
New Multitarget Hybrids Bearing Tacrine and Phenylbenzothiazole Motifs as Potential Drug Candidates for Alzheimer’s Disease

 , , and
, , and
Abstract
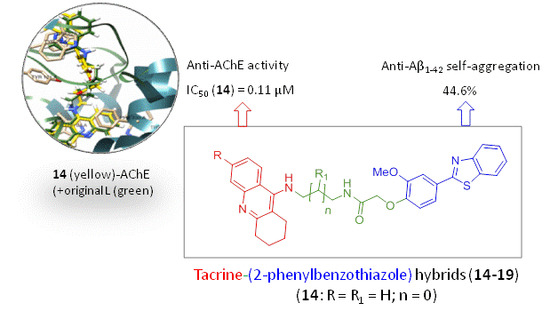
:
1. Introduction
2. Results and Discussion
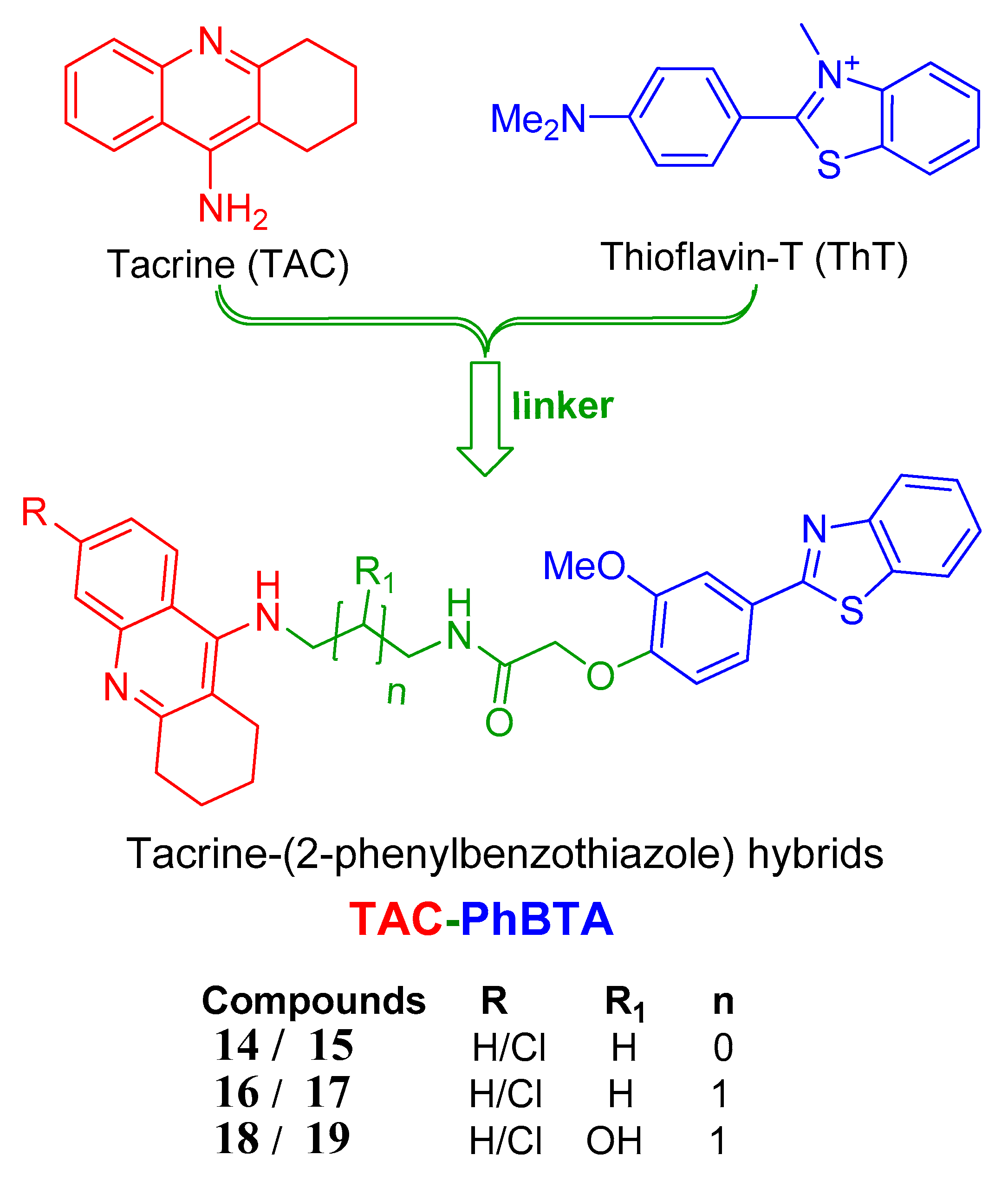
2.1. Molecular Design and Modelling
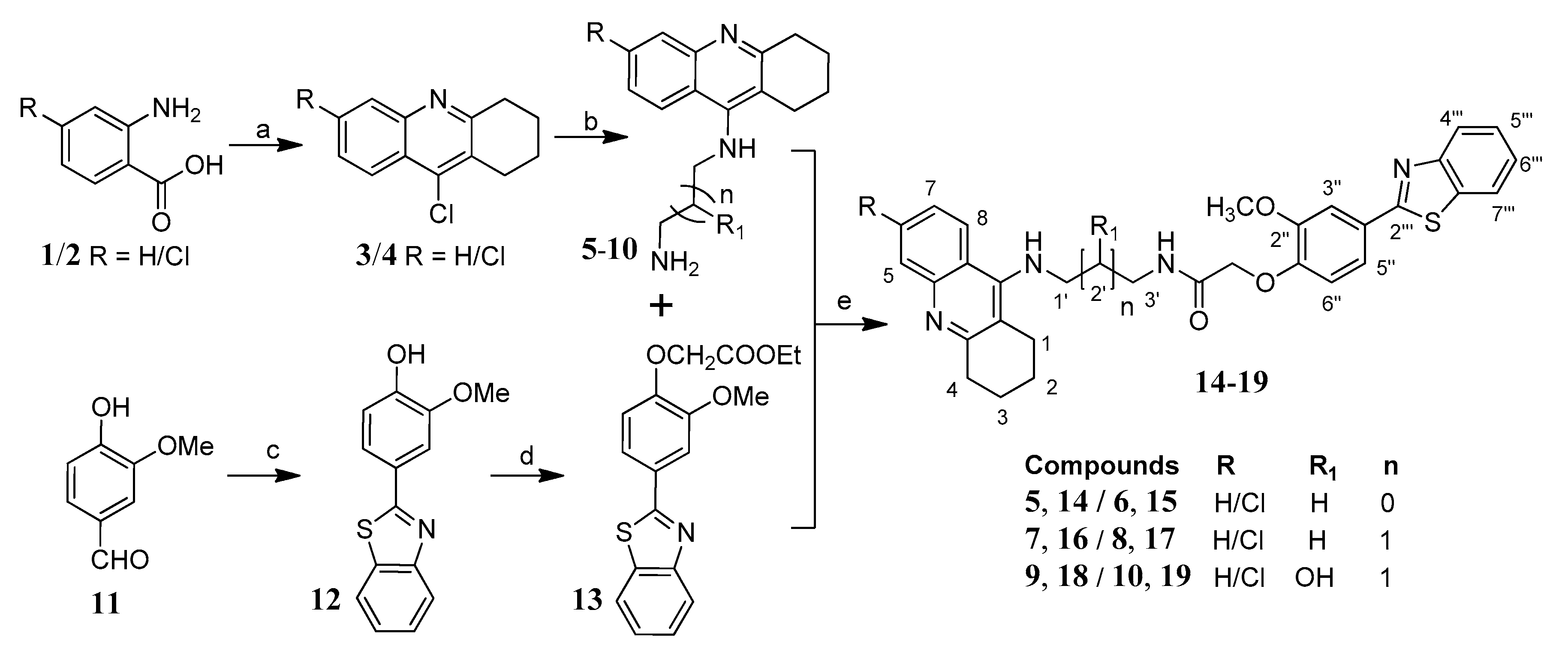
2.2. Chemistry
2.3. Biological Studies
2.3.1. Acetylcholinesterase Inhibition
2.3.2. Inhibition of Amyloid β Self-Aggregation
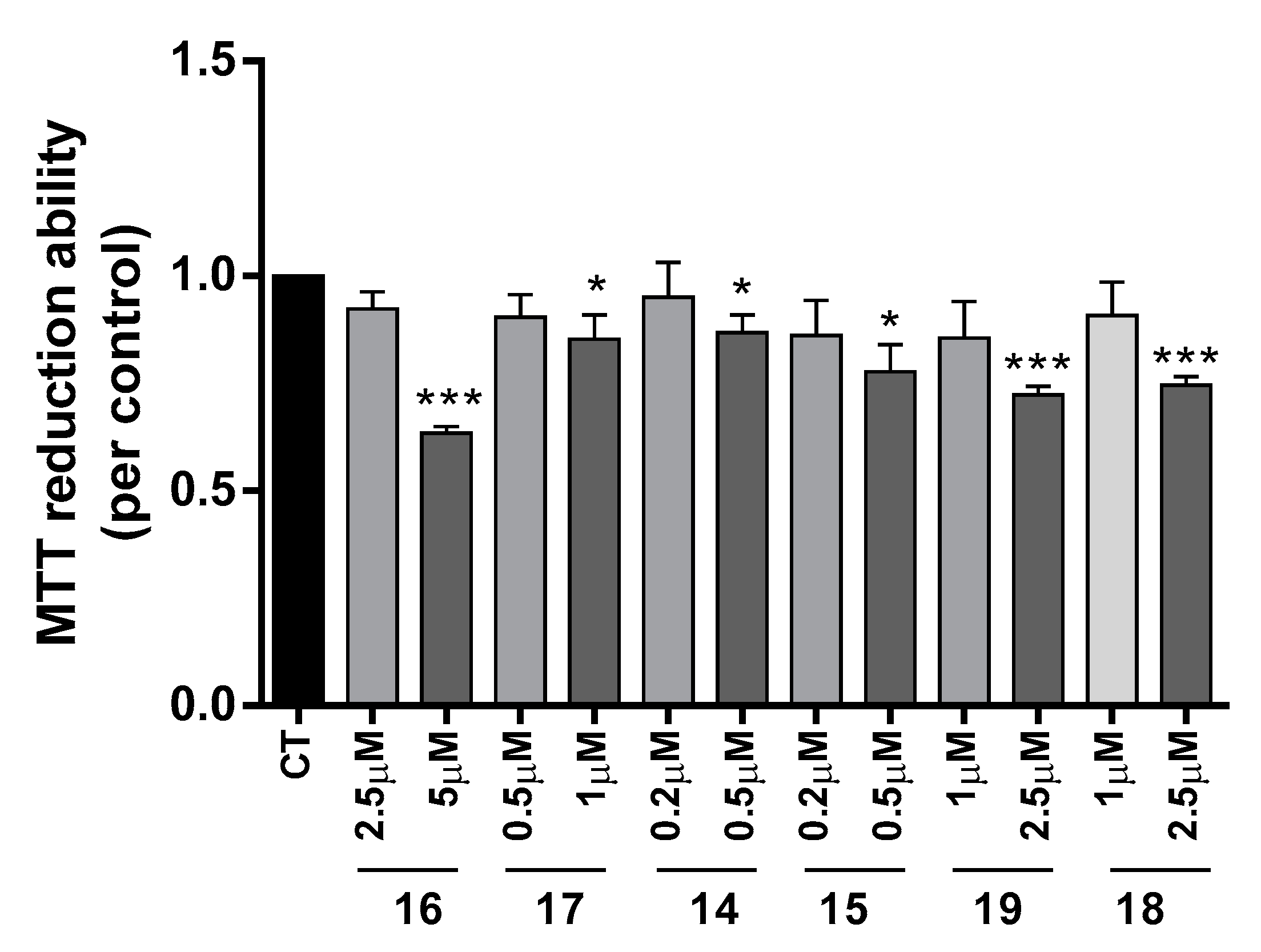
2.3.3. Neuroprotective Effect of Tacrine-Phenylbenzothiazole Hybrids
2.3.4. Pharmacokinetic Characterization
3. Materials and Methods
3.1. Experimental
3.1.1. Equipment/Reagents
3.1.2. Acetylcholinesterase Assay
3.1.3. Aβ Aggregation Assay
3.1.4. Cell Viability and Neuroprotection
3.1.5. Molecular Modelling and Pharmacokinetic Properties
3.1.6. Synthesis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Association. Alzheimer’s disease facts and figures. Alzheimers Dement. 2017, 13, 325–373. [Google Scholar]
- GBD 2015 Neurological Disorders Collaborator Group. Global, regional, and national burden of neurological disorders during 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 2017, 16, 877–897. [Google Scholar] [CrossRef]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alz. Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, J.; Aisen, P.S.; DuBois, B.; Frölich, L.; Jack, C.R.; Jones, R.W.; Morris, J.C.; Raskin, J.; Dowsett, S.A.; Scheltens, P. Drug development in Alzheimer’s disease: the path to 2025. Alzheimers Res. Ther. 2016, 8, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Small, G.; Bullock, R. Defining optimal treatment with cholinesterase inhibitors in Alzheimer’s disease. Alzheimer Dement. 2011, 7, 177–184. [Google Scholar] [CrossRef]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-target-directed ligands to combat neurodegenerative diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef] [PubMed]
- Guzior, N.; Więckowska, A.; Panek, D.; Malawska, B. Recent Development of Multifunctional Agents as Potential Drug Candidates for the Treatment of Alzheimer’s Disease. Curr. Med. Chem. 2015, 22, 373–404. [Google Scholar] [CrossRef]
- Ismaili, L.; Refouvelet, B.; Benchekroun, M.; Brogi, S.; Brindisi, M.; Gemma, S.; Campiani, G.; Filipic, S.; Agbaba, D.; Esteban, G.; et al. Multitarget compounds bearing tacrine- and donepezil-like structural and functional motifs for the potential treatment of Alzheimer’s disease. Prog. Neurobiol. 2017, 151, 4–34. [Google Scholar] [CrossRef]
- Santos, M.A.; Chand, K.; Chaves, S. Recent progress in repositioning Alzheimer’s disease drugs based on a multitarget strategy. Fut. Med. Chem. 2016, 8, 2113–2142. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 73, 1–14. [Google Scholar] [CrossRef]
- Spilovska, K.; Korabecny, J.; Nepovimova, E.; Dolezal, R.; Mezeiova, E.; Soukup, O.; Kuca, K. Multitarget Tacrine Hybrids with Neuroprotective Properties to Confront Alzheimer’s Disease. Curr. Top Med. Chem. 2017, 17, 1006–1026. [Google Scholar] [CrossRef] [PubMed]
- Spilovska, K.; Korabecny, J.; Sepsova, V.; Jun, D.; Hrabinova, M.; Jost, P.; Muckova, L.; Soukup, O.; Janockova, J.; Kucera, T.; et al. Novel Tacrine-Scutellarin Hybrids as Multipotent Anti-Alzheimer’s Agents: Design, Synthesis and Biological Evaluation. Molecules. 2017, 22, 1006. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.A.; Chand, K.; Chaves, S. Recent progress in multifunctional metal chelators as potential drugs for Alzheimer’s disease. Coord. Chem. Rev. 2016, 327–328, 287–303. [Google Scholar] [CrossRef]
- Hiremathad, A.; Keri, R.S.; Esteves, A.R.; Cardoso, S.M.; Chaves, S.; Santos, M.A. Novel tacrine-hydroxyphenylbenzimidazole hybrids as potential multitarget drug candidates for Alzheimer’s disease. Eur. J. Med. Chem. 2018, 148, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Piemontese, L.; Tomás, D.; Hiremathad, A.; Capriati, V.; Candeias, E.; Cardoso, S.M.; Chaves, S.; Santos, M.A. Donepezil structure-based hybrids as potential multifunctional anti-Alzheimer’s drug candidates. J. Enz. Inhib. Med. Chem. 2018, 33, 1212–1224. [Google Scholar] [CrossRef] [PubMed]
- Keri, R.S.; Quintanova, C.; Marques, S.M.; Cardoso, S.M.; Santos, M.A. Design, synthesis and neuroprotective evaluation of novel tacrine-benzothiazole hybrids as multi-targeted compounds against Alzheimer’s disease. Bioorg. Med. Chem. 2013, 21, 4559–4569. [Google Scholar] [CrossRef] [PubMed]
- Hroch, L.; Aitken, L.; Benek, O.; Dolezal, M.; Kuca, K.; Gunn-Moore, F.; Musilek, K. Benzothiazoles-scaffold of interest for CNS targeted drugs. Curr. Med. Chem. 2015, 22, 730–747. [Google Scholar] [CrossRef]
- Jia, J.; Zhou, K.; Dai, J.; Liu, B. Cui, M. 2-Arylbenzothiazoles labeled with [CpRe/Tc-99m(CO)(3)] and evaluated as beta-amyloid imaging probes. Eur. J. Med. Chem. 2016, 124, 763–772. [Google Scholar] [CrossRef]
- Kiritsis, C.; Mavroidi, B.; Shegani, A.; Palamaris, L.; Loudos, G.; Sagnou, M.; Pirmettis, I.; Papadopoulos, M.; Pelecanou, M. 2-(4′-Aminophenyl)benzothiazole Labeled with 99mTc-Cyclopentadienyl for Imaging β-Amyloid Plaques. ACS Med. Chem. Lett. 2017, 8, 1089–1092. [Google Scholar] [CrossRef]
- PDB, entry, 1ODC. Available online: http://www.rcsb.org/pdb/explore/explore.do?structureId=1ODC (accessed on 3 February 2017).
- Sebestik, J.; Marques, S.M.; Fale, P.L.; Santos, S.; Arduino, D.M.; Cardoso, S.M.; Oliveira, C.R.; Serralheiro, M.L.; Santos, M.A. Bifunctional phenolic-choline conjugates as anti-oxidants and acetylcholinesterase inhibitors. J. Enzyme Inhib. Med. Chem. 2011, 26, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, M.R.; Dyrager, C.; Hoarau, M.; Korshavn, K.J.; Lim, M.H.; Ramamoorthy, A.; Storr, T. Multifunctional quinoline-triazole derivatives as potential modulators of Aβ peptide aggregation. Inorg. Bio. Chem. 2016, 158, 131–138. [Google Scholar] [CrossRef]
- Rodríguez-Santiago, L.; Alí-Torres, J.; Vidossich, P.; Sodupea, M. Coordination properties of a metal chelator clioquinol to Zn2+ studied by static DFT and ab initio molecular dynamics. Phys. Chem. Chem. Phys. 2015, 17, 13582. [Google Scholar] [CrossRef] [PubMed]
- Chand, K.; Rajeshwari; Candeias, E.; Cardoso, S.M.; Chaves, S.; Santos, M.A. Tacrine-deferiprone hybrids as multi-target-directed metal chelators against Alzheimer’s disease: two in one drug. Metallomics 2018, 10, 1460–1475. [Google Scholar] [CrossRef] [PubMed]
- QikProp, version 2.5; Schrodinger, LLC: New York, NY, USA, 2005.
- Maestro, version 9.3; Schrodinger Inc.: Portland, OR, USA, 2012.
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Featherstone, R.M. A new and rapid colorimetric determi- nation of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Nunes, A.; Marques, S.M.; Quintanova, C.; Silva, D.F.; Cardoso, S.M.; Chaves, S.; Santos, M.A. Multifunctional iron-chelators with protective roles against neurodegenerative diseases. Dalton Trans. 2013, 42, 6058–6073. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Su, T.; Shan, W.; Luo, Z.; Sun, Y.; He, F.; Li, X. Inhibition of cholinesterase activity and amyloid aggregation by berberine-phenyl-benzoheterocyclic and tacrine-phenyl-benzoheterocyclic hybrids. Bioorg. Med. Chem. 2012, 20, 3038–3048. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Perring, D.D. Purification of Laboratory Chemicals, 4nd ed.; Butterworth Heinemann Press: Oxford, UK, 1999. [Google Scholar]
- Bartolini, M.; Bertucci, C.; Bolognesi, M.L.; Cavalli, A.; Melchiorre, C.; Andrisano, V. Insight into the kinetic of amyloid beta (1-42) peptide self-aggregation: elucidation of inhibitors’ mechanism of action. Chem. Biochem. 2007, 8, 2152–2161. [Google Scholar]
- Chao, X.; He, X.; Yang, Y.; Zhou, X.; Jin, M.; Liu, S.; Cheng, Z.; Liu, P.; Wang, Y.; Yu, J.; et al. Design, synthesis and pharmacological evaluation of novel tacrine–caffeic acid hybrids as multi-targeted compounds against Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2012, 22, 6498–6502. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Hassinen, T.; Perakyla, M. New energy terms for reduced protein models implemented in an off-lattice force field. J. Comput. Chem. 2001, 22, 1229–1242. [Google Scholar] [CrossRef]
- Hiremathad, A.; Chand, K.; Esteves, A.R.; Cardoso, S.M.; Ramsay, R.R.; Chaves, S.; Keri, R.S.; Santos, M.A. Tacrine-allyl/propargylcysteine-benzothiazole trihybrids as potential anti-Alzheimer’s drug. RSC Adv. 2016, 6, 53519–53532. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp | R | R1 | n | Inhibition of AChE (IC50, µM) b | Inhibition of Aβ-self-aggregation (%) c,d | MW | clog P e | log BB f | Caco-2 Permeability (nm/sec) |
|---|---|---|---|---|---|---|---|---|---|
| 14 | H | H | 0 | 0.11 | 44.6 | 538.7 | 6.200 | −0.927 | 1086 |
| 15 | Cl | H | 0 | 0.06 | 27.0 | 573.1 | 5.445 | −0.914 | 722 |
| 16 | H | H | 1 | 0.15 | 33.7 | 552.7 | 6.237 | −0.975 | 1057 |
| 17 | Cl | H | 1 | 0.13 | 32.5 | 587.1 | 6.726 | −0.739 | 1022 |
| 18 | H | OH | 1 | 0.27 | 31.1 | 568.6 | 5.222 | −1.592 | 388 |
| 19 | Cl | OH | 1 | 0.14 | 31.3 | 603.1 | 5.692 | −1.268 | 496 |
| TAC | - | - | - | 0.30 | 11 | 198.1 | - | - | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajeshwari, R.; Chand, K.; Candeias, E.; Cardoso, S.M.; Chaves, S.; Santos, M.A. New Multitarget Hybrids Bearing Tacrine and Phenylbenzothiazole Motifs as Potential Drug Candidates for Alzheimer’s Disease. Molecules 2019, 24, 587. https://doi.org/10.3390/molecules24030587
Rajeshwari R, Chand K, Candeias E, Cardoso SM, Chaves S, Santos MA. New Multitarget Hybrids Bearing Tacrine and Phenylbenzothiazole Motifs as Potential Drug Candidates for Alzheimer’s Disease. Molecules. 2019; 24(3):587. https://doi.org/10.3390/molecules24030587
Chicago/Turabian StyleRajeshwari, Rajeshwari, Karam Chand, Emanuel Candeias, Sandra M. Cardoso, Sílvia Chaves, and M. Amélia Santos. 2019. "New Multitarget Hybrids Bearing Tacrine and Phenylbenzothiazole Motifs as Potential Drug Candidates for Alzheimer’s Disease" Molecules 24, no. 3: 587. https://doi.org/10.3390/molecules24030587