
Ring Expansion of Alkylidenecarbenes Derived from Lactams, Lactones, and Thiolactones into Strained Heterocyclic Alkynes: A Theoretical Study

 and
and
Abstract
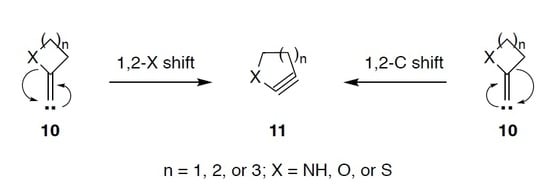
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
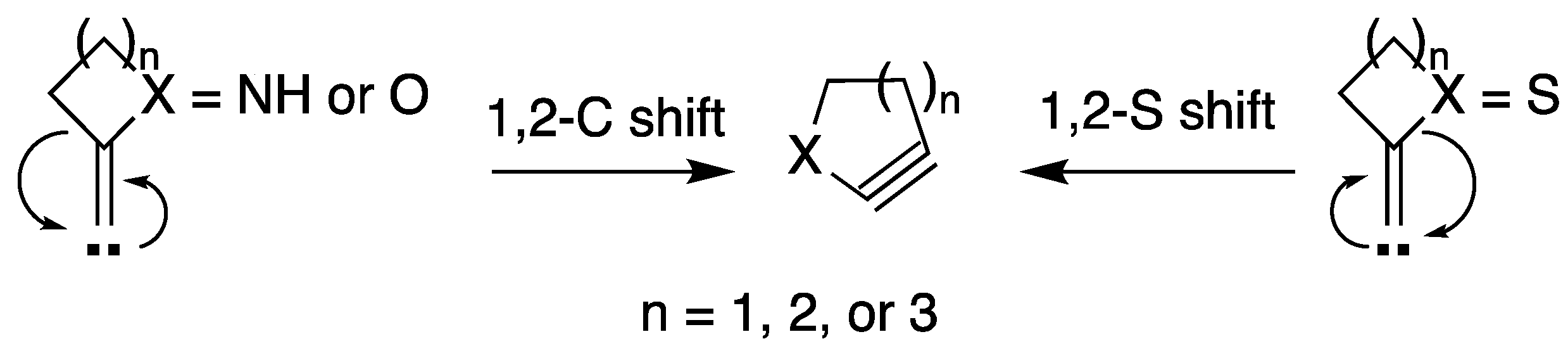
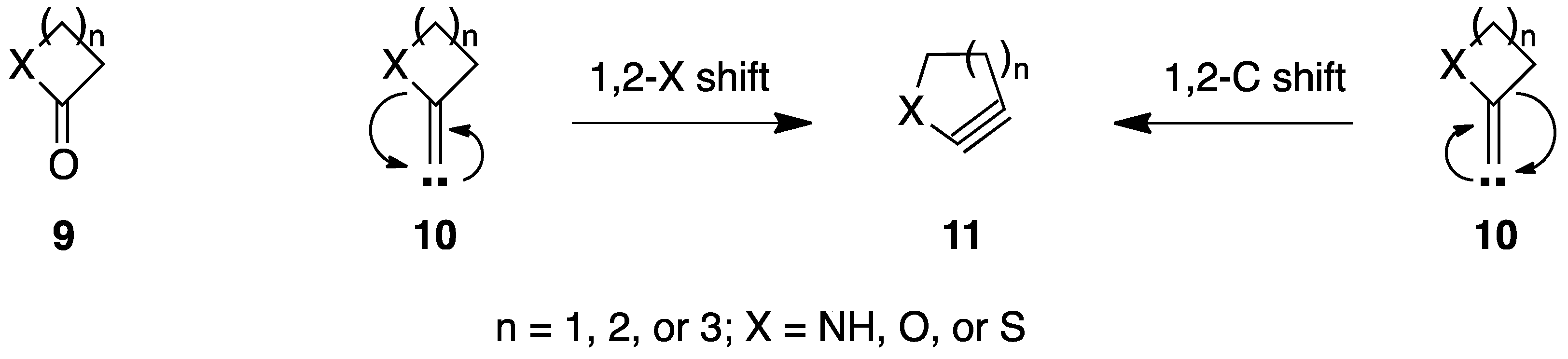
1. Introduction
2. Results and Discussion
2.1. Alkylidenecarbenes Derived from β-Lactam, β-Lactone, and β-Thiolactone
2.1.1. Ring Expansion of 2-(1-Azacyclobutylidene)carbene (12) into 1-Azacyclopent-2-yne (13)
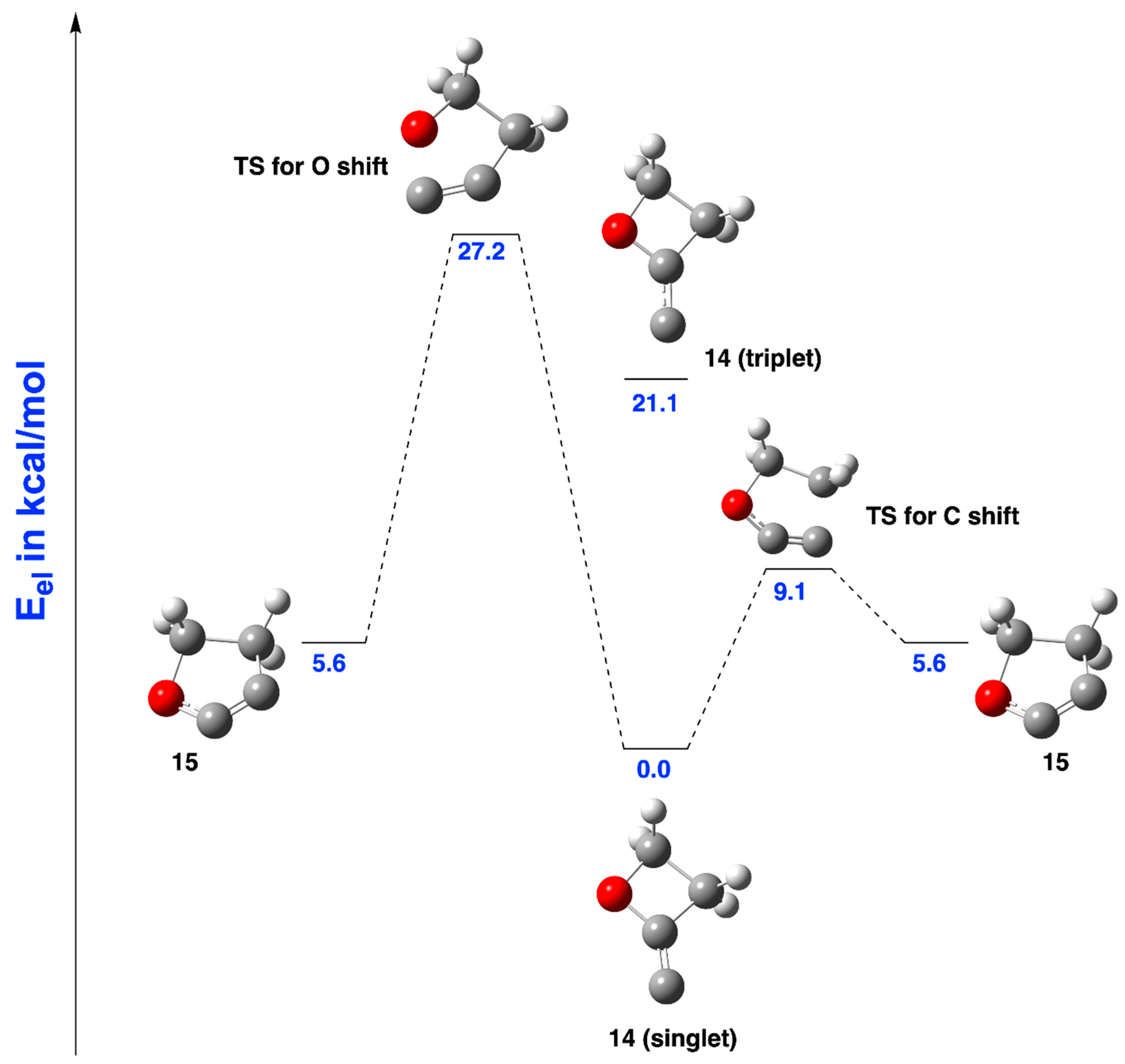
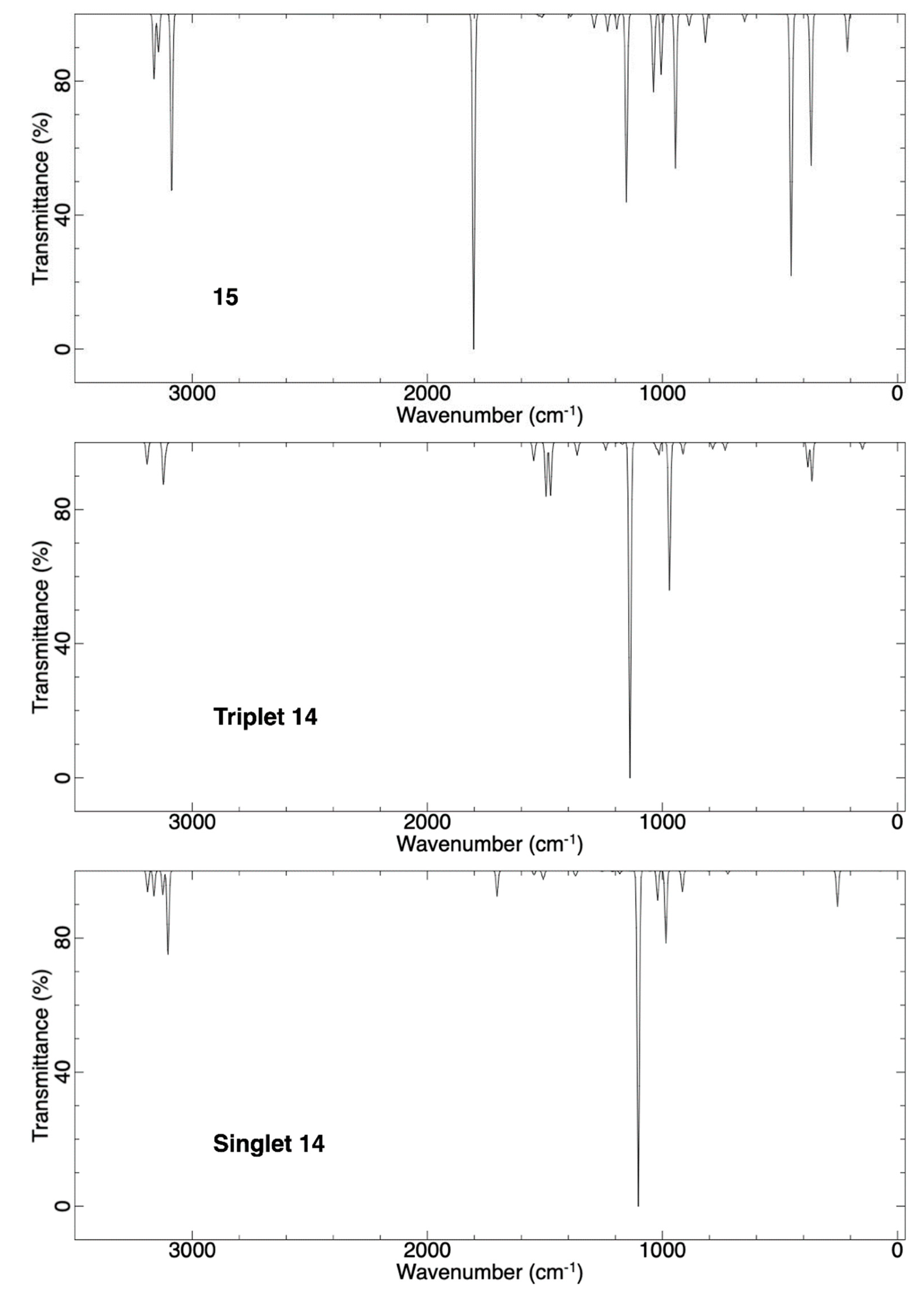
2.1.2. Ring Expansion of 2-(1-Oxacyclobutylidene)carbene (14) into 1-Oxacyclopent-2-yne (15)
2.1.3. Ring Expansion of 2-(1-Thiocyclobutylidene)carbene (16) into 1-Thiocyclopent-2-yne (17)
2.2. Alkylidenecarbenes Derived from γ-Lactam, γ-Lactone, and γ-Thiolactone
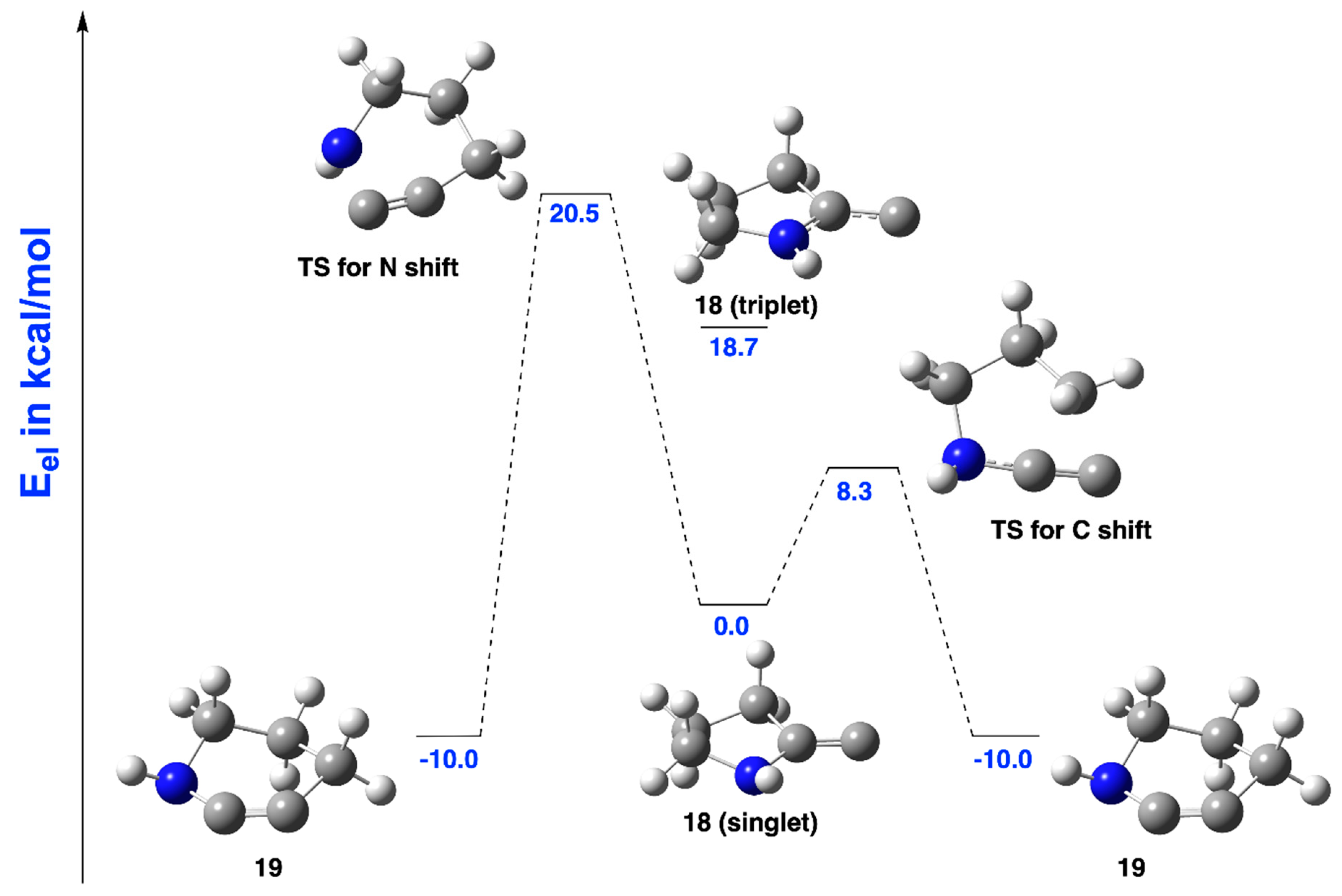
2.2.1. Ring Expansion of 2-(1-Azacyclopentylidene)carbene (18) into 1-Azacyclohex-2-yne (19)
2.2.2. Ring Expansion of 2-(1-Oxacyclopentylidene)carbene (20) into 1-Oxacyclohex-2-yne (21)
2.2.3. Ring Expansion of 2-(1-Thiacyclopentylidene)carbene (22) into 1-Thiacyclohex-2-yne (23)
2.3. Alkylidenecarbenes Derived from δ-Lactam, δ-Lactone, and δ-Thiolactone
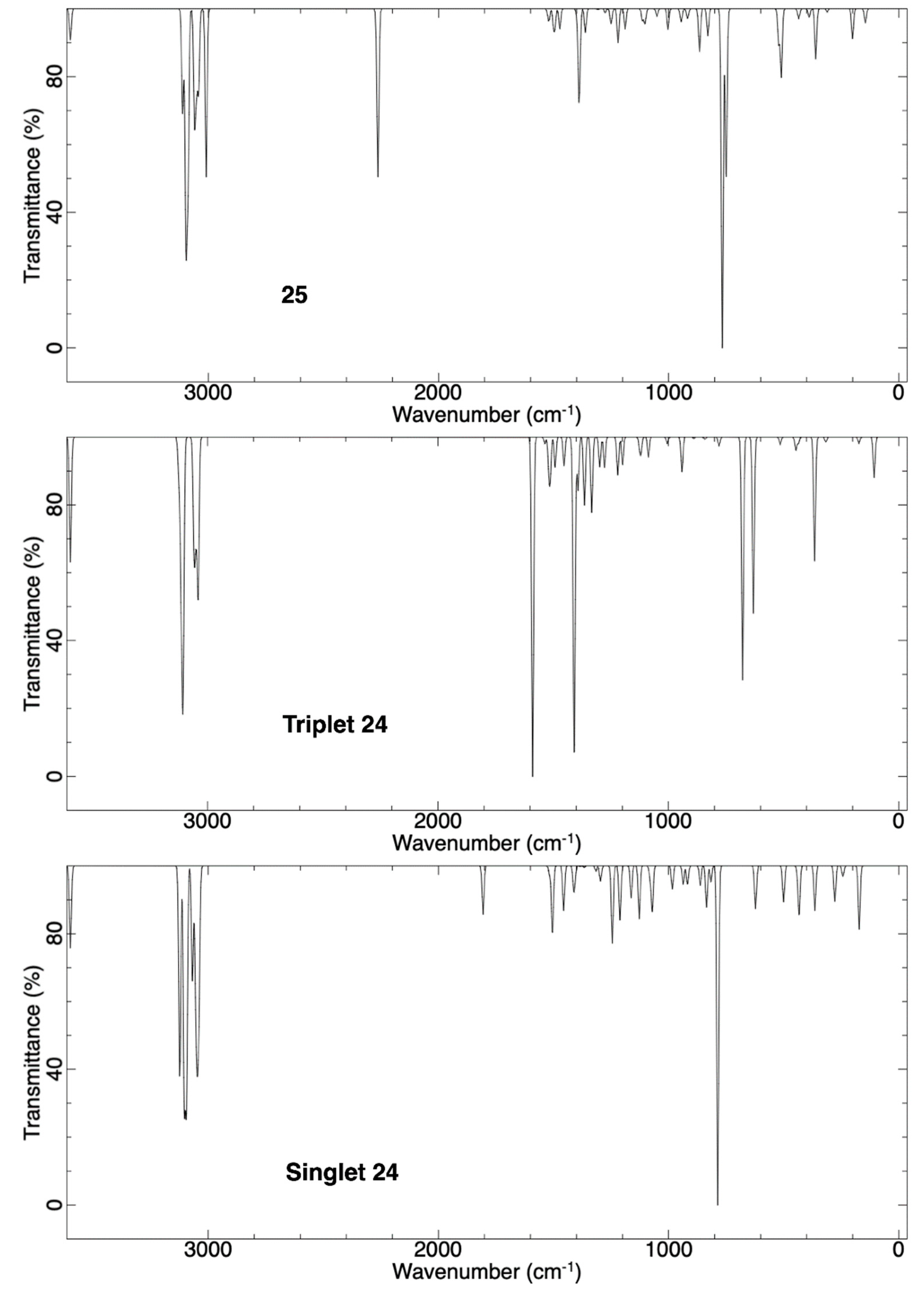
2.3.1. Ring Expansion of 2-(1-Azacyclohexylidene)carbene (24) into 1-Azacyclohept-2-yne (25)
2.3.2. Ring Expansion of 2-(1-Oxacyclohexylidene)carbene (26) into 1-Oxacyclohept-2-yne (27)
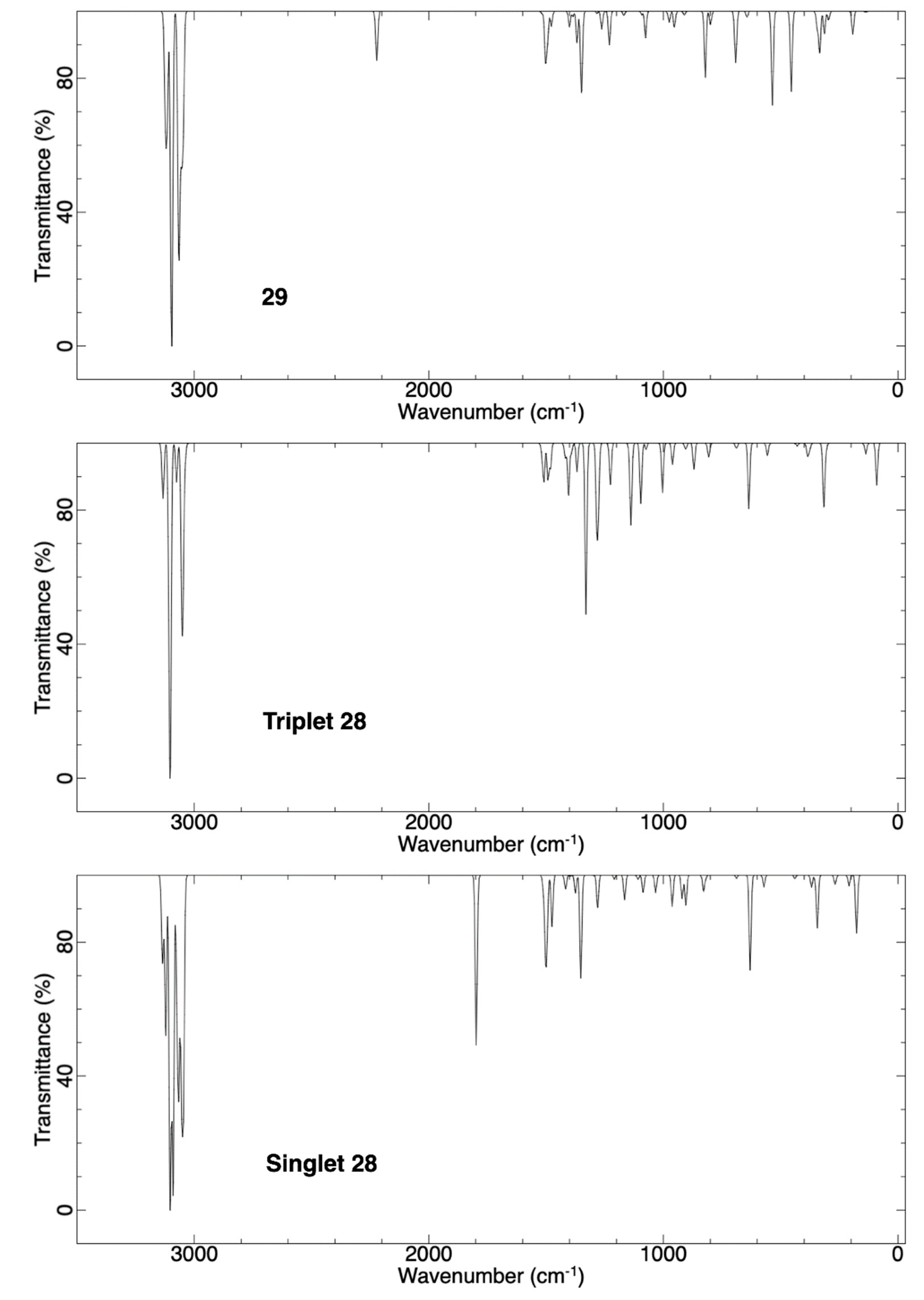
2.3.3. Ring Expansion of 2-(1-Thiocyclohexylidene)carbene (28) into 1-Thiocyclohept-2-yne (29)
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fritsch, P. On the Preparation of Diphenylacetaldehyde and a New Synthesis of Tolene Derivatives. Justus Liebigs Ann. Chem. 1894, 279, 319–323. [Google Scholar] [CrossRef]
- Buttenberg, W.P. Condensation of the Dichloroacetal with Phenol and Toluene. Justus Liebigs Ann. Chem. 1894, 279, 324–337. [Google Scholar] [CrossRef]
- Wiechell, H. Condensation of Dichloroacetate with Anisole and Phenetol. Justus Liebigs Ann. Chem. 1894, 279, 337–344. [Google Scholar] [CrossRef]
- Knorr, R. Alkylidenecarbenes, Alkylidenecarbenoids, and Competing Species: Which Is Responsible for Vinylic Nucleophilic Substitution, [1 + 2] Cycloadditions, 1,5-CH Insertions, and the Fritsch–Buttenberg–Wiechell Rearrangement? Chem. Rev. 2004, 104, 3795–3849. [Google Scholar] [CrossRef] [PubMed]
- Jahnke, E.; Tykwinski, R.R. The Fritsch–Buttenberg–Wiechell rearrangement: Modern applications for an old reaction. Chem. Commun. 2010, 46, 3235–3249. [Google Scholar]
- Grainger, R.S.; Munro, K.R. Recent advances in alkylidene carbene chemistry. Tetrahedron 2015, 71, 7795–7835. [Google Scholar] [CrossRef]
- Hopf, H.; Grunenberg, J. Angle-strained cycloalkynes. In Strained Hydrocarbons; Dodziuk, H., Ed.; Wiley-VCH: Weinheim, Germany, 2009; pp. 375–397. [Google Scholar]
- Detert, H. Angle-strained heterocyclic alkynes with five to ten ring atoms. Targets Heterocycl. Syst. 2011, 15, 1–49. [Google Scholar]
- Sahu, B.; Gururaja, G.N.; Kumar, T.; Chatterjee, A.; Ganguly, B.; Mobin, S.M.; Namboothiri, I.N.N. Generation and Trapping of a Cage Annulated Vinylidenecarbene and Approaches to Its Cycloalkyne Isomer. J. Org. Chem. 2012, 77, 6998–7004. [Google Scholar] [CrossRef]
- Moore, K.A.; Vidaurri-Martinez, J.S.; Thamattoor, D.M. The Benzylidenecarbene-Phenylacetylene Rearrangement: An Experimental and Computational Study. J. Am. Chem. Soc. 2012, 134, 20037–20040. [Google Scholar] [CrossRef]
- Hardikar, T.S.; Warren, M.A.; Thamattoor, D.M. Photochemistry of 1-(propan-2-ylidene)-1a,9b-dihydro-1H-cyclopropa[l]phenanthrene. Tetrahedron Lett. 2015, 56, 6751–6753. [Google Scholar] [CrossRef]
- Yang, X.; Languet, K.; Thamattoor, D.M. An Experimental and Computational Investigation of (α-Methylbenzylidene)carbene. J. Org. Chem. 2016, 81, 8194–8198. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Lan, X.; Phillips, D.L.; Coldren, W.H.; Hadad, C.M.; Yang, X.; Thamattoor, D.M. Direct Observation of an Alkylidenecarbene by Ultrafast Transient Absorption Spectroscopy. J. Phys. Chem. A 2018. [Google Scholar] [CrossRef] [PubMed]
- Maurer, D.P.; Fan, R.; Thamattoor, D.M. Photochemical Generation of Strained Cycloalkynes from Methylenecyclopropanes. Angew. Chem. Int. Ed. 2017, 56, 4499–4501. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Wen, Y.; Thamattoor, D.M. Photochemical generation and trapping of 3-oxacyclohexyne. Org. Biomol. Chem. 2017, 15, 8270–8275. [Google Scholar] [CrossRef] [PubMed]
- Liebman, J.F.; Greenberg, A. A survey of strained organic molecules. Chem. Rev. 1976, 76, 311–365. [Google Scholar] [CrossRef]
- Krebs, A.; Wilke, J. Angle strained cycloalkynes. Top. Curr. Chem. 1983, 109, 189–233. [Google Scholar]
- Meier, H. Cyclic alkynes, enynes and dienynes. A synthetic challenge. Adv. Strain Organic Chem. 1991, 1, 215–272. [Google Scholar]
- Debets, M.F.; van Berkel, S.S.; Dommerholt, J.; Dirks, A.J.; Rutjes, F.P.J.T.; van Delft, F.L. Bioconjugation with Strained Alkenes and Alkynes. Acc. Chem. Res. 2011, 44, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Gampe, C.M.; Carreira, E.M. Arynes and Cyclohexyne in Natural Product Synthesis. Angew. Chem. Int. Ed. 2012, 51, 3766–3778. [Google Scholar] [CrossRef] [PubMed]
- Medina, J.M.; McMahon, T.C.; Jimenez-Oses, G.; Houk, K.N.; Garg, N.K. Cycloadditions of cyclohexynes and cyclopentyne. J. Am. Chem. Soc. 2014, 136, 14706–14709. [Google Scholar] [CrossRef] [PubMed]
- Tlais, S.F.; Danheiser, R.L. N-Tosyl-3-Azacyclohexyne. Synthesis and Chemistry of a Strained Cyclic Ynamide. J. Am. Chem. Soc. 2014, 136, 15489–15492. [Google Scholar] [CrossRef] [PubMed]
- Shah, T.K.; Medina, J.M.; Garg, N.K. Expanding the Strained Alkyne Toolbox: Generation and Utility of Oxygen-Containing Strained Alkynes. J. Am. Chem. Soc. 2016, 138, 4948–4954. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S. Controlled reactive intermediates enabling facile molecular conjugation. Bull. Chem. Soc. Jpn. 2018, 91, 1293–1318. [Google Scholar] [CrossRef]
- Qiu, D.; Shi, J.; Guo, Q.; Xu, Q.; Li, B.; Li, Y. Cyclohexenynone Precursors: Preparation via Oxidative Dearomatization Strategy and Reactivity. J. Am. Chem. Soc. 2018, 140, 13214–13218. [Google Scholar] [CrossRef] [PubMed]
- Picazo, E.; Anthony, S.M.; Giroud, M.; Simon, A.; Miller, M.A.; Houk, K.N.; Garg, N.K. Arynes and Cyclic Alkynes as Synthetic Building Blocks for Stereodefined Quaternary Centers. J. Am. Chem. Soc. 2018, 140, 7605–7610. [Google Scholar] [CrossRef] [PubMed]
- Agard, N.J.; Prescher, J.A.; Bertozzi, C.R. A Strain-Promoted [3 + 2] Azide−Alkyne Cycloaddition for Covalent Modification of Biomolecules in Living Systems. J. Am. Chem. Soc. 2004, 126, 15046–15047. [Google Scholar] [CrossRef] [PubMed]
- Sletten, E.M.; Bertozzi, C.R. Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew. Chem. Int. Ed. 2009, 48, 6974–6998. [Google Scholar] [CrossRef]
- Jewett, J.C.; Sletten, E.M.; Bertozzi, C.R. Rapid Cu-Free Click Chemistry with Readily Synthesized Biarylazacyclooctynones. J. Am. Chem. Soc. 2010, 132, 3688–3690. [Google Scholar] [CrossRef]
- De Almeida, G.; Sletten, E.M.; Nakamura, H.; Palaniappan, K.K.; Bertozzi, C.R. Thiacycloalkynes for Copper-Free Click Chemistry. Angew. Chem. Int. Ed. 2012, 51, 2443–2447. [Google Scholar] [CrossRef]
- McNitt, C.D.; Popik, V.V. Photochemical generation of oxa-dibenzocyclooctyne (ODIBO) for metal-free click ligations. Org. Biomol. Chem. 2012, 10, 8200–8202. [Google Scholar] [CrossRef]
- Gold, B.; Dudley, G.B.; Alabugin, I.V. Moderating Strain without Sacrificing Reactivity: Design of Fast and Tunable Noncatalyzed Alkyne–Azide Cycloadditions via Stereoelectronically Controlled Transition State Stabilization. J. Am. Chem. Soc. 2013, 135, 1558–1569. [Google Scholar] [CrossRef] [PubMed]
- Gold, B.; Batsomboon, P.; Dudley, G.B.; Alabugin, I.V. Alkynyl Crown Ethers as a Scaffold for Hyperconjugative Assistance in Noncatalyzed Azide–Alkyne Click Reactions: Ion Sensing through Enhanced Transition-State Stabilization. J. Org. Chem. 2014, 79, 6221–6232. [Google Scholar] [CrossRef] [PubMed]
- Hagendorn, T.; Bräse, S. A new route to dithia- and thiaoxacyclooctynes via Nicholas reaction. RSC Adv. 2014, 4, 15493–15495. [Google Scholar] [CrossRef]
- Debets, M.F.; Prins, J.S.; Merkx, D.; van Berkel, S.S.; van Delft, F.L.; van Hest, J.C.M.; Rutjes, F.P.J.T. Synthesis of DIBAC analogues with excellent SPAAC rate constants. Org. Biomol. Chem. 2014, 12, 5031–5037. [Google Scholar] [CrossRef] [PubMed]
- Ni, R.; Mitsuda, N.; Kashiwagi, T.; Igawa, K.; Tomooka, K. Heteroatom-embedded Medium-Sized Cycloalkynes: Concise Synthesis, Structural Analysis, and Reactions. Angew. Chem. Int. Ed. 2015, 54, 1190–1194. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.; Gomes, G.D.P.; Ayad, S.; Clark, R.J.; Lobodin, V.V.; Tuscan, M.; Hanson, K.; Alabugin, I.V. Twisted Cycloalkynes and Remote Activation of “Click” Reactivity. Chemisty 2017, 3, 629–640. [Google Scholar] [CrossRef]
- Del Grosso, A.; Galanopoulos, L.D.; Chiu, C.K.C.; Clarkson, G.J.; O′ Connor, P.B.; Wills, M. Strained alkynes derived from 2,2′-dihydroxy-1,1′-biaryls; synthesis and copper-free cycloaddition with azides. Org. Biomol. Chem. 2017, 15, 4517–4521. [Google Scholar] [CrossRef] [PubMed]
- Nainar, S.; Kubota, M.; McNitt, C.; Tran, C.; Popik, V.V.; Spitale, R.C. Temporal Labeling of Nascent RNA Using Photoclick Chemistry in Live Cells. J. Am. Chem. Soc. 2017, 139, 8090–8093. [Google Scholar] [CrossRef]
- Lyapunova, A.G.; Danilkina, N.A.; Rumyantsev, A.M.; Khlebnikov, A.F.; Chislov, M.V.; Starova, G.L.; Sambuk, E.V.; Govdi, A.I.; Bräse, S.; Balova, I.A. Relative Reactivity of Benzothiophene-Fused Enediynes in the Bergman Cyclization. J. Org. Chem. 2018, 83, 2788–2801. [Google Scholar] [CrossRef] [PubMed]
- Lyapunova, A.G.; Danilkina, N.A.; Khlebnikov, A.F.; Köberle, B.; Bräse, S.; Balova, I.A. Oxaenediynes through the Nicholas-Type Macrocyclization Approach. Eur. J. Org. Chem. 2016, 2016, 4842–4851. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A Gen. Phys. 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Bartlett, R.J.; Purvis, G.D., III. Many-body perturbation theory, coupled-pair many-electron theory, and the importance of quadrupole excitations for the correlation problem. Int. J. Quantum Chem. 1978, 14, 561–581. [Google Scholar] [CrossRef]
- Purvis, G.D., III; Bartlett, R.J. A full coupled-cluster singles and doubles model: The inclusion of disconnected triples. J. Chem. Phys. 1982, 76, 1910–1918. [Google Scholar] [CrossRef]
- Scuseria, G.E.; Janssen, C.L.; Schaefer, H.F., III. An efficient reformulation of the closed-shell coupled cluster single and double excitation (CCSD) equations. J. Chem. Phys. 1988, 89, 7382–7387. [Google Scholar] [CrossRef]
- Jacox, M.E. The spectroscopy of molecular reaction intermediates trapped in the solid rare gases. Chem. Soc. Rev. 2002, 31, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Andrews, L. IR and Raman Spectroscopies, Matrix Isolation Studies. In Encyclopedia of Spectroscopy and Spectrometry, 3rd ed.; Lindon, J.C., Tranter, G.E., Koppenaal, D.W., Eds.; Academic Press: Oxford, UK, 2017; pp. 359–364. [Google Scholar]
- Bally, T. Matrix isolation. In Research on Chemical Intermediates; Moss, R.A., Platz, M.S., Jones, M., Jr., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2004; pp. 797–845. [Google Scholar]
- Allen, F.; Watson, D.; Brammer, L.; Orpen, A.; Taylor, R. Typical interatomic distances: Organic compounds. In International Tables for Crystallography; Prince, E., Ed.; Springer: Dordrecht, The Netherlands, 2006; pp. 790–811. [Google Scholar]
- Nickon, A. New perspectives on carbene rearrangements: Migratory aptitudes, bystander assistance, and geminal efficiency. Acc. Chem. Res. 1993, 26, 84–89. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, Version 6; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Avogadro: An Open-Source Molecular Builder and Visualization Tool. Available online: http://avogadro.cc/ (accessed on 6 February 2019).
- Lee, T.J.; Taylor, P.R. A diagnostic for determining the quality of single-reference electron correlation methods. Int. J. Quantum Chem. 1989, 36, 199–207. [Google Scholar] [CrossRef]
- Dykstra, C.E. An examination of the Brueckner condition for the selection of molecular orbitals in correlated wavefunctions. Chem. Phys. Lett. 1977, 45, 466–469. [Google Scholar] [CrossRef]
- Handy, N.C.; Pople, J.A.; Head-Gordon, M.; Raghavachari, K.; Trucks, G.W. Size-consistent Brueckner theory limited to double substitutions. Chem. Phys. Lett. 1989, 164, 185–192. [Google Scholar] [CrossRef]
- Raghavachari, K.; Pople, J.A.; Replogle, E.S.; Head-Gordon, M. Fifth order Moeller-Plesset perturbation theory: Comparison of existing correlation methods and implementation of new methods correct to fifth order. J. Phys. Chem. 1990, 94, 5579–5586. [Google Scholar] [CrossRef]
- Lee, T.J.; Kobayashi, R.; Handy, N.C.; Amos, R.D. Comparison of the Brueckner and coupled-cluster approaches to electron correlation. J. Chem. Phys. 1992, 96, 8931–8937. [Google Scholar] [CrossRef]
- Robson, J.H.; Shechter, H. Effects of neighboring heteroatoms in rearrangement to divalent carbon. J. Am. Chem. Soc. 1967, 89, 7112–7114. [Google Scholar] [CrossRef]



























© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, N.N.T.; Just, J.; Pankauski, J.M.; Rablen, P.R.; Thamattoor, D.M. Ring Expansion of Alkylidenecarbenes Derived from Lactams, Lactones, and Thiolactones into Strained Heterocyclic Alkynes: A Theoretical Study. Molecules 2019, 24, 593. https://doi.org/10.3390/molecules24030593
Le NNT, Just J, Pankauski JM, Rablen PR, Thamattoor DM. Ring Expansion of Alkylidenecarbenes Derived from Lactams, Lactones, and Thiolactones into Strained Heterocyclic Alkynes: A Theoretical Study. Molecules. 2019; 24(3):593. https://doi.org/10.3390/molecules24030593
Chicago/Turabian StyleLe, Nguyen Nhat Thu, Josefine Just, Jonathan M. Pankauski, Paul R. Rablen, and Dasan M. Thamattoor. 2019. "Ring Expansion of Alkylidenecarbenes Derived from Lactams, Lactones, and Thiolactones into Strained Heterocyclic Alkynes: A Theoretical Study" Molecules 24, no. 3: 593. https://doi.org/10.3390/molecules24030593