), 3.71 (s, 3H), 3.60 ppm (s, 2H); 13C-NMR (100 MHz, CD3Cl): δ (ppm) = 171.4, 136.1, 132.4, 130.3, 130.1, 128.0, 122.6, 52.2, 40.7.
4.7. Diethyl 2,2′-([1,1′-biphenyl]-3,3′-diyl)diacetate (10)
The halogenated compound 9 (90 mg, 0.40 mmol, 1.0 eq), XPhos-Pd-G2 (3.1 mg, 0.004 mmol, 0.01 eq), XPhos (3.8 mg, 0.008 mmol, 0.02 eq), B2(OH)4 (106.2 mg, 1.20 mmol, 3.0 eq), and KOAc (116.3 mg, 1.20 mmol, 3.0 eq) were dissolved in ethanol under inert conditions. The reaction mixture was stirred for 30 min at 80 °C until the color changed to yellow, and then cooled down to room temperature. K2CO3 (272.9 mg, 2.0 mmol, 5.0 eq) and 9 (90 mg, 0.40 mmol, 1.0 eq) were added and the reaction mixture was stirred for 16 h at 80 °C. After cooling to room temperature, the reaction mixture was filtered with Celite and the solvent evaporated. The crude product was purified by silica gel chromatography (PE:EtOAc/9:1) yielding the biphenyl ester 10 as yellowish oil (40 mg, 0.014 mmol, 34%). Rf (PE:EtOAc): 0.64. 1H-NMR (400 MHz, CDCl3): δ (ppm) = 7.49 (d, J = 7.6 Hz, 4H), 7.39 (t, J = 7.8 Hz, 2H), 7.28 (d, J = 7.5 Hz, 2H), 4.17 (q, J = 7.1 Hz, 4H), 3.68 (s, 4H), 1.27 (t, J = 7.1 Hz, 6H). 13C-NMR (100 MHz, CDCl3): δ (ppm) = 171.5, 141.2, 134.6, 128.9, 128.2, 128.2, 1256.0, 60.9, 41.5, 14.2. HRMS (ESI) m/z calculated for C20H22O4 [M + H]+ = 327.1591, found: 327.1619.
4.8. Colorimetric Lipase Activity Screening
Lipases were purchased from Sigma Aldrich (Lipase basic kit). In every well of a 96-well microtiter plate protein (final concentration: 10 U· mL-1) was 180 µL phosphate buffer (10 mM, pH 7.4) with 0.01% bromothymol blue (BTB) mixed with 20 µL compound (100 mM in DMSO, CH3CN or toluene). The final concentration of the organic solvent in each well was 10% (v/v). For every lipase a negative control, containing only protein and BTB in phosphate buffer, as well as a positive control using glyceryl tributyrate (TAG (13), 100 mM in DMSO) were prepared. The plate was shaken gently for 28 h at room temperature and the color was monitored at 0, 1, 2, 4, 24, and 28 h, respectively. The assay was performed in duplicates. With lipases RNL, MML, PFL, and CRL, the assay was additionally performed in the presence of 40% DMSO (v/v).
4.9. Analysis of Lipase Activity and Selectivity by Analytical LC-MS
In total, 1 mg lipase in 180 µL phosphate buffer (50 mM, pH 7.4/8.0/9.0 (AMP-buffer)) was mixed with 20 µL compound (100 mM in DMSO) for all experiments with 10% DMSO (v/v). For experiments in the presence of 40% DMSO (v/v), 80 µL compound (25 mM in DMSO) was used. The reaction mixtures were shaken at 25 or 37 °C, and 20 µL samples were taken at the indicated time points for analytical LC-MS measurements. The reaction in the samples was stopped by the addition of 5 µL 2M HCl and diluted with 20 µL CH3CN. Analytical LC-MS measurements were performed as mentioned before. The product–reactant ratios were determined by comparing the UV signals from a diode array, where the ratio of peak areas was proportional to the ratio of concentrations.
4.10. Benzyl 2-(2′,6-dihydroxy-5′-(2-methoxy-2-oxoethyl)-[1,1′-biphenyl]-3-yl)acetate (14) and 2-(2′,6-Bis(benzyloxy)-5′-(2-methoxy-2-oxoethyl)-[1,1′-biphenyl]-3-yl)acetic acid (16)
10.3 mg of CALB were dissolved in phosphate buffer (4 mL, 50 mM, pH 8.0) and biphenyl ester 2 (15 mg, 0.045 mmol, 1.0 eq) in DMSO (450 µL) was added to the mixture. The solution was stirred for 2 h at room temperature. To terminate the reaction 1 M HCl was added to adjust the pH to 1, and the aqueous layer was washed three times with EtOAc. The solvent was evaporated, and the residue was directly used for benzylation without any purification steps. The crude product was dissolved in CH3CN (10 mL), and 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) (1.5 mg, 0.01 mmol, 0.22 eq) and benzyl bromide (90 µL, 0.757 mmol, 16.8 eq) were added. The reaction mixture was stirred for 2 h at 84 °C. After cooling down dH2O was added and the aqueous layer was washed three times with EtOAc. The combined organic layer was dried over Na2SO4 and the solvent was evaporated. Purification of the crude product was performed using a Büchi Reverlis Prep equipped with a 4 g silica column (Ecoflex) and PE/EtOAc gradient; 14 eluted at 5% EtOAc and 16 at 50% EtOAc. The unsymmetric ester 14 was obtained as brown oil with 20% yield (3.6 mg, 0.009 mmol), and the acid 16 with a 45% yield (9.2 mg, 0.019 mmol).
14: Rf (PE:EtOAc/4:1): 0.646. 1H-NMR (400 MHz, CDCl3): δ (ppm) = 7.31 (dd, J = 10.8, 4.7 Hz, 5H), 7.22 (d, J = 7.5 Hz, 4H), 7.20–7.14 (m, 2H), 5.92 (ddt, J = 15.9, 11.0, 5.6 Hz, 1H), 5.16 (dd, J = 11.0, 1.4 Hz, 3H), 5.14–5.09 (m, 2H), 4.22 (dd, J = 4.2, 1.4 Hz, 4H). 13C-NMR (100 MHz, CDCl3): δ (ppm) = 178.4, 134.4, 134.4, 128.7, 128.7, 125.8, 120.2, 120.2, 116.5, 116.5, 116.5. HRMS [ESI] m/z calculated for C24H22O6 [M + Na]+ = 429.1314, found: 429.1311.
16: Rf (PE:EtOAc/4:1): 0.132. 1H-NMR (400 MHz, CDCl3): δ (ppm) = 7.37–7.26 (m, 10H), 7.26–7.09 (m, 4H), 7.06 (d, J = 8.2 Hz, 1H), 6.97 (d, J = 8.2 Hz, 1H), 6.28 (t, J = 5.4 Hz, 1H; OH), 5.14 (s, 2H), 5.10 (d, J = 4.2 Hz, 2H), 3.70 (s, 3H), 3.67–3.59 (m, 4H).13C-NMR (100 MHz, CDCl3): δ (ppm) = 172.5, 172.1, 154.1, 153.0, 133.6, 132.2, 130.3, 130.2, 128.8, 128.7, 128.7, 128.3, 128.3, 127.5, 117.9, 114.5, 72.1, 72.0, 66.9, 66.7, 52.3, 52.2, 40.5, 40.0. HRMS (ESI) m/z calculated for C31H28O6 [M + Na]+ = 519.1784, found: 519.1782 and calculated for C31H28O6 [M + H]− = 495.1808, found: 495.1810.
4.11. 2-(2′,6-Dimethoxy-5′-(2-methoxy-2-oxoethyl)-[1,1′-biphenyl]-3-yl)acetic acid (5)
A total of 29.7 mg of PFL was dissolved in phosphate buffer (3.36 mL, 50 mM, pH 8.0) and 3 (15 mg, 0.042 mmol, 1.0 eq) in DMSO (2.23 mL) was added. The solution was stirred for 48 h at 37 °C. To terminate the reaction 1 M HCl was added to receive pH 1 and the aqueous layer was washed three times with EtOAc. The solvent was evaporated, and the residue was purified with preparative LC–MS as mentioned above. Compound 5 was obtained as a colorless oil with 41% yield (5.9 mg, 0.017 mmol).
1H-NMR (400 MHz, CDCl3): δ (ppm) = 7.26–22 (m, 2H), 7.15 (dd, J = 4.1, 2.3 Hz, 2H), 6.92 (dd, J = 8.4, 4.1 Hz, 2H), 3.75 (d, J = 3.6 Hz, 6H), 3.69 (s, 3H), 3.62 (s, 2H), 3.59 (s, 2H).13C-NMR (100 MHz, CDCl3): δ (ppm) = 176.6, 172.4, 156.3, 156.1, 132.5, 132.3, 129.5, 129.4, 127.6, 127.3, 125.7, 125.0, 111.2, 111.2, 55.8, 55.8, 52.0, 40.2, 40.0. HRMS (ESI) m/z calculated for C19H20O6 [M + Na]+ = 367.1158, found: 367.1161.
4.12. Benzyl 2-(2′,6-dimethoxy-5′-(2-methoxy-2-oxoethyl)-[1,1′-biphenyl]-3-yl)acetate (15)
Firstly, 5 (4 mg, 0.012 mmol, 1 eq) was dissolved in CH3CN (3 mL). DBU (0.5 mg, 0.003 mmol, 0.2 eq) and benzyl bromide (4 µL, 0.024 mmol, 2.0 eq) were added. The reaction mixture was stirred at 84 °C for 2 h. After cooling down, H2O was added, and the aqueous layer was washed three times with EtOAc. The combined organic layers were dried over Na2SO4 and the solvent was evaporated. The residue was purified using silica gel chromatography using a PE:EtOAc gradient (start: 100% PE, 2CV; 50%:50% PE:EtOAc, 2 CV; 100% EtOAc, 15 eluted at PE:EtOAc 50%:50%). Compound 15 was obtained as a colorless oil with 81% yield (4.1 mg, 0.009 mmol).
1H-NMR (400 MHz, CDCl3): δ (ppm) = 7.36–7.29 (m, 5H), 7.26–7.22 (m, 2H), 7.14 (dd, J = 13.1, 2.3 Hz, 2H), 6.91 (d, J = 8.4 Hz, 2H), 5.13 (s, 2H), 3.76 (s, 3H), 3.74 (s, 3H), 3.69 (s, 3H), 3.63 (s, 2H), 3.59 (s, 2H).13C-NMR (100 MHz, CDCl3): δ (ppm) = 172.34, 171.72, 162.43, 162.01, 158.74, 156.20, 156.17, 132.37, 132.31, 129.44, 129.38, 128.50, 128.14, 128.09, 127.58, 127.57, 125.65, 125.58, 111.67, 111.16, 77.32, 77.00, 76.68, 66.52, 55.80, 55.78, 51.99, 40.47, 40.32. HRMS (ESI) m/z calculated for C26H26O6 [M + Na]+ = 457.1627, found: 457.1624.
4.13. Molecular Docking
All compounds were sketched in Schrödinger Maestro (Schrödinger Release 2019-3: Maestro; Protein Preparation Wizard; Epik; Impact, Schrödinger, LLC, New York, NY, USA, 2019), and energy minimized with Schrödinger MacroModel (Schrödinger Release 2019-3: MacroModel, Schrödinger, LLC, New York, NY, USA, 2019) and the OPLS3 force field [
40] to a gradient of 10
-4 kJ·mol
−1Å
−1. Molecular docking was performed using Autodock 4.2 (MGL Tools) [
41] and the Lamarckian genetic algorithm [
42]. Protein coordinates were obtained from the pdb database (PDB codes: CRL: 1TRH [
43], MML: 4TGL [
44], RNL: 1LGY [
45], PCL: 3LIP [
46]) and preprocessed with the Protein Preparation Wizard of the Schrödinger Suite and AutodockTools. The search space covered the catalytic center of the proteins.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}