
Licofelone-DPPC Interactions: Putting Membrane Lipids on the Radar of Drug Development

 ,
,  , ,
, , 
Abstract
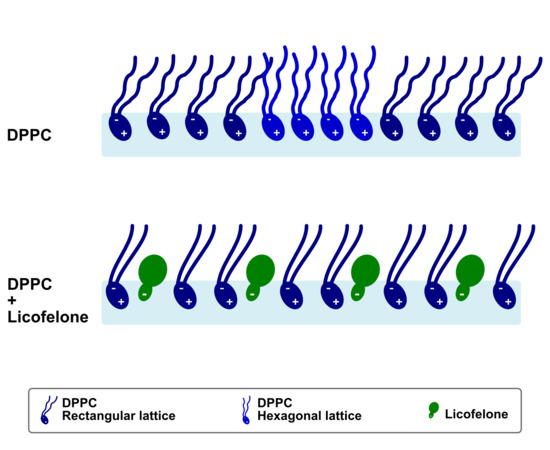
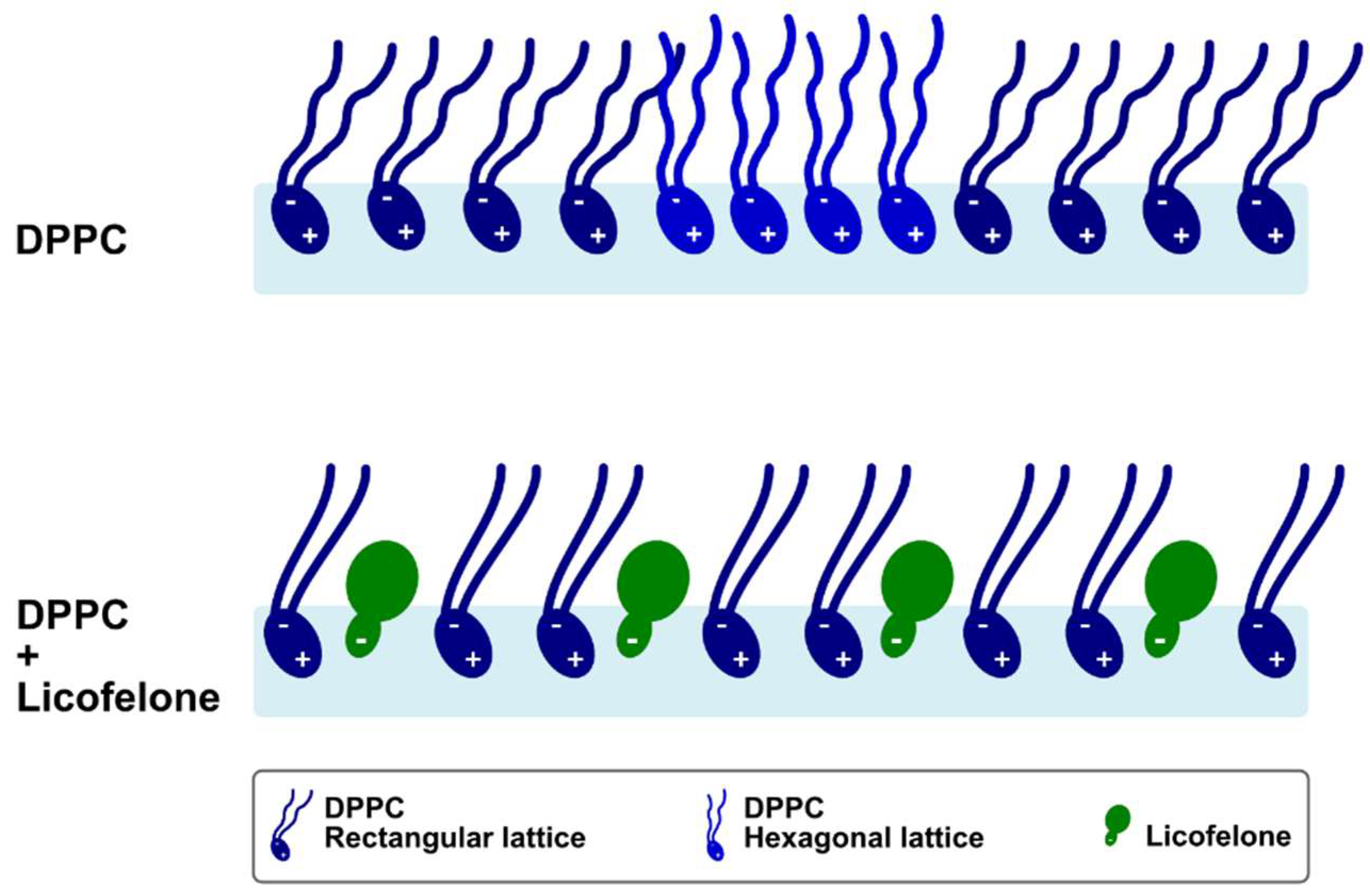
:
1. Introduction
2. Results
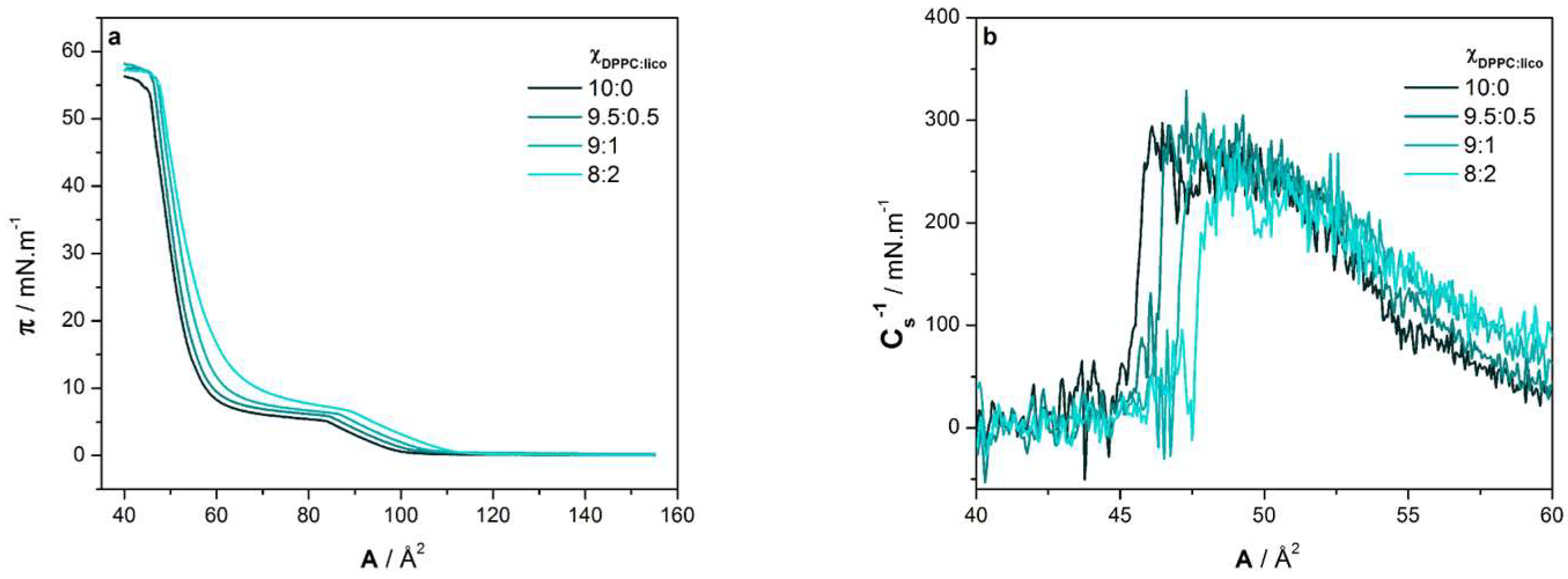
2.1. Langmuir Isotherms
2.2. Brewster Angle Microscopy
2.3. Polarization-Modulation Infrared Reflection-Absorption Spectroscopy
2.4. Grazing-Incidence X-Ray Diffraction
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Monolayers Preparation
4.3. Langmuir Isotherms
4.4. Brewster Angle Microscopy
4.5. Polarization-Modulation Infrared Reflection-Absorption Spectroscopy
4.6. Grazing-Incidence X-ray Diffraction
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Blass, B.E. Chapter 1-Drug Discovery and Development: An Overview of Modern Methods and Principles. In Basic Principles of Drug Discovery and Development; Blass, B.E., Ed.; Academic Press: Boston, MA, USA, 2015; pp. 1–34. [Google Scholar]
- Wang, D.; Gao, G. State-of-the-art human gene therapy: Part II. Gene therapy strategies and clinical applications. Discov. Med. 2014, 18, 151–161. [Google Scholar] [PubMed]
- Burnett, J.C.; Rossi, J.J. RNA-based Therapeutics- Current Progress and Future Prospects. Chem. Biol. 2012, 19, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Escriba, P.V.; Busquets, X.; Inokuchi, J.; Balogh, G.; Torok, Z.; Horvath, I.; Harwood, J.L.; Vigh, L. Membrane lipid therapy: Modulation of the cell membrane composition and structure as a molecular base for drug discovery and new disease treatment. Prog. Lipid Res. 2015, 59, 38–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sethi, S.; Brietzke, E. Recent advances in lipidomics: Analytical and clinical perspectives. Prostaglandins Other Lipid Mediators 2017, 128–129, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, L.M.; Nunes, C.; Lucio, M.; Segundo, M.A.; Reis, S.; Lima, J.L.F.C. High-throughput microplate assay for the determination of drug partition coefficients. Nat. Protoc. 2010, 5, 1823–1830. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Leite, C.; Nunes, C.; Reis, S. Interaction of nonsteroidal anti-inflammatory drugs with membranes: In vitro assessment and relevance for their biological actions. Prog. Lipid Res. 2013, 52, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Lopes, D.; Jakobtorweihen, S.; Nunes, C.; Sarmento, B.; Reis, S. Shedding light on the puzzle of drug-membrane interactions: Experimental techniques and molecular dynamics simulations. Prog. Lipid Res. 2017, 65, 24–44. [Google Scholar] [CrossRef]
- Blass, B.E. Chapter 5-Medicinal Chemistry. In Basic Principles of Drug Discovery and Development; Blass, B.E., Ed.; Academic Press: Boston, MA, USA, 2015; pp. 203–243. [Google Scholar]
- Gaspar, D.; Lucio, M.; Rocha, S.; Lima, J.L.F.C.; Reis, S. Changes in PLA(2) activity after interacting with anti-inflammatory drugs and model membranes: Evidence for the involvement of tryptophan residues. Chem. Phys. Lipids 2011, 164, 292–299. [Google Scholar] [CrossRef]
- Pereira-Leite, C.; Nunes, C.; Grahl, D.; Bozelli, J.C.; Schreier, S.; Kamma-Lorger, C.S.; Cuccovia, I.M.; Reis, S. Acemetacin-phosphatidylcholine interactions are determined by the drug ionization state. Phys. Chem. Chem. Phys. 2018, 20, 14398–14409. [Google Scholar]
- Lopes, D.; Nunes, C.; Fontaine, P.; Sarmento, B.; Reis, S. Proof of pore formation and biophysical perturbations through a 2D amoxicillin-lipid membrane interaction approach. Biochim. Biophys. Acta 2017, 1859, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Leite, C.; Nunes, C.; Jamal, S.K.; Cuccovia, I.M.; Reis, S. Nonsteroidal Anti-Inflammatory Therapy: A Journey Toward Safety. Med. Res. Rev. 2017, 37, 802–859. [Google Scholar] [CrossRef] [PubMed]
- Stefaniu, C.; Brezesinski, G.; Möhwald, H. Langmuir monolayers as models to study processes at membrane surfaces. Adv. Colloid Interface Sci. 2014, 208, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Peetla, C.; Stine, A.; Labhasetwar, V. Biophysical interactions with model lipid membranes: Applications in drug discovery and drug delivery. Mol. Pharm. 2009, 6, 1264–1276. [Google Scholar] [CrossRef]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Deleu, M.; Paquot, M.; Nylander, T. Fengycin interaction with lipid monolayers at the air-aqueous interface-implications for the effect of fengycin on biological membranes. J. Colloid Interface Sci. 2005, 283, 358–365. [Google Scholar] [CrossRef]
- Kouzayha, A.; Besson, F. GPI-alkaline phosphatase insertion into phosphatidylcholine monolayers: Phase behavior and morphology changes. Biochem. Biophys. Res. Commun. 2005, 333, 1315–1321. [Google Scholar] [CrossRef]
- Vollhardt, D.; Fainerman, V.B. Characterisation of phase transition in adsorbed monolayers at the air/water interface. Adv. Colloid Interface Sci. 2010, 154, 1–19. [Google Scholar] [CrossRef]
- Kaganer, V.M.; Möhwald, H.; Dutta, P. Structure and phase transitions in Langmuir monolayers. Rev. Mod. Phys. 1999, 71, 779–819. [Google Scholar] [CrossRef] [Green Version]
- Alves, A.C.; Nunes, C.; Lima, J.; Reis, S. Daunorubicin and doxorubicin molecular interplay with 2D membrane models. Colloids Surf., B 2017, 160, 610–618. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, S. Effects of fullerenes on phospholipid membranes: A langmuir monolayer study. ChemPhysChem 2009, 10, 2284–2289. [Google Scholar] [CrossRef] [PubMed]
- McConlogue, C.W.; Vanderlick, T.K. A Close Look at Domain Formation in DPPC Monolayers. Langmuir 1997, 13, 7158–7164. [Google Scholar] [CrossRef]
- Pinheiro, M.; Arede, M.; Giner-Casares, J.J.; Nunes, C.; Caio, J.M.; Moiteiro, C.; Lucio, M.; Camacho, L.; Reis, S. Effects of a novel antimycobacterial compound on the biophysical properties of a pulmonary surfactant model membrane. Int. J. Pharm. 2013, 450, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Castro, C.M.; Pinheiro, M.; Lucio, M.; Giner-Casares, J.J.; Camacho, L.; Lima, J.L.; Reis, S.; Segundo, M.A. Insights about alpha-tocopherol and Trolox interaction with phosphatidylcholine monolayers under peroxidation conditions through Brewster angle microscopy. Colloids Surf., B 2013, 111, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, R.; Mao, G.; Flach, C.R. Infrared reflection–absorption spectroscopy: Principles and applications to lipid–protein interaction in Langmuir films. Biochim. Biophys. Acta, Biomembr. 2010, 1798, 788–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefaniu, C.; Brezesinski, G. X-ray investigation of monolayers formed at the soft air/water interface. Curr. Opin. Colloid Interface Sci. 2014, 19, 216–227. [Google Scholar] [CrossRef]
- Stefaniu, C.; Brezesinski, G. Grazing incidence X-ray diffraction studies of condensed double-chain phospholipid monolayers formed at the soft air/water interface. Adv. Colloid Interface Sci. 2014, 207, 265–279. [Google Scholar] [CrossRef]
- Miller, C.E.; Busath, D.D.; Strongin, B.; Majewski, J. Integration of ganglioside GT1b receptor into DPPE and DPPC phospholipid monolayers: An X-ray reflectivity and grazing-incidence diffraction study. Biophys. J. 2008, 95, 3278–3286. [Google Scholar] [CrossRef]
- Neville, F.; Cahuzac, M.; Konovalov, O.; Ishitsuka, Y.; Lee, K.Y.C.; Kuzmenko, I.; Kale, G.M.; Gidalevitz, D. Lipid Headgroup Discrimination by Antimicrobial Peptide LL-37: Insight into Mechanism of Action. Biophys. J. 2006, 90, 1275–1287. [Google Scholar] [CrossRef] [Green Version]
- Hąc-Wydro, K.; Flasiński, M.; Broniatowski, M.; Dynarowicz-Łątka, P.; Majewski, J. Properties of β-sitostanol/DPPC monolayers studied with Grazing Incidence X-ray Diffraction (GIXD) and Brewster Angle Microscopy. J. Colloid Interface Sci. 2011, 364, 133–139. [Google Scholar] [CrossRef]
- Gzyl-Malcher, B.; Filek, M.; Brezesinski, G. Mixed DPPC/DPTAP Monolayers at the Air/Water Interface: Influence of Indolilo-3-acetic Acid and Selenate Ions on the Monolayer Morphology. Langmuir 2011, 27, 10886–10893. [Google Scholar] [CrossRef]
- Bialkowska, K.; Bobrowska-Hagerstrand, M.; Hagerstrand, H. Expansion of phosphatidylcholine and phosphatidylserine/phosphatidylcholine monolayers by differently charged amphiphiles. Z. Naturforsch. C 2001, 56, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Nobre, T.M.; Pavinatto, F.J.; Caseli, L.; Barros-Timmons, A.; Dynarowicz-Łątka, P.; Oliveira, O.N. Interactions of bioactive molecules & nanomaterials with Langmuir monolayers as cell membrane models. Thin Solid Films 2015, 593, 158–188. [Google Scholar]
- Ariga, K.; Nakanishi, T.; Hill, J.P.; Shirai, M.; Okuno, M.; Abe, T.; Kikuchi, J. Tunable pK of amino acid residues at the air-water interface gives an L-zyme (langmuir enzyme). J. Am. Chem. Soc. 2005, 127, 12074–12080. [Google Scholar] [CrossRef] [PubMed]
- Wellen, B.A.; Lach, E.A.; Allen, H.C. Surface pKa of octanoic, nonanoic, and decanoic fatty acids at the air-water interface: Applications to atmospheric aerosol chemistry. Phys. Chem. Chem. Phys. 2017, 19, 26551–26558. [Google Scholar]
- Yang, H.; Imanishi, Y.; Harata, A. Estimating pH at the Air/Water Interface with a Confocal Fluorescence Microscope. Anal. Sci. 2015, 31, 1005–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes, C.; Brezesinski, G.; Pereira-Leite, C.; Lima, J.L.F.C.; Reis, S.; Lucio, M. NSAIDs Interactions with Membranes: A Biophysical Approach. Langmuir 2011, 27, 10847–10858. [Google Scholar] [CrossRef] [PubMed]
- Boggara, M.B.; Mihailescu, M.; Krishnamoorti, R. Structural association of nonsteroidal anti-inflammatory drugs with lipid membranes. J. Am. Chem. Soc. 2012, 134, 19669–19676. [Google Scholar] [CrossRef]
- Lichtenberger, L.M.; Zhou, Y.; Jayaraman, V.; Doyen, J.R.; O’Neil, R.G.; Dial, E.J.; Volk, D.E.; Gorenstein, D.G.; Boggara, M.B.; et al. Insight into NSAID-induced membrane alterations, pathogenesis and therapeutics: Characterization of interaction of NSAIDs with phosphatidylcholine. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2012, 1821, 994–1002. [Google Scholar] [CrossRef] [Green Version]
- Smith, W.L.; DeWitt, D.L.; Garavito, R.M. Cyclooxygenases: Structural, cellular, and molecular biology. Annu. Rev. Biochem. 2000, 69, 145–182. [Google Scholar] [CrossRef]
- Smith, W.L.; Urade, Y.; Jakobsson, P.J. Enzymes of the Cyclooxygenase Pathways of Prostanoid Biosynthesis. Chem. Rev. 2011, 111, 5821–5865. [Google Scholar] [CrossRef]
- Conaghan, P.G. A turbulent decade for NSAIDs: Update on current concepts of classification, epidemiology, comparative efficacy, and toxicity. Rheumatol. Int. 2012, 32, 1491–1502. [Google Scholar] [CrossRef] [PubMed]
- Pande, A.H.; Qin, S.; Tatulian, S.A. Membrane fluidity is a key modulator of membrane binding, insertion, and activity of 5-lipoxygenase. Biophys. J. 2005, 88, 4084–4094. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, W.E. t.; Robinson, J.M.; Kniss, D.A. Despite transcriptional and functional coordination, cyclooxygenase-2 and microsomal prostaglandin E synthase-1 largely reside in distinct lipid microdomains in WISH epithelial cells. J. Histochem. Cytochem. 2005, 53, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, P.; Ciatto, G.; Aubert, N.; Goldmann, M. Soft Interfaces and Resonant Investigation on Undulator Source: A Surface X-ray Scattering Beamline to Study Organic Molecular Films at the SOLEIL Synchrotron. Sci. Adv. Mater. 2014, 6, 2312–2316. [Google Scholar] [CrossRef]
- Lucio, M.; Bringezu, F.; Reis, S.; Lima, J.L.F.C.; Brezesinski, G. Binding of nonsteroidal anti-inflammatory drugs to DPPC: Structure and thermodynamic aspects. Langmuir 2008, 24, 4132–4139. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ΧDPPC:lico | Amin (Å2) | A30 (Å2) | Cs−1 (mN m−1) | πcollapse (mN m−1) |
|---|---|---|---|---|
| 10:0 | 56 ± 1 | 57 ± 1 | 255 ± 21 | 54 ± 1 |
| 9.5:0.5 | 56 ± 2 | 57 ± 2 | 256 ± 20 | 54 ± 1 |
| 9:1 | 57 ± 1 | 59 ± 1 | 241 ± 20 | 54 ± 2 |
| 8:2 | 59 ± 2 | 62 ± 2 | 232 ± 20 | 55 ± 1 |
| ΧDPPC:lico | θ (°) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 10:0 | 4.46 | 4.29 | 4.14 | 1.8 | 12.6 | 9.2 | 0.522 | 0.857 | 30 |
| 9:1 | 4.53 | 4.28 | - | 3.1 | 15.4 | - | 0.534 | 0.856 | 29 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereira-Leite, C.; Lopes-de-Campos, D.; Fontaine, P.; Cuccovia, I.M.; Nunes, C.; Reis, S. Licofelone-DPPC Interactions: Putting Membrane Lipids on the Radar of Drug Development. Molecules 2019, 24, 516. https://doi.org/10.3390/molecules24030516
Pereira-Leite C, Lopes-de-Campos D, Fontaine P, Cuccovia IM, Nunes C, Reis S. Licofelone-DPPC Interactions: Putting Membrane Lipids on the Radar of Drug Development. Molecules. 2019; 24(3):516. https://doi.org/10.3390/molecules24030516
Chicago/Turabian StylePereira-Leite, Catarina, Daniela Lopes-de-Campos, Philippe Fontaine, Iolanda M. Cuccovia, Cláudia Nunes, and Salette Reis. 2019. "Licofelone-DPPC Interactions: Putting Membrane Lipids on the Radar of Drug Development" Molecules 24, no. 3: 516. https://doi.org/10.3390/molecules24030516