
Fast Amide Bond Cleavage Assisted by a Secondary Amino and a Carboxyl Group—A Model for yet Unknown Peptidases?

 , and
, and
Abstract
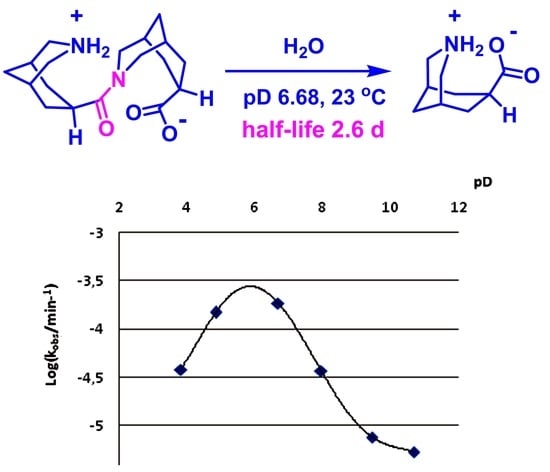
:
1. Introduction
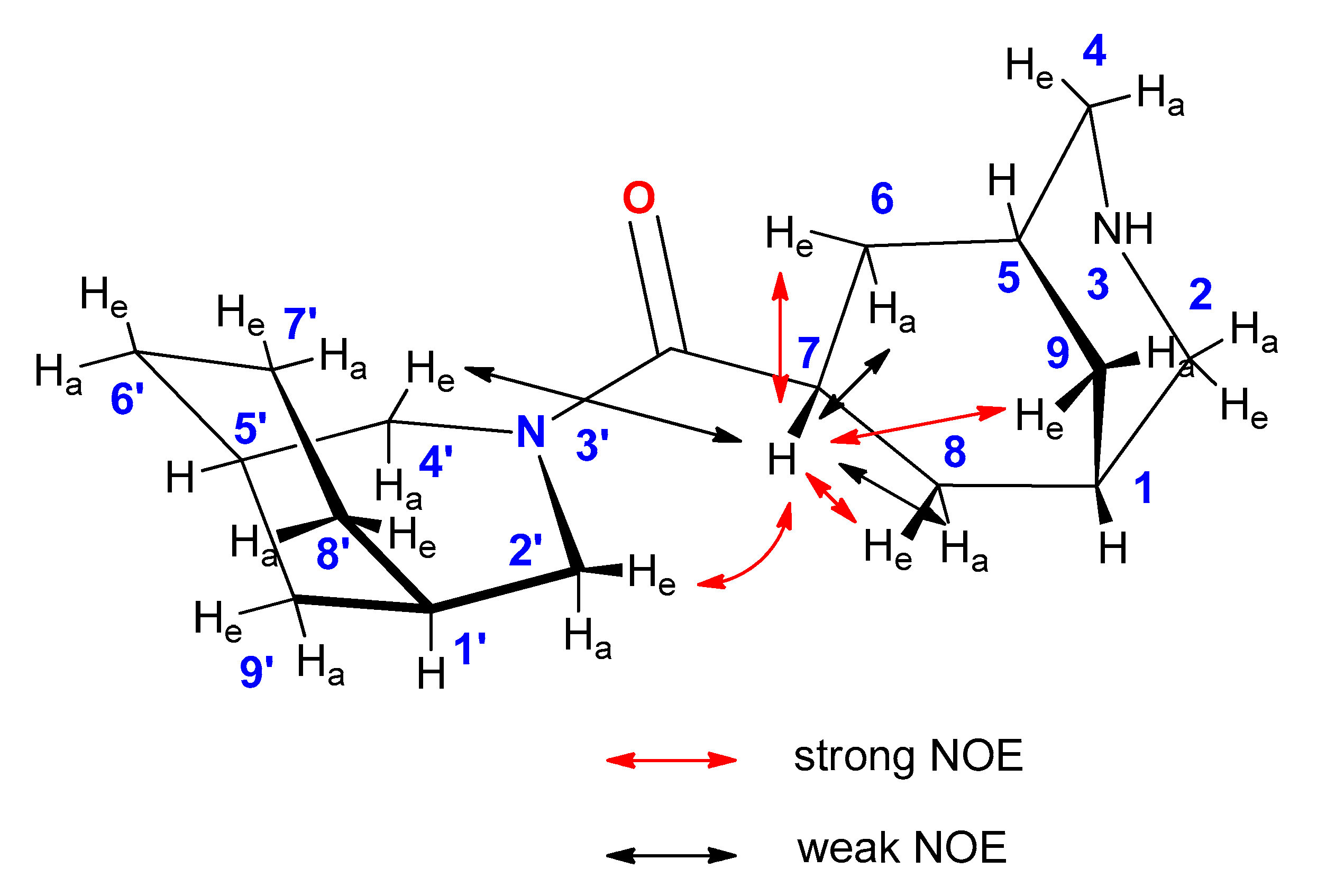
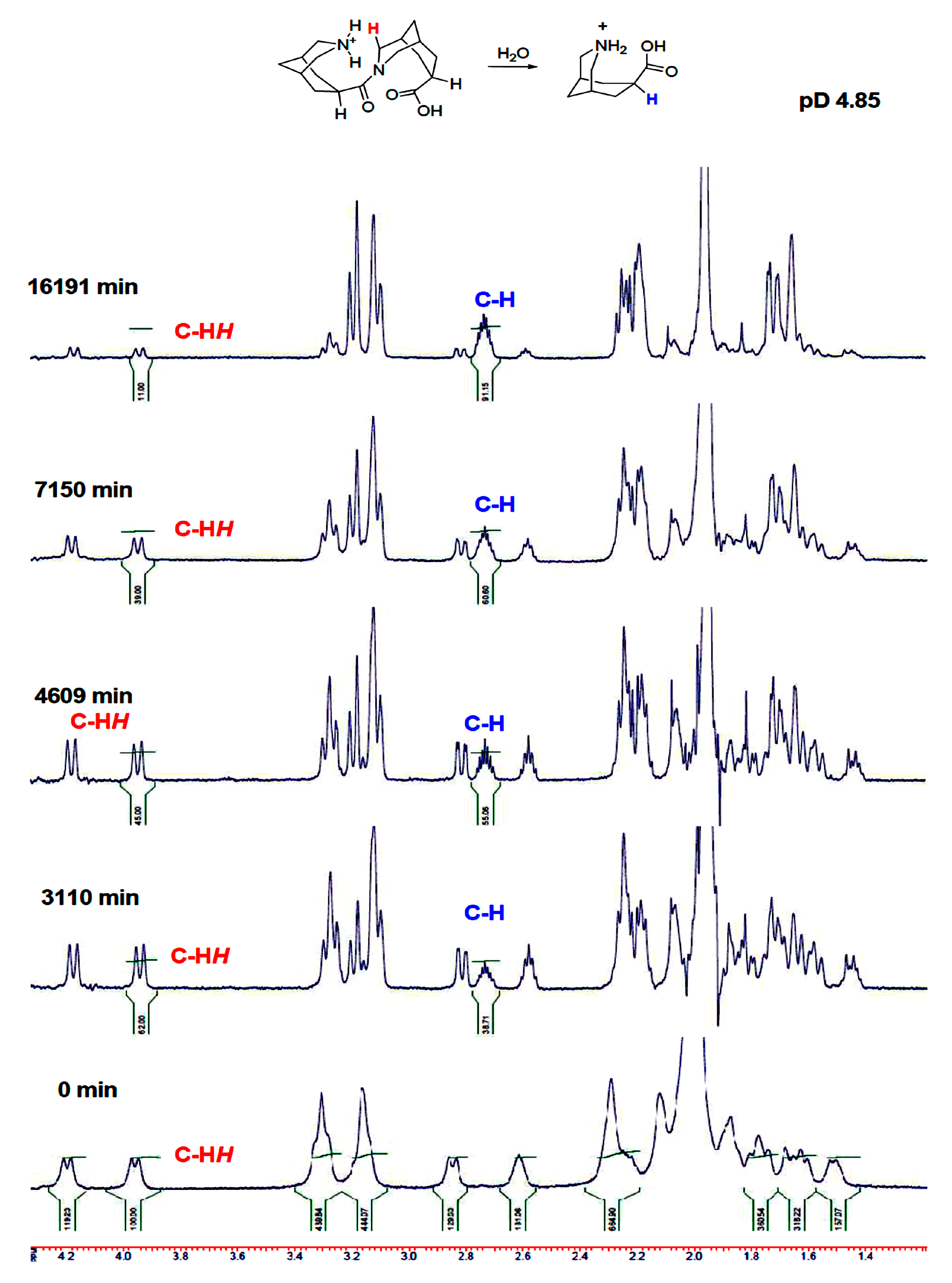
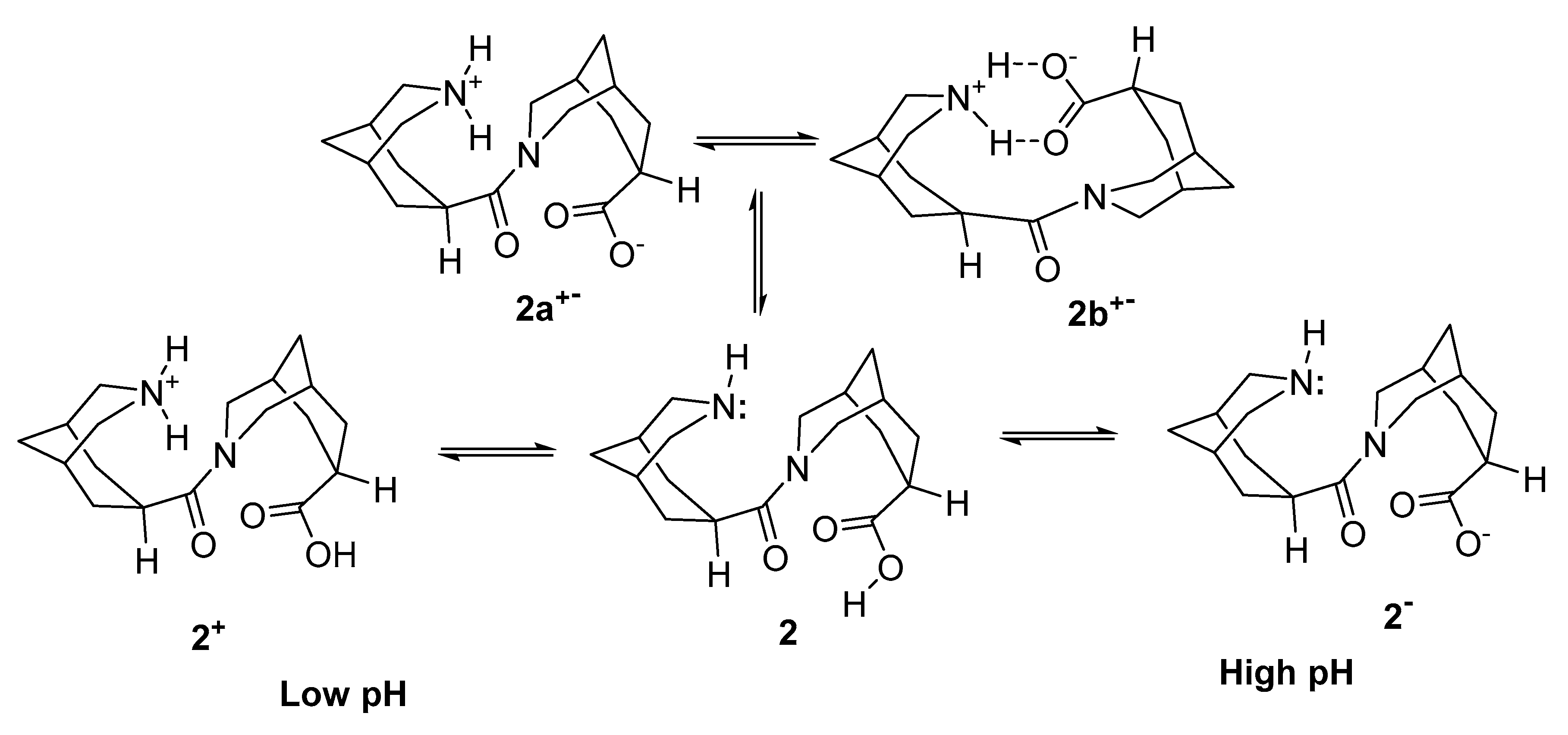
2. Results
3. Discussion
4. Materials and Methods
4.1. General
4.2. Synthetic Procedures
4.2.1. Kinetic Measurements for Hydrolysis Reaction of Compound 2 in D2O
4.2.2. Estimation of the Kinetic Isotope Effect by Comparison of the Hydrolysis Rates of 2·HCl in D2O and H2O
4.2.3. X-ray Data for Compound 13
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Komarov, I.V.; Yanik, S.; Ishchenko, A.Y.; Davies, J.E.; Goodman, J.M.; Kirby, A.J. The Most Reactive Amide As a Transition-State Mimic For cis–trans Interconversion. J. Am. Chem. Soc. 2015, 137, 926–930. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Szostak, M. Twisted Amides: From Obscurity to Broadly Useful Transition-Metal-Catalyzed Reactions by N–C Amide Bond Activation. Chem. Eur. J. 2017, 23, 7157–7173. [Google Scholar] [CrossRef] [PubMed]
- Szostak, M.; Aube, J. Chemistry of bridged lactams and related heterocycles. Chem. Rev. 2013, 113, 5701–5765. [Google Scholar] [CrossRef] [PubMed]
- Clayden, J.; Moran, W.J. The Twisted Amide 2-Quinuclidone: 60 Years in the Making. Angew. Chem. Int. Ed. 2006, 45, 7118–7120. [Google Scholar] [CrossRef] [PubMed]
- Bender, M.L. General acid-base catalysis in the intramolecular hydrolysis of phthalamic acid. J. Am. Chem. Soc. 1957, 79, 1258–1259. [Google Scholar] [CrossRef]
- Kirby, A.J.; Lancaster, P.W. Structure and efficiency in intramolecular and enzymic catalysis. Catalysis of amide hydrolysis by the carboxy-group of substituted maleamic acids. J. Chem. Soc. Perkin Trans. 2 1972, 9, 1206–1214. [Google Scholar] [CrossRef]
- Kirby, A.J.; McDonald, R.S.; Smith, C.R. Intramolecular catalysis of amide hydrolysis by two carboxy-groups. J. Chem. Soc. Perkin Trans. 2 1974, 12, 1495–1504. [Google Scholar] [CrossRef]
- Groves, J.T.; Olson, J.R. Models of zinc-containing proteases. Rapid amide hydrolysis by an unusually acidic Zn2+-OH2 complex. Inorg.Chem. 1985, 24, 2715–2717. [Google Scholar] [CrossRef]
- Menger, F.M.; Ladika, M. Fast hydrolysis of an aliphatic amide at neutral pH and ambient temperature. A peptidase model. J. Am. Chem. Soc. 1988, 110, 6794–6796. [Google Scholar] [CrossRef]
- Curran, T.P.; Borysenko, C.W.; Abelleira, S.M.; Messier, R.J. Intramolecular acylolysis of amide derivatives of Kemp’s triacid: Strain effects and reaction rates. J. Org. Chem. 1994, 59, 3522–3529. [Google Scholar] [CrossRef]
- Dougan, M.L.; Chin, J.L.; Solt, K.; Hansen, D.E. Rapid cleavage of cyclic tertiary amides of Kemp’s triacid: Effects of ring structure. Bioorg. Med. Chem. Lett. 2004, 14, 4153–4156. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, N.M.; Fache, F.; Rosen, M.; Nguyen, P.L.; Hansen, D.E. Rapid Cleavage of Unactivated, Unstrained Amide Bonds at Neutral pH. J. Org. Chem. 2008, 73, 6413–6416. [Google Scholar] [CrossRef] [PubMed]
- Souza, B.S.; Mora, J.R.; Wanderlind, E.H.; Clementin, R.M.; Gesser, J.C.; Fiedler, H.D.; Nome, F.; Menger, F.M. Transforming a stable amide into a highly reactive one: Capturing the essence of enzymatic catalysis. Angew. Chem. Int. Ed. 2017, 56, 5345–5348. [Google Scholar] [CrossRef] [PubMed]
- Kirby, A.J. Enzyme Mechanisms, Models, and Mimics. Angew. Chem. Int. Ed. 1996, 35, 707–724. [Google Scholar] [CrossRef]
- Bender, M.L.; Chow, Y.-L.; Chloupek, F. Intramolecular Catalysis of Hydrolytic Reactions. II. The Hydrolysis of Phthalamic Acid. J. Am. Chem. Soc. 1958, 80, 5380–5384. [Google Scholar] [CrossRef]
- Jindal, G.; Warshel, A. Misunderstanding the preorganization concept can lead to confusions about the origin of enzyme catalysis. Proteins 2017, 85, 2157–2161. [Google Scholar] [CrossRef]
- Kubyshkin, V.; Budisa, N. Amide rotation trajectories probed by symmetry. Org. Biomol. Chem. 2017, 15, 6764–6772. [Google Scholar] [CrossRef] [Green Version]
- Hu, F.; Nareddy, P.; Lalancette, R.; Jordan, F.; Szostak, M. σ N–C Bond Difunctionalization in Bridged Twisted Amides: Sew-and-Cut Activation Approach to Functionalized Isoquinolines. Org. Lett. 2017, 19, 2386–2389. [Google Scholar] [CrossRef]
- Meng, G.; Shi, S.; Lalancette, R.; Szostak, R.; Szostak, M. Reversible Twisting of Primary Amides via Ground State N–C (O) Destabilization: Highly Twisted Rotationally Inverted Acyclic Amides. J. Am. Chem. Soc. 2018, 140, 727–734. [Google Scholar] [CrossRef]
- Meng, G.; Shi, S.; Szostak, M. Cross-coupling of amides by N–C bond activation. Synlett 2016, 27, 2530–2540. [Google Scholar] [CrossRef]
- Dander, J.E.; Garg, N.K. Breaking amides using nickel catalysis. ACS Catal. 2017, 7, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Adachi, S.; Kumagai, N.; Shibasaki, M. Conquering amide planarity: Structural distortion and its hidden reactivity. Tetrahedron Lett. 2018, 59, 1147–1158. [Google Scholar] [CrossRef]
- Wybon, C.C.; Mensch, C.; Hollanders, K.; Gadais, C.; Herrebout, W.A.; Ballet, S.; Maes, B.U. Zn-catalyzed tert-butyl nicotinate-directed amide cleavage as a biomimic of metallo-exopeptidase activity. ACS Catal. 2018, 8, 203–218. [Google Scholar] [CrossRef]
- Balachandra, C.; Sharma, N.K. Instability of Amide Bond Comprising the 2-Aminotropone Moiety: Cleavable under Mild Acidic Conditions. Org. Lett. 2015, 17, 3948–3951. [Google Scholar] [CrossRef] [PubMed]
- Hutchby, M.; Houlden, C.E.; Haddow, M.F.; Tyler, S.N.; Lloyd-Jones, G.C.; Booker-Milburn, K.I. Switching Pathways: Room-Temperature Neutral Solvolysis and Substitution of Amides. Angew. Chem. Int. Ed. 2012, 51, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Milović, N.M.; Kostić, N.M. Palladium (II) complexes, as synthetic peptidases, regioselectively cleave the second peptide bond “upstream” from methionine and histidine side chains. J. Am. Chem. Soc. 2002, 124, 4759–4769. [Google Scholar] [CrossRef]
- Hohage, O.; Sheldrick, W.S. Cisplatin mediates selective downstream hydrolytic cleavage of Met-(Gly) n-His segments (n= 1, 2) in methionine-and histidine-containing peptides: The role of ammine loss trans to the initial Pt-S (Met) anchor in facilitating amide hydrolysis. J. Inorg. Biochem. 2006, 100, 1506–1513. [Google Scholar] [CrossRef] [PubMed]
- Rajković, S.; Živković, M.D.; Kállay, C.; Sóvágó, I.; Djuran, M.I. A study of the reactions of a methionine-and histidine-containing tetrapeptide with different Pd (II) and Pt (II) complexes: Selective cleavage of the amide bond by platination of the peptide and steric modification of the catalyst. Dalton Trans. 2009, 39, 8370–8377. [Google Scholar] [CrossRef]
- Barrios, A.M.; Lippard, S.J. Interaction of Urea with a Hydroxide-Bridged Dinuclear Nickel Center: An Alternative Model for the Mechanism of Urease. J. Am. Chem. Soc. 2000, 122, 9172–9177. [Google Scholar] [CrossRef]
- Uprety, B.; Arderne, C.; Bernal, I. Catalytic Cleavage of the Amide Bond in Urea Using a Cobalt(III) Amino-Based Complex. Eur. J. Inorg. Chem. 2018, 5058–5067. [Google Scholar] [CrossRef]
- MacDonald, M.J.; Lavis, L.D.; Hilvert, D.; Gellman, S.H. Evaluation of the Ser-His dipeptide, a putative catalyst of amide and ester hydrolysis. Org. Lett. 2016, 18, 3518–3521. [Google Scholar] [CrossRef] [PubMed]
- Menger, F.M.; Ladika, M. Remote enzyme-coupled amine release. J. Org. Chem. 1990, 55, 3006–3007. [Google Scholar] [CrossRef]
- Blagoeva, I.B.; Kirby, A.J. Intramolecular nucleophilic catalysis of anilide hydrolysis by pyrimidine nitrogen. J. Chem. Soc. Perkin Trans. 2 1985, 7, 1017–1020. [Google Scholar] [CrossRef]
- McEuten, J.M.; Nelson, R.P.; Lawton, R.G. The α,α′ Annelation of Cyclic Ketones. Synthesis and Conformational Properties of Bicyclo[3.3.l]nonanone Derivatives. J. Org. Chem. 1970, 35, 690–696. [Google Scholar] [CrossRef]
- Ishchenko, A.Y.; Yanik, S.; Rusanov, E.B.; Komarov, I.V.; Kirby, A.J. An Expedient and Practical Approach to Functionalized 3-Aza-, 3-Oxa-, and 3-Thiabicyclo [3.3.1] nonane Systems. Synthesis 2015, 47, 367. [Google Scholar] [CrossRef]
- Komppa, G. Über ein neues bicyclisches Imin, das Iso-granatanin. Ber. Dtsch. Chem. Ges. 1932, 65, 792–793. [Google Scholar] [CrossRef]
- Rossi, S.; Valvo, C. Several derivatives of 3-azabicyclo-(3, 3, 1)-nonane; pharmacological activity. I. Il Farmaco 1957, 12, 1008–1015. [Google Scholar]
- Wimley, W.C.; Gawrisch, K.; Creamer, T.P.; White, S.H. Direct measurement of salt-bridge solvation energies using a peptide model system: Implications for protein stability. Proc. Natl. Acad. Sci. USA 1996, 93, 2985–2990. [Google Scholar] [CrossRef]
- Webb, E.C. Enzyme Nomenclature 1992. Recommendations of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology on the Nomenclature and Classification of Enzymes, 6th ed.; Academic Press: San Diego, CA, USA, 1992; p. 863. ISBN 0122271653. [Google Scholar]
- Price, N.; Stevens, L. Fundamentals of Enzymology: The Cell and Molecular Biology of Catalytic Proteins, 3rd ed.; OUP Oxford: Oxford, UK, 2000; p. 496. ISBN 9780198502296. [Google Scholar]
- Seemuller, E.; Lupas, A.; Stock, D.; Lowe, J.; Huber, R.; Baumeister, W. Proteasome from Thermoplasma acidophilum: A threonine protease. Science 1995, 268, 579–582. [Google Scholar] [CrossRef]
- Fujinaga, M.; Cherney, M.M.; Oyama, H.; Oda, K.; James, M.N. The molecular structure and catalytic mechanism of a novel carboxyl peptidase from Scytalidium lignicolum. Proc. Natl. Acad. Sci. USA 2004, 101, 3364–3369. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Bateman, A. Asparagine peptide lyases: A seventh catalytic type of proteolytic enzymes. J. Biol. Chem. 2011, 286, 38321–38328. [Google Scholar] [CrossRef] [PubMed]
- Paulus, H. Protein splicing and related forms of protein autoprocessing. Annu. Rev. Biochem. 2000, 69, 447–496. [Google Scholar] [CrossRef] [PubMed]
- Noren, C.J.; Wang, J.; Perler, F.B. Dissecting the chemistry of protein splicing and its applications. Angew. Chem. Int. Ed. 2000, 39, 450–466. [Google Scholar] [CrossRef]
- Sun, P.; Ye, S.; Ferrandon, S.; Evans, T.C.; Xu, M.Q.; Rao, Z. Crystal structures of an intein from the split dnaE gene of Synechocystis sp. PCC6803 reveal the catalytic model without the penultimate histidine and the mechanism of zinc ion inhibition of protein splicing. J. Mol. Biol. 2005, 353, 1093–1105. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Frutos, S.; Bick, M.J.; Vila-Perelló, M.; Debelouchina, G.T.; Darst, S.A.; Muir, T.W. Structure of the branched intermediate in protein splicing. Proc. Natl. Acad. Sci. USA 2014, 111, 8422–8427. [Google Scholar] [CrossRef] [PubMed]
- Kirby, A.J.; Komarov, I.V.; Feeder, N. Synthesis, structure and reactions of the most twisted amide. J. Chem. Soc. Perkin Trans. 2 2001, 4, 522–529. [Google Scholar] [CrossRef]
- Lizak, C.; Gerber, S.; Numao, S.; Aebi, M.; Locher, K.P. X-ray structure of a bacterial oligosaccharyltransferase. Nature 2011, 474, 350. [Google Scholar] [CrossRef] [PubMed]
- Cleland, W.W.; Andrews, T.J.; Gutteridge, S.; Hartman, F.C.; Lorimer, G.H. Mechanism of Rubisco: The carbamate as general base. Chem. Rev. 1998, 98, 549–562. [Google Scholar] [CrossRef]
- Komiya, C.; Aihara, K.; Morishita, K.; Ding, H.; Inokuma, T.; Shigenaga, A.; Otaka, A. Development of an intein-inspired amide cleavage chemical device. J. Org. Chem. 2015, 81, 699–707. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C. Purification of Laboratory Chemicals, 7th ed.; Butterworth-Heinemann: Oxford, UK, 2013; p. 1024. ISBN 9780123821614. [Google Scholar]
- Peters, J.A.; Van Der Toorn, J.M.; Van Bekkum, H. 3, 7-disubstituted bicyclo [3.3.1] nonanes—III: Synthesis and conformation of bicyclo[3.3.1]nonane-3α, 7α-dicarboxylic acid, its dimethyl ester and some other 3,7-disubstituted bicyclo[3.3.1]nonanes; adamantane as an integrated holding system. Tetrahedron 1975, 31, 2273–2281. [Google Scholar] [CrossRef]
- Połoński, T.; Pham, M.; Milewska, M.J. Structure, Conformation, and Stereodynamics of N-Nitroso-2,4-diaryl-3-azabicyclo[3.3.1]nonanes and N-Nitroso-2,4-diaryl-3-azabicyclo[3.3.1]nonan-9- ones. J. Org. Chem. 1996, 61, 3766–3772. [Google Scholar] [CrossRef]
- Hwang, T.-L.; Shaka, A.J. Water Suppression that Works. Excitation Sculpting Using Arbitrary Waveform and Pulsed Field Gradient. J. Magn. Res. Ser. A 1995, 112, 275–279. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 13, 14 are available from the authors. |
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
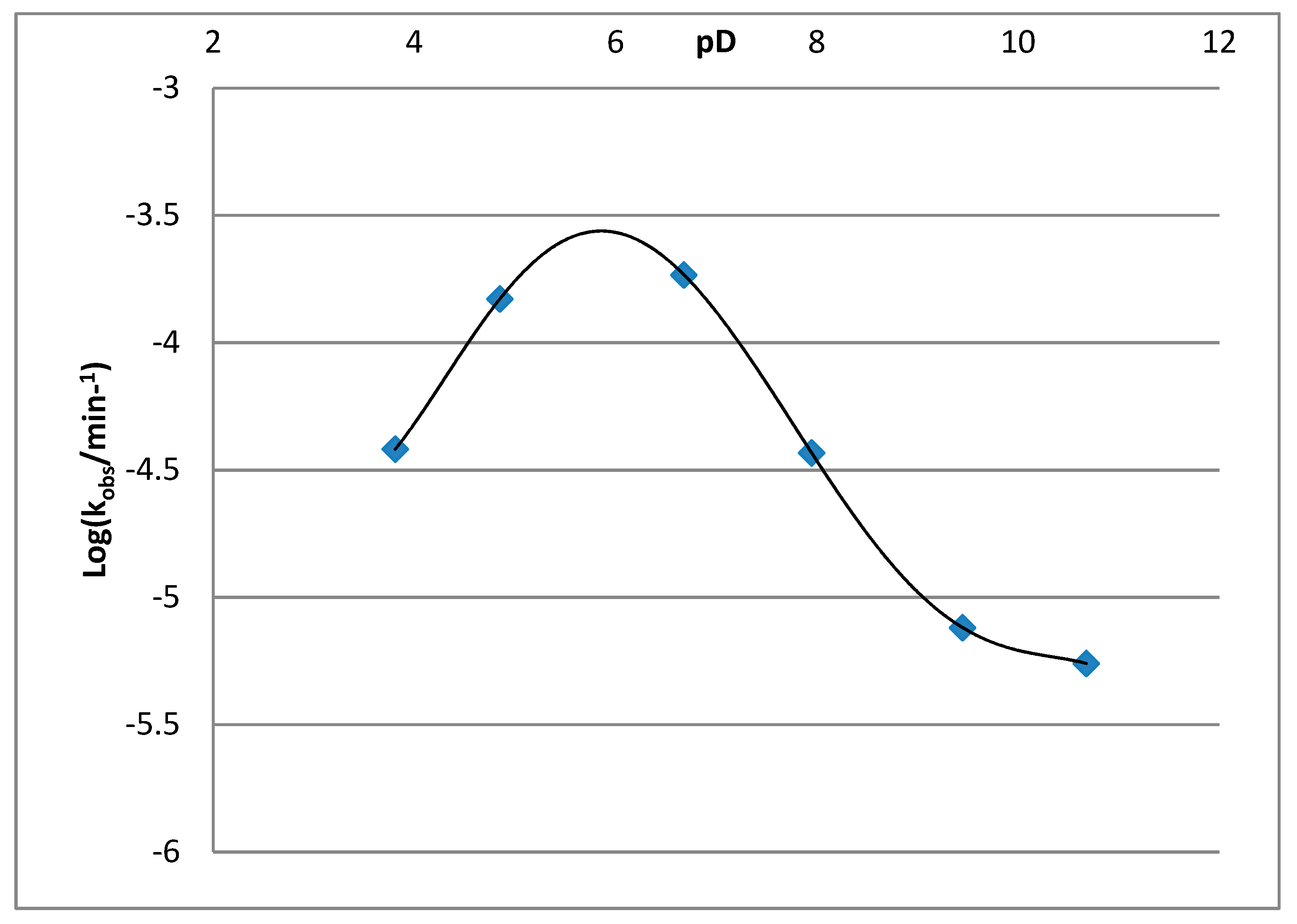
| pD Value | Half-life of 2·HCl, min | kobs, min−1 | Log(kobs) |
|---|---|---|---|
| 3.81 | 18,138 | 3.821 × 10−5 | −4.418 |
| 4.85 | 4668 | 1.485 × 10−4 | −3.828 |
| 6.68 | 3748 | 1.849 × 10−4 | −3.733 |
| pD Value | Half-life of 2·HCl, min | kobs, min−1 | Log(kobs) |
|---|---|---|---|
| 7.95 | 18,802 | 3.686 × 10−5 | −4.433 |
| 9.45 | 91,125 | 7.605 × 10−6 | −5.119 |
| 10.68 | 126,114 | 5.496 × 10−6 | −5.260 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
V. Komarov, I.; Yu. Ishchenko, A.; Hovtvianitsa, A.; Stepanenko, V.; Kharchenko, S.; D. Bond, A.; J. Kirby, A. Fast Amide Bond Cleavage Assisted by a Secondary Amino and a Carboxyl Group—A Model for yet Unknown Peptidases? Molecules 2019, 24, 572. https://doi.org/10.3390/molecules24030572
V. Komarov I, Yu. Ishchenko A, Hovtvianitsa A, Stepanenko V, Kharchenko S, D. Bond A, J. Kirby A. Fast Amide Bond Cleavage Assisted by a Secondary Amino and a Carboxyl Group—A Model for yet Unknown Peptidases? Molecules. 2019; 24(3):572. https://doi.org/10.3390/molecules24030572
Chicago/Turabian StyleV. Komarov, Igor, Aleksandr Yu. Ishchenko, Aleksandr Hovtvianitsa, Viacheslav Stepanenko, Serhii Kharchenko, Andrew D. Bond, and Anthony J. Kirby. 2019. "Fast Amide Bond Cleavage Assisted by a Secondary Amino and a Carboxyl Group—A Model for yet Unknown Peptidases?" Molecules 24, no. 3: 572. https://doi.org/10.3390/molecules24030572