
Synthesis, Characterization, Absorption Properties, and Electronic Structures of Paddlewheel-Type Dirhodium(II) Tetra-μ-(n-naphthoate) Complexes: An Experimental and Theoretical Study


Abstract
:
1. Introduction
2. Results and Discussions
2.1. Synthesis and Characterization of [1(OCMe2)2] and [2(OCMe2)2]
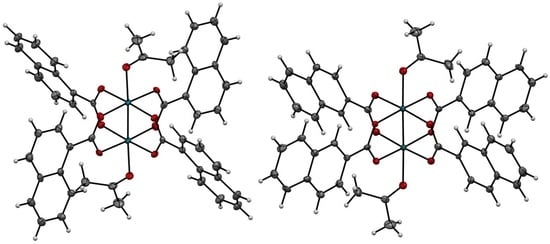
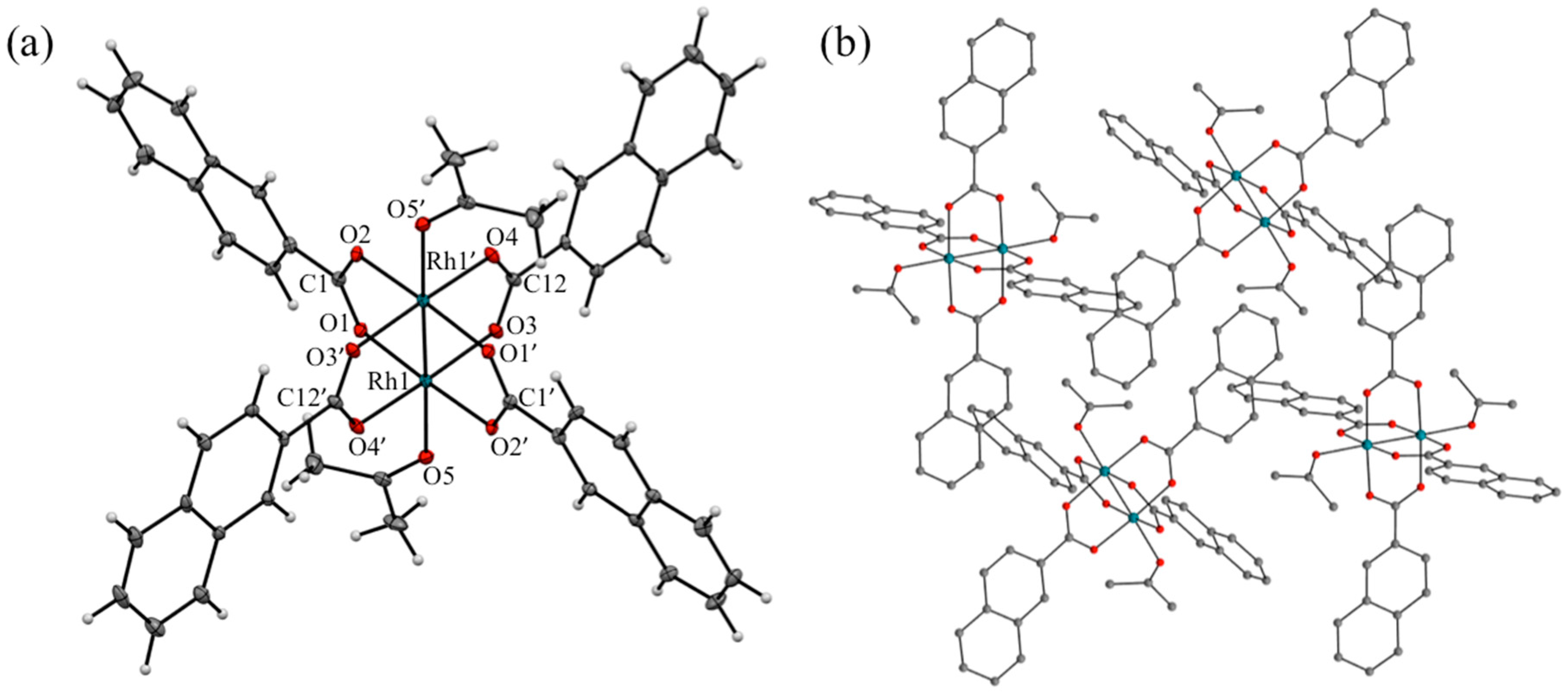
2.2. X-ray Diffraction Analyses of [1(OCMe2)2] and [2(OCMe2)2]
2.3. Optimized Geometries and Electronic Structures of [1(OCMe2)2] and [2(OCMe2)2]
2.4. Absorption Spectral Properties of [1(OCMe2)2] and [2(OCMe2)2]
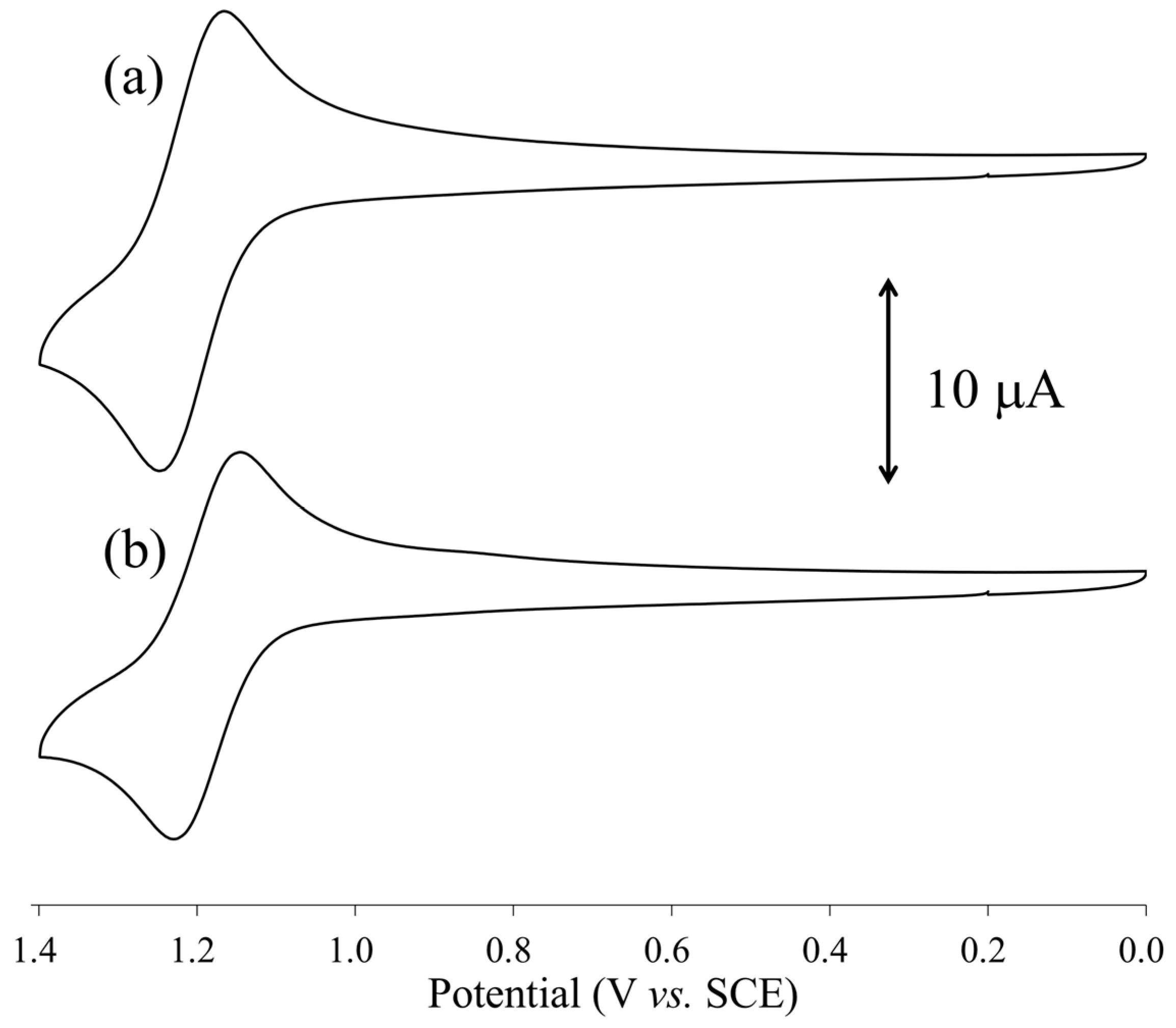
2.5. Redox Properties of [1(OCMe2)2] and [2(OCMe2)2]
3. Experimental
3.1. Materials and Instruments
3.2. Materials and Instruments Synthesis of [Rh2(1-NC)4(OCMe2)2] ([1(OCMe2)2])
3.3. Synthesis of [Rh2(2-NC)4(OCMe2)2] ([2(OCMe2)2])
3.4. Single Crystal X-Ray Diffraction Analyses
3.5. Details of Theoretical Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cotton, F.A.; Murillo, C.A.; Walton, R.A. Multiple Bonds between Metal Atoms, 3rd ed.; Springer Science and Business Media: New York, NY, USA, 2005. [Google Scholar]
- Schiavo, S.L.; Piraino, P.; Bonavita, A.; Micali, G.; Rizzo, G.; Neri, G. A dirhodium(II,II) molecular species as a candidate material for resistive carbon monoxide gas sensors. Sens. Actuator B Chem. 2008, 129, 772–778. [Google Scholar] [CrossRef]
- Hilderbrand, S.A.; Lim, M.H.; Lippard, S.J. Dirhodium Tetracarboxylate Scaffolds as Reversible Fluorescence-Based Nitric Oxide Sensors. J. Am. Chem. Soc. 2004, 126, 4972–4978. [Google Scholar] [CrossRef]
- Chifotides, H.T.; Dunber, K.R. Interactions of Metal−Metal-Bonded Antitumor Active Complexes with DNA Fragments and DNA. Acc. Chem. Res. 2005, 38, 146–156. [Google Scholar] [CrossRef]
- Hansen, J.; Davies, H.M.L. High Symmetry Dirhodium(II) Paddlewheel Complexes as Chiral Catalysts. Coord. Chem. Rev. 2008, 252, 545–555. [Google Scholar] [CrossRef]
- Fiori, K.W.; Du Bois, J. Catalytic Intermolecular Amination of C−H Bonds: Method Development and Mechanistic Insights. J. Am. Chem. Soc. 2007, 129, 562–568. [Google Scholar] [CrossRef]
- Kataoka, Y.; Sato, K.; Miyazaki, Y.; Suzuki, Y.; Tanaka, H.; Kitagawa, Y.; Kawakami, T.; Okumura, M.; Mori, W. Photocatalytic Hydrogen Production from Water Using Heterogeneous Two-dimensional Rhodium Coordination Polymer [Rh2(p-BDC)2]n. Chem. Lett. 2010, 39, 358–359. [Google Scholar] [CrossRef]
- Norman, J.G.; Kolari, H.J. Strength and trans influence of the rhodium-rhodium bond in rhodium(II) carboxylate dimers. J. Am. Chem. Soc. 1978, 100, 791–799. [Google Scholar] [CrossRef]
- Kataoka, Y.; Kitagawa, Y.; Saito, T.; Nakanishi, Y.; Matsui, T.; Sato, K.; Miyazaki, Y.; Kawakami, T.; Okumura, M.; Mori, W.; et al. Theoretical Study on the Electronic Configurations and Nature of Chemical Bonds of Dirhodium Tetraacetato Complexes [Rh2(CH3COO)4(L)2] (L = H2O, Free): Broken Symmetry Approach. Bull. Chem. Soc. Jpn. 2010, 83, 1481–1488. [Google Scholar] [CrossRef]
- Cotton, F.A.; Murillo, C.A.; Stiriba, S.-E.; Wang, X.; Yu, R. Chiral Organometallic Triangles with Rh−Rh Bonds. 2. Compounds Prepared from Enantiopure cis-Rh2(C6H4PPh2)2(OAc)2(HOAc)2 and Their Catalytic Potentials. Inorg. Chem. 2005, 44, 8223–8233. [Google Scholar] [CrossRef]
- Bickley, J.F.; Bonar-Law, R.P.; Femoni, C.; MacLean, E.J.; Steiner, A.; Teat, S.J. Dirhodium(II) carboxylate complexes as building blocks. Synthesis and structures of square boxes with tilted walls. J. Chem. Soc. Dalton Trans. 2000, 22, 4025–4027. [Google Scholar] [CrossRef]
- Kataoka, Y.; Arakawa, K.; Ueda, H.; Yano, N.; Kawamoto, T.; Handa, M. Experimental and Theoretical Study for Dimer-of-Dimers-type Tetrarhodium(II) Complexes Bridged by 1,4-Benzenedicarboxylate Linkers. Dalton Trans. 2018, 47, 17233–17242. [Google Scholar] [CrossRef]
- Cotton, F.A.; Lei, P.; Lin, C.; Murillo, C.A.; Wang, X.; Yu, S.-Y.; Zhang, Z.-X. A Calix[4]arene Carceplex with Four Rh24+ Fasteners. J. Am. Chem. Soc. 2004, 124, 1518–1525. [Google Scholar] [CrossRef]
- Tong, L.H.; Guénée, L.; Williams, A.F. Pentasubstituted Ferrocene and Dirhodium(II) Tetracarboxylate as Building Blocks for Discrete Fullerene-Like and Extended Supramolecular Structures. Inorg. Chem. 2011, 50, 2450–2457. [Google Scholar] [CrossRef]
- Takamizawa, S.; Nakata, E.; Yokoyama, H.; Mochizuki, K.; Mori, W. Carbon Dioxide Inclusion Phases of a Transformable 1D Coordination Polymer Host [Rh2(O2CPh)4(pyz)]n. Angew. Chem. Int. Ed. 2003, 42, 4331–4334. [Google Scholar] [CrossRef]
- Uemura, K.; Ebihara, M. One-Dimensionally Extended Paddlewheel Dirhodium Complexes from Metal–Metal Bonds with Diplatinum Complexes. Inorg. Chem. 2011, 50, 7919–7921. [Google Scholar] [CrossRef]
- Handa, M.; Muraki, Y.; Mikuriya, M.; Azuma, H.; Kasuga, K. Polymer Complexes of Rhodium(II) Trifluoroacetamidate with Pyrazine, 4,4′-Bipyridine, and 1,4-Diazabicyclo[2.2.2]octane. Bull. Chem. Soc. Jpn. 2002, 75, 1755–1756. [Google Scholar] [CrossRef]
- Kataoka, Y.; Yano, N.; Shimodaira, T.; Yan, Y.-N.; Yamasaki, M.; Tanaka, H.; Omata, K.; Kawamoto, T.; Handa, M. Paddlewheel-Type Dirhodium Tetrapivalate Based Coordination Polymer: Synthesis, Characterization, and Self-Assembly and Disassembly Transformation Properties. Eur. J. Inorg. Chem. 2016, 17, 2810–2815. [Google Scholar] [CrossRef]
- Cotton, F.A.; Dikarev, E.V.; Petrukhina, M.A. Neutral Cyclooctasulfur as a Polydentate Ligand: Supramolecular Structures of [Rh2(O2CCF3)4]n(S8)m (n:m=1:1, 3:2). Angew. Chem. Int. Ed. 2001, 40, 1521–1523. [Google Scholar] [CrossRef]
- Naito, S.; Tanibe, T.; Saito, E.; Miyao, T.; Mori, W. A Novel Reaction Pathway in Olefin-Deuterium Exchange Reaction inside the Micropores of Rh(II) Dicarboxylate Polymer Complexes. Chem. Lett. 2001, 30, 1178–1179. [Google Scholar] [CrossRef]
- Kataoka, Y.; Kataoka, K.S.; Murata, H.; Handa, M.; Mori, W.; Kawamoto, T. Synthesis and characterizations of a paddlewheel-type dirhodium-based photoactive porous metal-organic framework. Inorg. Chem. Commun. 2016, 68, 37–41. [Google Scholar] [CrossRef]
- Yano, N.; Kataoka, Y.; Tanaka, H.; Kawamoto, T.; Handa, M. A New Paddlewheel-Type Dirhodium-Based Metal-Organic Framework with Deprotonated 2,6-Bis(2-benzimidazolyl)pyridine. Chem. Sel. 2016, 1, 2571–2575. [Google Scholar] [CrossRef]
- Reger, D.L.; Debreczeni, A.; Reinecke, B.; Rassolov, V.; Smith, M.D.; Semeniuc, R.F. Highly Organized Structures and Unusual Magnetic Properties of Paddlewheel Copper(II) Carboxylate Dimers Containing the π−π Stacking, 1,8-Naphthalimide Synthon. Inorg. Chem. 2009, 48, 8911–8924. [Google Scholar] [CrossRef]
- Reger, D.L.; Debreczeni, A.; Smith, M.D. Synthesis and structure of a zinc(II)-carboxylate trimer containing the π···π stacking, 1,8-naphthalimide synthon: A supramolecular metal–organic framework. Inorg. Chim. Acta 2010, 364, 10–15. [Google Scholar] [CrossRef]
- Dikarev, E.V.; Andreini, K.W.; Petrukhina, M.A. On the Road to a Termolecular Complex with Acetone: A Heterometallic Supramolecular Network {[Rh2(O2CCF3)4]·μ2-OCMe2·[Cu4(O2CCF3)4]}. Inorg. Chem. 2004, 43, 3219–3224. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision C.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Legzdins, P.; Mitchell, R.W.; Rempel, G.L.; Ruddick, J.D.; Wilkinson, G. The protonation of ruthenium- and rhodium-bridged carboxylates and their use as homogeneous hydrogenation catalysts for unsaturated substances. J. Chem. Soc. A 1970, 0, 3322–3326. [Google Scholar] [CrossRef]
- Kataoka, Y.; Yano, N.; Kawamoto, T.; Handa, M. Isolation of the Intermediate in the Synthesis of Paddlewheel-type Dirhodium Tetraacetate. Eur. J. Inorg. Chem. 2015, 34, 5650–5655. [Google Scholar] [CrossRef]
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; De Caro, L.; Polidori, G.; Spagna, R. SIR2004: An improved tool for crystal structure determination and refinement. J. Appl. Cryst. 2005, 38, 381–388. [Google Scholar] [CrossRef]
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; De Caro, L.; Giacovazzo, C.; Polidori, G.; Siliqi, D.; Spagna, R. IL MILIONE: A suite of computer programs for crystal structure solution of proteins. J. Appl. Cryst. 2007, 40, 609–613. [Google Scholar] [CrossRef]
- A Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | [1(OCMe2)2] | [2(OCMe2)2] |
|---|---|---|
| Crystal size (mm3) | 0.18 × 0.14 × 0.14 | 0.14 × 0.04 × 0.04 |
| Chemical formula | C50H40O10Rh2 | C50H40O10Rh2 |
| T (K) | 150 | 150 |
| Formula weight (g/mol) | 1006.64 | 1006.64 |
| Crystal system | Triclinic | Monoclinic |
| Space group | P-1 | P 21/c |
| a (Å) | 10.480(5) | 10.195(7) |
| b (Å) | 10.960(5) | 17.537(10) |
| c (Å) | 11.441(6) | 13.264(9) |
| (°) | 98.215(4) | 90 |
| (°) | 115.975(3) | 110.979(10) |
| (°) | 110.223(2) | 90 |
| V (Å3) | 1038.9(9) | 2214(2) |
| Z | 1 | 2 |
| Dcalc(g/cm3) | 1.609 | 1.510 |
| F(000) | 510 | 1020 |
| Final R1 indices [I > 2σ(I)] | R1 = 0.0345, wR2 = 0.0795 | R1 = 0.0947, wR2 = 0.2225 |
| R indices (all data) | R1 = 0.0368, wR2 = 0.0810 | R1 = 0.1195, wR2 = 0.2445 |
| Goodness of fit (GOF) on F2 | 1.094 | 1.215 |
| [1(OCMe2)2] | [2(OCMe2)2] | [Rh2(O2CCF3)4(O2CMe)2] | |
|---|---|---|---|
| Rh–Rh | 2.374 | 2.395 | 2.406 |
| Rh–Oequatorial | 2.035 | 2.040 | 2.036 |
| Rh–Oaxial | 2.296 | 2.303 | 2.252 |
| Rh–Rh–Oequatorial | 88.18 | 88.19 | 87.97 |
| Rh–Rh–Oaxial | 175.51 | 178.54 | 175.67 |
| 125.52 | 125.03 | 128.93 | |
| References | This study | This study | [25] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kataoka, Y.; Fukumoto, R.; Yano, N.; Atarashi, D.; Tanaka, H.; Kawamoto, T.; Handa, M. Synthesis, Characterization, Absorption Properties, and Electronic Structures of Paddlewheel-Type Dirhodium(II) Tetra-μ-(n-naphthoate) Complexes: An Experimental and Theoretical Study. Molecules 2019, 24, 447. https://doi.org/10.3390/molecules24030447
Kataoka Y, Fukumoto R, Yano N, Atarashi D, Tanaka H, Kawamoto T, Handa M. Synthesis, Characterization, Absorption Properties, and Electronic Structures of Paddlewheel-Type Dirhodium(II) Tetra-μ-(n-naphthoate) Complexes: An Experimental and Theoretical Study. Molecules. 2019; 24(3):447. https://doi.org/10.3390/molecules24030447
Chicago/Turabian StyleKataoka, Yusuke, Raiki Fukumoto, Natsumi Yano, Daiki Atarashi, Hidekazu Tanaka, Tatsuya Kawamoto, and Makoto Handa. 2019. "Synthesis, Characterization, Absorption Properties, and Electronic Structures of Paddlewheel-Type Dirhodium(II) Tetra-μ-(n-naphthoate) Complexes: An Experimental and Theoretical Study" Molecules 24, no. 3: 447. https://doi.org/10.3390/molecules24030447