Synthesis, Physical Properties, and Reactivity of Stable, π-Conjugated, Carbon-Centered Radicals


Department of Chemistry, Graduate School of Science, Osaka University, Toyonaka, Osaka 560-0043, Japan
Molecules 2019, 24(4), 665; https://doi.org/10.3390/molecules24040665
Submission received: 26 January 2019
/
Revised: 8 February 2019
/
Accepted: 11 February 2019
/
Published: 13 February 2019
(This article belongs to the Special Issue Open-Shell Systems for Functional Materials)
Abstract
:Recently, long-lived, organic radical species have attracted much attention from chemists and material scientists because of their unique electronic properties derived from their magnetic spin and singly occupied molecular orbitals. Most stable and persistent organic radicals are heteroatom-centered radicals, whereas carbon-centered radicals are generally very reactive and therefore have had limited applications. Because the physical properties of carbon-centered radicals depend predominantly on the topology of the π-electron array, the development of new carbon-centered radicals is key to new basic molecular skeletons that promise novel and diverse applications of spin materials. This account summarizes our recent studies on the development of novel carbon-centered radicals, including phenalenyl, fluorenyl, and triarylmethyl radicals.
1. Introduction
Organic radical species are generally recognized as highly reactive, intermediate species. However, recently, functional materials taking advantage of the feature of open-shell electronic structure have attracted much attention from chemists and material scientists; therefore, the development of novel, long-lived, organic, radical species becomes more important [1,2,3,4,5,6,7]. Nitronyl nitroxides, galvinoxyl, and DPPH are well known as stable, organic, radical species, which are commercially available chemicals. These stable radical species are “heteroatom-centered radicals”, in which unpaired electrons are mainly distributed on heteroatoms. On the other hand, “carbon-centered radicals”, in which unpaired electrons exist only on carbon atoms, are highly reactive and difficult to handle and therefore have had limited applications. However, because the physical properties of carbon-centered radicals depend predominantly on the topology of the π-electron array, the development of new carbon-centered radicals is key to new basic molecular skeletons that promise novel and diverse applications of spin materials. By investigating the physical properties and reactivity of newly developed radical species, it becomes possible to find new applications, leading to the creation of new functional materials.
There are two definitions of stability of radicals: thermodynamic stability and kinetic stability. Thermodynamic stability is related to the degree of generation of radical species; for instance, it determines how many monomers are formed when the radical dimer is cleaved into a monomer. On the other hand, kinetic stability is related to the lifetime of radical species. The thermodynamic stability of radical species can be evaluated by the indices such as bond dissociation energy (BDE) and radical stabilization energy (RSE) [8]. The BDE is the energy required for homolytic cleavage of R–H into R• and H•, whereas the RSE is expressed by the difference between the BDE of a radical species of interest and the BDE of the methyl radical. Either BDEs or RSEs may be used for evaluating the thermodynamic stability of radical species. However, RSE might be easier to evaluate the stability of radicals because RSEs indicate the relative stability to the methyl radical. Table 1 shows RSE values of selected carbon-centered radicals calculated at the second-order, restricted, open-shell, Møller–Plesset (ROMP2) level of theory. Generally, the larger the delocalization of unpaired electrons, the more thermodynamically stabilized the radical species, and the RSE shows a larger negative value.
Research on stable organic radicals started with the triphenylmethyl radical (1), which was discovered by Gomberg in 1900 [9]. Although 1 reacts promptly with molecular oxygen, the radical exists for a long period of time under degassed conditions or in an inert gas atmosphere. In solution, 1 is in equilibrium with a dimer and dissociates mostly into a monomeric radical in dilute solution (about 10−5 M), indicating high thermodynamic stability of 1. The RSE value of 1 is calculated to be −103.4 kJ mol−1.
2. Phenalenyl Radical
The phenalenyl radical (2) is a spin-delocalized hydrocarbon radical with D3h symmetry. Due to its relatively long lifespan, 2 has attracted attention from organic chemists in terms of its chemical reactivity of radical species [10]. On the other hand, in 1973, Haddon proposed that a one-dimensional (1D) stack of organic radicals should show metallic behavior and also showed that 2 is a good model compound for his theoretical prediction [11]. After that, many researchers became interested in the physical properties of 1 [12,13,14,15,16]. However, organic radicals generally dimerize to form closed-shell compounds. Therefore, a considerable effort is required for the molecular design to obtain a one-dimensional chain of radicals. This section describes our recent studies on self-association behaviors of phenalenyl radicals, aiming at constructing a one-dimensional chain as a final goal.
2.1. Electronic Structure of Phenalenyl Radical
The phenalenyl radical (2) is an odd-alternant hydrocarbon radical, and owing to its highly symmetric structure, an unpaired electron is delocalized on six equivalent carbon atoms (1, 3, 4, 6, 7, and 9-positions: the α-position), as shown in Figure 1. This spin delocalization leads to the high thermodynamic stability of 2, and indeed, the RSE value of 2 is −201.6 kJ mol−1, which is almost twice that of 1. The Hückel molecular orbital (HMO) calculation shows a singly occupied molecular orbital (SOMO) with an energy of α + 0β. The SOMO of 2 is distributed only on the α-position, and the MO coefficients of the other atoms are zero, consistent with the resonance form shown in Figure 1. The feature of the SOMO of 2 is a hexagonal arrangement, which allows perfect orbital overlap in both eclipsed and staggered stacking motifs. The staggered stacking is more favorable to short π–π contact (face-to-face contact) than the eclipsed stacking due to smaller atom–atom repulsion.
2.2. Dimerizaion Behavior of Phenalenyl Radicals
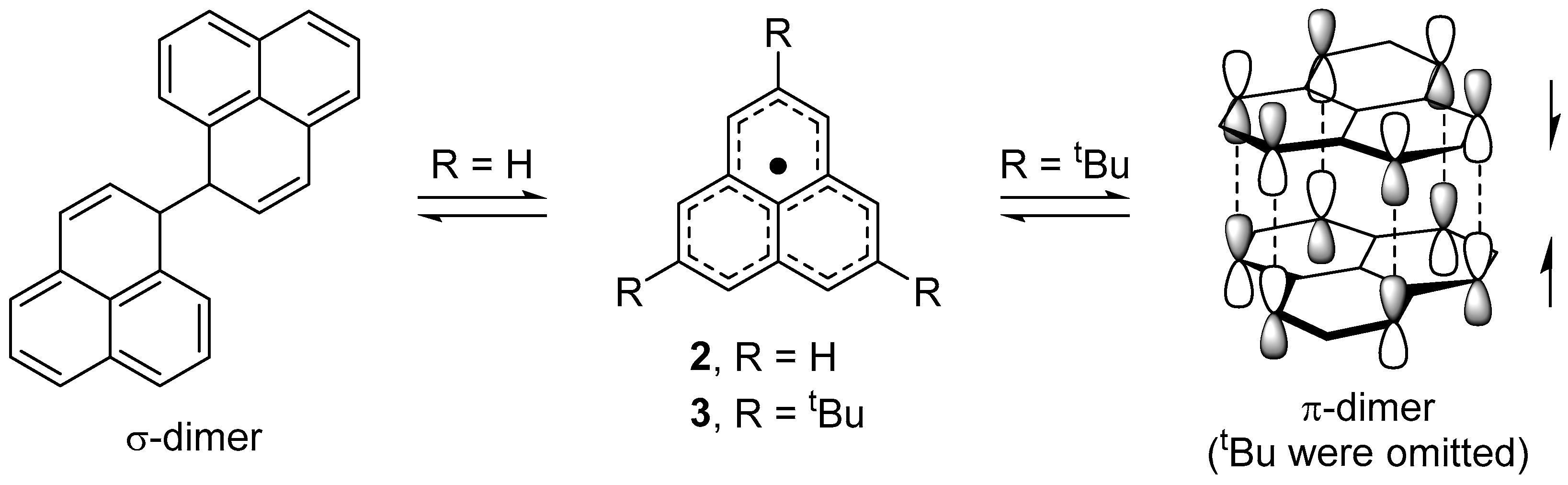
The parent phenalenyl radical 2 is a long-lived species in the absence of air and is in equilibrium with a σ-bonded dimer (σ-dimer) in solution state (Figure 2) [17]. The thermodynamically stabilized nature of 2 suggests that the structural modification, such as the introduction of substituent groups, may lead to another association mode other than a σ-dimer form. In fact, the introduction of bulky tert-butyl groups at 2,5,8-positions afforded a face-to-face π-dimer in the solid [18] and solution states [19] (Figure 2). Two phenalenyl planes are superimposed at the separation distance of 3.25 Å in the staggered stacking manner. The π-dimer of the tert-butyl derivative (3) shows a deep blue color and an intense absorption band is observed at 612 nm in the solid state. A quantum chemical calculation (ZINDO/S) for the π-dimer predicted a fully allowed absorption band around 600 nm, with the transition moment along the direction connecting the centers of the phenalenyl rings. The intense band can be assigned to an electronic transition from HOMO to LUMO, which are newly formed by the orbital interaction of a pair of SOMO in the π-dimer. The short π–π contact and the orbital splitting indicate the adequate covalent bonding interaction of two unpaired electrons [20,21].
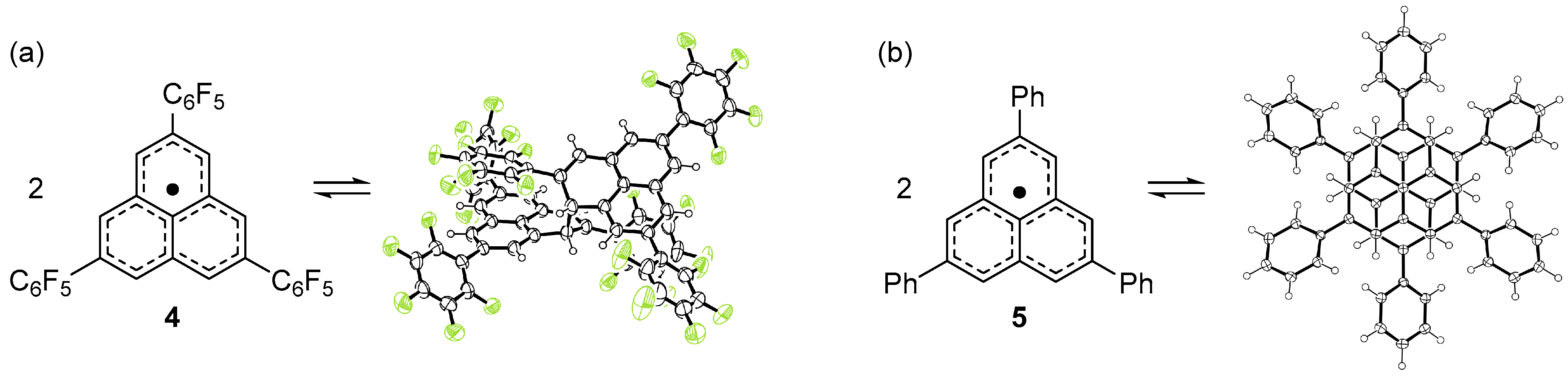
Because a π-dimer is the smallest unit of a 1D stack, we expected that a chemical modification such as the introduction of substituent groups could lead to an equidistantly stacked 1D chain [22]. As a first trial, we decided to introduce a pentafluorophenyl (C6F5) group at 2,5,8-positions [23]. Highly polarized C6F5 group would afford aggregates exceeding a π-dimer due to the strong electrostatic interaction between the C6F5 groups. Indeed, C6F5 groups serve as a supramolecular synthon, giving rise to a 1D stack of molecules [24,25]. The key precursor of the target radical (4) was synthesized from 2,7-dibromonaphthalene in nine steps. The dehydrogenation of the precursor with dichlorodicyano-p-benzoquinone (DDQ) afforded pale yellow crystals, which is in sharp contrast to the blue color of the π-dimer of 3. The X-ray crystallographic analysis of the yellow crystal showed that 4 adopts a σ-dimer form (42) in the solid state (Figure 3a). The 1H NMR spectrum of the pale yellow crystals in CDCl3 showed six sharp singlet signals in the temperature range 233–298 K, consistent with the σ-dimer form. Thus, 4 prefers a σ-dimer form over a π-dimer form in the solution and solid states. The σ-dimer features a long σ-bond of 1.636(7) Å connecting the two phenalenyl rings, indicating the weakness of the σ-bond. Indeed, a toluene solution of the σ-dimer at room temperature showed a well-resolved multiline ESR spectrum originating from monomeric 4. The enthalpy and entropy changes for the σ-dimerization of 4 were determined to be −64.0 kJ mol−1 and −135 J K−1 mol−1, respectively, from the temperature dependency of the ESR signal intensity.
We also prepared a phenyl derivative (5) of 2 [26]. The radical precursor was prepared from 2,7-diphenylnaphthalene in a similar manner as 4. The dehydrogenation of the precursor with p-chloranil afforded deep blue crystals, contrary to our expectation. The X-ray crystallographic analysis of the blue crystal showed that 5 forms a discreet face-to-face π-dimer (52) with a C(α)–C(α) separation of 3.067 Å at 300 K (Figure 3b), much shorter than that of 32. To elucidate the structure of the dimeric species in the solution state, the variable-temperature 1H NMR spectra of 5 was measured in CD2Cl2. No signal was observed at 273 K for the solution of 5, whereas at 203 K, two broad signals were observed at δ 7.5 ppm (phenyl protons) and 6.8 ppm (phenalenyl protons). Considering that a similar behavior was observed for the π-dimerization of 3, the phenyl derivative 5 prefers a π-dimer form over a σ-dimer form in the solution state. The dissolution of 52 in toluene showed a well-resolved multiline ESR spectrum corresponding to monomeric 5 at room temperature. The intensity of the ESR signals of 5 decreased with decreasing temperature and almost disappeared at 170 K due to the formation of a diamagnetic π-dimer. The enthalpy and entropy changes for the dimerization were determined to be −40 kJ mol−1 and −75 J K−1 mol−1, respectively, by the variable-temperature ESR measurements.
The steric size of the phenyl group is smaller than that of the C6F5 group. The preference of π-dimerization for the phenalenyl radical with smaller substituents indicates that the dimerization mode of phenalenyl radicals is not simply determined by a steric factor. Therefore, we decided to prepare a phenalenyl radical derivative with small substituent groups, 2,5,8-trimethyl phenalenyl radical (6), in order to investigate the intrinsic dimerization nature of the phenalenyl radical [26,27]. The precursor of 6 was prepared from 2,7-dibromonaphthalene in ten steps. The dehydrogenation reaction of the precursor with p-chloranil produced a pink-violet solution, indicating the generation of a π-dimer. However, the concentration of the violet solution yielded colorless platelet crystals, which turned out to be a σ-dimer form by the X-ray crystallographic analysis (Figure 4a). When the colorless plates were heated at 573 K in a sealed, degassed tube, melting occurred, accompanied by a color change from colorless to purple. Subsequent cooling of the purple liquid afforded purple platelet crystals together with colorless plates. The X-ray crystallographic analysis of the purple plate at 100 K showed that the purple, crystalline state adopts a π-dimer form (Figure 4b) with a short contact of α-carbons (av. 3.054 Å). Thus, 6 can exist in both σ- and π-dimer forms in the solid state. The 1D 1H NMR spectrum of 6 recorded in degassed THF-d8 at 173 K showed a relatively complicated spectral pattern. The spectrum consisted of three sets of signals derived from three dimeric isomers (62-σ-chiral, 62-σ-meso, and 62-π, shown in Figure 4c), which were unambiguously characterized by the 2D-NMR analyses. From the integration ratio of the 1D 1H NMR signals, the most stable isomer was found to be the σ-dimer chiral form (62-σ-chiral), and the other two (62-σ-meso and 62-π) were metastable forms. However, the energy differences, estimated from the ratio of the NMR signal intensities and Boltzmann distribution, between the metastable forms relative to the most stable 62-σ-chiral were very small: 0.33 kJ mol−1 for 62-σ-meso and 2.8 kJ mol−1 for 62-π.
From the results of the dimerization behavior of 6, we concluded that the σ-dimer and π-dimer of the phenalenyl radical are close in energy, and that its dimerization mode can be changed with a slight perturbation of substituents either by a steric or electronic factor. For instance, in 42, the attractive interaction between the C6F5 group and naphthalene ring makes the σ-dimer form preferable in energy due to the electrostatic interaction (perfluoroarene–arene interaction), as shown in Figure 5a. On the other hand, 5 adopts the π-dimer form due to a cumulative CH–π interaction among the phenyl substituents (Figure 5b). The M05-2X/6-31G** calculation showed that the total stabilization energies by the substituents in 42 and 52 are 35 kJ mol−1 and 43 kJ mol−1, respectively.
2.3. One-Dimensional Stack of Phenalenyl Radicals
As mentioned above, phenalenyl radicals can adopt σ-dimer and π-dimer forms. Therefore, we attempted to transform the σ-dimer form of 4 into a π-dimer form by external stimulus [23]. The white powder of 42 was placed in a sealed, degassed tube and heated at 573 K, resulting in the melting of the power and a color change from white to purple. Subsequent cooling of the resulting purple liquid afforded dark purple needles suitable for the X-ray crystallographic analysis. Surprisingly, 4 was found to be equidistantly stacked with the interplanar distance of 3.503 Å to form a 1D chain (Figure 6a,b). No Peierls transition (that is, dimerization in the 1D stack) was observed even at 10 K. Each 1D chain was surrounded by six adjacent chains, and the 1D chains stick together by a weak C–H•••F–C hydrogen-bonding interaction. Considering that an unpaired electron resides only on the phenalenyl moiety, the C6F5 groups serve as spacers to separate neighboring radical moieties; thus, an ideal 1D chain of spin-1/2 was achieved. The reason for the regular stack of 4 is the close stacking of polarized C6F5 groups; the interplanar distance between the C6F5 groups is 3.252 Å on average. Thus, the C6F5 substituent groups dominantly control the structure of the 1D chain. Because of the large interplanar distance (3.503 Å) of the phenalenyl radical site, the overlapping of the wave functions between the radicals was small. The band width calculated by the extended HMO method was as small as about 0.29 eV. As a result, the 1D chain of 4 was an insulator.
Pentafluorophenyl and phenyl groups alternately stack in the solid state and are used as supramolecular synthons [24,25]. We heated the mixed powder of 42 and 52 at 573 K in a sealed, degassed tube, and subsequent cooling gave rise to brown needles. The X-ray crystallographic analysis of the brown needle at 200 K showed that 4 and 5 stack alternately to form a 1D chain with an interplanar distance of 3.69 Å by virtue of the attractive interaction between C6F5 and phenyl groups (Figure 6c).
3. Fluorenyl Radical
Fluorenyl radical (7), which possesses the same sp2 carbon number as the phenalenyl radical (2), is also a spin-delocalized hydrocarbon radical. However, unlike 2, about half of the spin density resides on the 9-position (Figure 7). Therefore, 7 dimerizes promptly, and its σ-dimer does not dissociate into a monomeric radical even in solution [28,29]. Because the RSE of 7 is −93 kJ mol−1, the thermodynamic stability of 7 is much lower than that of 2.
3.1. π-Extended Fluorenyl Radicals
To stabilize 7, which shows remarkable reactivity, a considerable device must be made in the molecular design. It would be possible to prolong the lifetime of 7 by sterically protecting only the spin-localized part by taking advantage of the situation where an unpaired electron is mainly localized on the carbon atom at the 9-position. We decided to introduce an anthryl group at the 9-position, and to expand the π-skeleton to ensure extraordinary stability in air. In addition, three types of dibenzo-fluorenyl radicals (8, 9, 10), which possess an anthryl substituent group at the central five-membered ring, were prepared by short step syntheses [30]. Interestingly, the difference in the ring-condensation position caused a big difference in lifespan, dimerization mode, and electronic properties of the radicals.
The radicals 8, 9, and 10 showed relatively long half-life times of 7, 3.5, and 43 days, respectively, in air in the solution state. As for dimerization, 8 formed a σ-bond on the fluorenyl ring, whereas 10 formed a σ-bond on a bent anthracene ring (Figure 8). In contrast, no dimerization reaction was observed in 9. Regarding physical properties such as redox potentials and UV-Vis-NIR absorptions, 8 and 10 showed similar properties, whereas 9 showed behavior different from those radicals. For instance, the redox waves of 8 and 10 in the cyclic voltammogram appeared on the higher potential side (that is, not easily oxidized and easily reduced) compared with 9. Regarding the UV-Vis-NIR absorption spectra, 8 and 10 showed weak absorptions in the NIR region, but 9 did not absorb light beyond the visible region. These differences arise from the difference in the manner of condensation of the naphthalene ring with respect to the five-membered ring (Figure 9). It is well-known that naphthalene possesses a larger double-bond character at the 1,2-bond than at the 2,3-bond. Therefore, the five-membered ring condensation to the 1,2 or 2,3 region of naphthalene leads to endo- or exo-double bonds at the five-membered ring, respectively. This difference in the condensation affects the manner of spin delocalization, as shown in Figure 10. Thus, 8 and 10 behave like a cyclopentadienyl radical, whereas 9 behaves like a linear pentadienyl radical. The higher redox potentials of 8 and 10 are attributable to the antiaromatic character of the cyclopentadienyl cation in the cation state and the aromatic character of the cyclopentadienyl anion in the anion state.
As shown in Figure 8b, σ-dimerization of 10 occurs not on the dibenzofluorenyl moiety, where an unpaired electron predominantly distributes, but on the anthryl group that is introduced as a sterically protecting group. This σ-dimerization is accompanied by a large structural change of the anthryl group from the planar form to the butterfly form. We estimated the energy profile for the structural change upon the σ-dimerization of 10 by quantum chemical calculations (Figure 11). Although the most stable form of the monomeric 10 is the twisted form, 10 can adopt a metastable folded form, where an unpaired electron resides mostly on the 10-positon of the anthryl group. The structure of the transition state (10*) for the most stable twisted form ⇄ the metastable folded form is close to the folded form, except that the anthryl group is contorted. The calculated enthalpy of activation (ΔH‡) for the twisted form to 10* was 95.5 kJ mol−1 (Figure 11). Although a σ-dimer (102) of 10 is more stable than the twisted form, the energy difference is just 0.6 kJ mol−1. The low barrier height for the dissociation of 102 into the monomeric 10 is in line with the fact that the dissolution of the solid of 102 in a solvent gradually resulted in the equilibrium with the monomeric 10. The facile dissociation of 102 suggests the weakness of the σ-bond connecting two anthryl groups. In reality, the solid of 102 showed mechanochromic behavior. Hard grinding of the crystals of 102 led to a drastic color change from orange to purple. The purple color corresponds to the color of the monomeric 10.
3.2. Ring Expansion of Fluorenyl Radical
We also investigated the effect of ring size of the fluorenyl radical on chemical and physical properties. Three kinds of anthracene-attached tricyclic radicals (11, 12, and 13) were designed and synthesized (Figure 12) [31]. It is well-known that five- and seven-membered rings have electron-deficient and electron-rich properties, respectively. Additionally, the size of the central polygon in the tricyclic system affects steric congestion in the fjord region. These electronic and steric factors would cause the difference in the properties among the tricyclic radicals.
From the decay of the ESR signal intensities in air-saturated toluene, the half-life time of 11 at room temperature was found to be 5.6 days. Considering that the 9-phenylfluorenyl radical rapidly decomposes [32], it seems that the effect of steric protection by an anthryl group on the stability is unexpectedly large. 12 showed a longer half-life time (9 days) than 11 presumably due to the perpendicular conformation between the anthryl group and the tricyclic scaffold [33]. In contrast, 13, which also adopts the perpendicular conformation, was a short-lived species with a half-life time of about 5 min and was easily oxidized. This high reactivity toward oxygen would be related to the high SOMO level originating from the electron-rich character of the seven-membered ring. The oxidation and reduction potentials (Eox and Ered, respectively) determined by cyclic voltammetry are summarized in Table 2. 13 is most easily oxidized among the three radicals, whereas 11 shows the highest electron-accepting ability.
Similar to 10, 11 also gradually gave rise to a colorless σ-dimer at room temperature in the solution state. In contrast, 12 showed no dimerization behavior for a long period of time. The ΔH‡ of 12 to a σ-dimer was calculated to be 138 kJ mol−1, which is much larger than that of 11 (90 kJ mol−1). Therefore, 12 could not exceed the transition state at room temperature. As the central polygon of the tricyclic scaffold becomes a six-membered ring, the steric hindrance of the fjord region became larger than 11, leading to a higher activation barrier. However, 13, where the fjord region was anticipated to be crowded more than 11 and 12, possessed a ΔH‡ of only 83 kJ mol−1. The reason for the unexpectedly low barrier is that in the transition state, the seven-membered ring moiety adopts a tub-like structure, whereby the tricyclic radical moiety greatly bends and the steric hindrance of the fjord region is alleviated (Figure 13). The flexibility of the seven-membered ring moiety is also related to the stability of the σ-dimer. The energy differences between the monomeric radical and the σ-dimer are 64 kJ mol−1 and 109 kJ mol−1 for 11 and 13, respectively (the σ-dimers are lower in energy than the monomeric species). Indeed, 13 afforded a σ-dimer quickly at room temperature in the solution state under the degassed condition; furthermore, no dissociation reaction was observed for the σ-dimer.
4. Trianthrylmethyl Radical
The stability of the triphenylmethyl radical (1) is largely dependent on the steric factor of the phenyl groups [34]. We decided to investigate the effect of larger aryl groups on the stability of the methyl radical and designed a new highly congested hydrocarbon radical, trianthrylmethyl radical (14) [35]. As shown in Figure 14, the central carbon atom of 14, which possesses the largest spin density, is more effectively protected by three anthryl groups than that of 1. We expected that 14 could be isolated in the solid state with no substituent groups on the anthryl group.
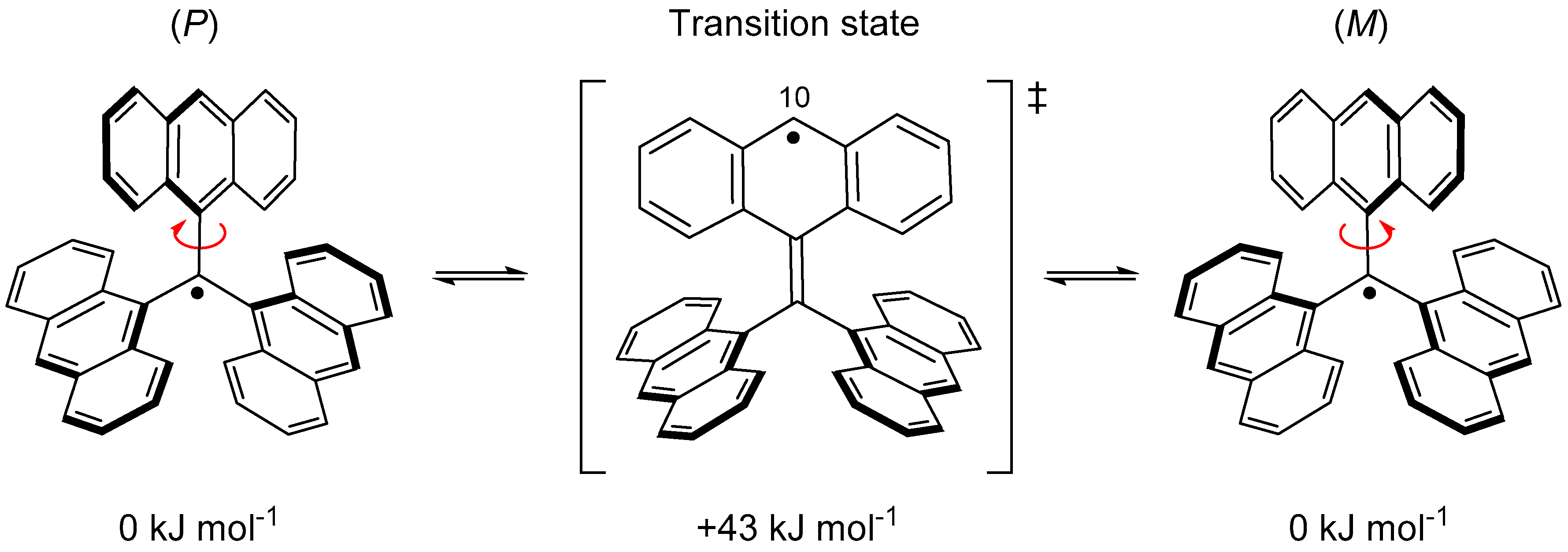
However, the radical that could be isolated in the solid state was not 14 but 15, where the carbon atom at the 10-position of all anthryl groups was sterically protected by a mesityl group. The stability of 15 was extraordinarily high and showed no decomposition even in air at room temperature. The absence of attack of molecular oxygen against the central carbon indicates that the steric protection by the anthryl groups works very effectively. However, it was found that 16, which removed one of the three mesityl groups, immediately undergoes a dimerization reaction at the 10-position of the non-substituted anthryl group (Figure 15). According to a UωB97X-D/6-31G** calculation, the flipping motion of the anthryl blade in 14 occurs very rapidly because of the small ΔH‡ (43 kJ mol−1), as shown in Figure 16. Near the transition state, an unpaired electron is mostly localized at the 10-position of the anthryl group, and this localized radical species shows high reactivity with the σ-dimer.
5. Conclusions
Our recent studies on stable, π-conjugated, carbon-centered, radical species were described. The π-conjugated radical species with high thermodynamic stability were found to be able to show various association modes due to steric and electronic effects of substituent groups. On the other hand, it was found that the anthryl group used for kinetic stabilization has not only a simple, steric, protective effect but also a role as a reaction site accompanied by a large structural change. By utilizing electronic and steric effects of substituent groups, organic radicals could offer a further possibility for developing new reactions [36] and new functionalities for optoelectronics [37], electronics [38], and spintronics [13,16] devices.
Acknowledgments
The author gratefully acknowledge all the collaborators for their extensive work, particularly Yasukazu Hirao (Osaka University), Tomohiko Nishiuchi (Osaka University), and Masayoshi Nakano (Osaka University).
Conflicts of Interest
The author declare no conflict of interest.
References
- Miller, J.S.; Glatzhofer, D.T.; Vazquez, C.; McLean, R.S.; Calabrese, J.C.; Marshall, W.J.; Raebiger, J.W. Electron-Transfer Salts of 1,2,3,4,5-Pentamethylferrocene, FeII(C5Me5)(C5H5). Structure and Magnetic Properties of Two 1:1 and Two 2:3 Fe(C5Me5)(C5H5) Electron-Transfer Salts of Tetracyanoethylene. Inorg. Chem. 2001, 40, 2058–2064. [Google Scholar] [CrossRef] [PubMed]
- Kothe, G.; Denkel, K.-H.; Sümmermann, W. Schlenk’s Biradical.—A Molecule in the Triplet Ground State. Angew. Chem. Int. Ed. Engl. 1970, 9, 906–907. [Google Scholar] [CrossRef]
- Hicks, R.G. What’s new in stable radical chemistry? Org. Biomol. Chem. 2007, 5, 1321–1338. [Google Scholar] [CrossRef] [PubMed]
- Rajca, A. Organic Diradicals and Polyradicals: From Spin Coupling to Magnetism? Chem. Rev. 1994, 94, 871–893. [Google Scholar] [CrossRef]
- Hicks, R.G. Stable Radicals: Fundamentals and Applied Aspects of Odd-Electron Compounds; Wiley-Blackwell: Hoboken, NJ, USA, 2010; ISBN 9780470666975. [Google Scholar]
- Gaudenzi, R.; Burzurí, E.; Reta, D.; Moreira, I.d.P.R.; Bromley, S.T.; Rovira, C.; Veciana, J.; van der Zant, H.S.J. Exchange Coupling Inversion in a High-Spin Organic Triradical Molecule. Nano Lett. 2016, 16, 2066–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasini, D.; Takeuchi, D. Cyclopolymerizations: Synthetic Tools for the Precision Synthesis of Macromolecular Architectures. Chem. Rev. 2018, 118, 8983–9057. [Google Scholar] [CrossRef] [PubMed]
- Hioe, J.; Zipse, H. Radical stability and its role in synthesis and catalysis. Org. Biomol. Chem. 2010, 8, 3609–3617. [Google Scholar] [CrossRef] [PubMed]
- Gomberg, M. AN INSTANCE OF TRIVALENT CARBON: TRIPHENYLMETHYL. J. Am. Chem. Soc. 1900, 22, 757–771. [Google Scholar] [CrossRef]
- Reid, D.H. The chemistry of the phenalenes. Q. Rev. Chem. Soc. 1965, 19, 274–302. [Google Scholar] [CrossRef]
- Haddon, R.C. Design of organic metals and superconductors. Nature 1975, 256, 394–396. [Google Scholar] [CrossRef]
- Morita, Y.; Nishida, S. Phenalenyls, Cyclopentadienyls, and Other Carbon-Centered Radicals. In Stable Radicals; Hicks, R.G., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2010; pp. 81–145. [Google Scholar]
- Raman, K.V.; Kamerbeek, A.M.; Mukherjee, A.; Atodiresei, N.; Sen, T.K.; Lazić, P.; Caciuc, V.; Michel, R.; Stalke, D.; Mandal, S.K.; et al. Interface-engineered templates for molecular spin memory devices. Nature 2013, 493, 509–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneda, K.; Nakano, M.; Fukuda, K.; Matsui, H.; Takamuku, S.; Hirosaki, Y.; Kubo, T.; Kamada, K.; Champagne, B. Third-Order Nonlinear Optical Properties of One-Dimensional Open-Shell Molecular Aggregates Composed of Phenalenyl Radicals. Chem. A Eur. J. 2014, 20, 11129–11136. [Google Scholar] [CrossRef] [PubMed]
- Salustro, S.; Maschio, L.; Kirtman, B.; Rérat, M.; Dovesi, R. Third-Order Electric Field Response of Infinite Linear Chains Composed of Phenalenyl Radicals. J. Phys. Chem. C 2016, 120, 6756–6761. [Google Scholar] [CrossRef]
- Mukherjee, A.; Sau, S.C.; Mandal, S.K. Exploring Closed-Shell Cationic Phenalenyl: From Catalysis to Spin Electronics. Acc. Chem. Res. 2017, 50, 1679–1691. [Google Scholar] [CrossRef] [PubMed]
- Reid, D.H. Stable π-electron systems and new aromatic structures. Tetrahedron 1958, 3, 339–352. [Google Scholar] [CrossRef]
- Goto, K.; Kubo, T.; Yamamoto, K.; Nakasuji, K.; Sato, K.; Shiomi, D.; Takui, T.; Kubota, M.; Kobayashi, T.; Yakusi, K.; et al. A Stable Neutral Hydrocarbon Radical: Synthesis, Crystal Structure, and Physical Properties of 2,5,8-Tri-tert-butyl-phenalenyl. J. Am. Chem. Soc. 1999, 121, 1619–1620. [Google Scholar] [CrossRef]
- Suzuki, S.; Morita, Y.; Fukui, K.; Sato, K.; Shiomi, D.; Takui, T.; Nakasuji, K. Aromaticity on the pancake-bonded dimer of neutral phenalenyl radical as studied by MS and NMR spectroscopies and NICS analysis. J. Am. Chem. Soc. 2006, 128, 2530–2531. [Google Scholar] [CrossRef]
- Small, D.; Zaitsev, V.; Jung, Y.; Rosokha, S.V.; Head-Gordon, M.; Kochi, J.K. Intermolecular pi-to-pi bonding between stacked aromatic dyads. Experimental and theoretical binding energies and near-IR optical transitions for phenalenyl radical/radical versus radical/cation dimerizations. J. Am. Chem. Soc. 2004, 126, 13850–13858. [Google Scholar] [CrossRef]
- Kolb, B.; Kertesz, M.; Thonhauser, T. Binding Interactions in Dimers of Phenalenyl and Closed-Shell Analogues. J. Phys. Chem. A 2013, 117, 3642–3649. [Google Scholar] [CrossRef]
- Koutentis, P.A.; Chen, Y.; Cao, Y.; Best, T.P.; Itkis, M.E.; Beer, L.; Oakley, R.T.; Cordes, A.W.; Brock, C.P.; Haddon, R.C. Perchlorophenalenyl radical. J. Am. Chem. Soc. 2001, 123, 3864–3871. [Google Scholar] [CrossRef]
- Uchida, K.; Hirao, Y.; Kurata, H.; Kubo, T.; Hatano, S.; Inoue, K. Dual association modes of the 2,5,8-tris(pentafluorophenyl)phenalenyl radical. Chem. Asian J. 2014, 9, 1823–1829. [Google Scholar] [CrossRef]
- Blanchard, M.D.; Hughes, R.P.; Concolino, T.E.; Rheingold, A.L. π-Stacking between Pentafluorophenyl and Phenyl Groups as a Controlling Feature of Intra- and Intermolecular Crystal Structure Motifs in Substituted Ferrocenes. Observation of Unexpected Face-to-Face Stacking between Pentafluorophenyl Rings. Chem. Mater. 2000, 12, 1604–1610. [Google Scholar] [CrossRef]
- Smith, C.E.; Smith, P.S.; Thomas, R.L.; Robins, E.G.; Collings, J.C.; Dai, C.; Scott, A.J.; Borwick, S.; Batsanov, A.S.; Watt, S.W.; et al. Arene-perfluoroarene interactions in crystal engineering: Structural preferences in polyfluorinated tolans. J. Mater. Chem. 2004, 413–420. [Google Scholar] [CrossRef]
- Mou, Z.; Uchida, K.; Kubo, T.; Kertesz, M. Evidence of σ- and π-Dimerization in a Series of Phenalenyls. J. Am. Chem. Soc. 2014, 136, 18009–18022. [Google Scholar] [CrossRef]
- Uchida, K.; Mou, Z.; Kertesz, M.; Kubo, T. Fluxional σ-Bonds of the 2,5,8-Trimethylphenalenyl Dimer: Direct Observation of the Sixfold σ-Bond Shift via a π-Dimer. J. Am. Chem. Soc. 2016, 138, 4665–4672. [Google Scholar] [CrossRef] [PubMed]
- Rakus, K.; Verevkin, S.P.; Schätzer, J.; Beckhaus, H.-D.; Rüchardt, C. Thermolabile Hydrocarbons, 33. Thermochemistry and Thermal Decomposition of 9,9′-Bifluorenyl and 9,9′-Dimethyl-9,9′-bifluorenyl—The Stabilization Energy of 9-Fluorenyl Radicals. Berichte der Deutschen Chemischen Gesellschaft 1994, 127, 1095–1103. [Google Scholar] [CrossRef]
- Font-Sanchis, E.; Aliaga, C.; Focsaneanu, K.-S.; Scaiano, J.C. Greatly attenuated reactivity of nitrile-derived carbon-centered radicals toward oxygen. Chem. Commun. 2002, 2, 1576–1577. [Google Scholar] [CrossRef]
- Tian, Y.; Uchida, K.; Kurata, H.; Hirao, Y.; Nishiuchi, T.; Kubo, T. Design and Synthesis of New Stable Fluorenyl-Based Radicals. J. Am. Chem. Soc. 2014, 136, 12784–12793. [Google Scholar] [CrossRef]
- Nishiuchi, T.; Ito, R.; Takada, A.; Yasuda, Y.; Nagata, T.; Stratmann, E.; Kubo, T. Anthracene-Attached Persistent Tricyclic Aromatic Hydrocarbon Radicals. Chem. Asian J. 2019. [Google Scholar] [CrossRef]
- Frenette, M.; Aliaga, C.; Font-Sanchis, E.; Scaiano, J.C. Bond Dissociation Energies for Radical Dimers Derived from Highly Stabilized Carbon-Centered Radicals. Org. Lett. 2004, 6, 2579–2582. [Google Scholar] [CrossRef]
- Zeng, Z.; Sung, Y.M.; Bao, N.; Tan, D.; Lee, R.; Zafra, J.L.; Lee, B.S.; Ishida, M.; Ding, J.; López Navarrete, J.T.; et al. Stable tetrabenzo-Chichibabin’s hydrocarbons: Tunable ground state and unusual transition between their closed-shell and open-shell resonance forms. J. Am. Chem. Soc. 2012, 134, 14513–14525. [Google Scholar] [CrossRef] [PubMed]
- Sabacky, M.J.; Johnson, C.S.; Smith, R.G.; Gutowsky, H.S.; Martin, J.C. Triarylmethyl Radicals. Synthesis and Electron Spin Resonance Studies of Sesquixanthydryl Dimer and Related Compounds. J. Am. Chem. Soc. 1967, 89, 2054–2058. [Google Scholar] [CrossRef]
- Nishiuchi, T.; Aibara, S.; Kubo, T. Synthesis and Properties of a Highly Congested Tri(9-anthryl)methyl Radical. Angew. Chem. Int. Ed. 2018, 57, 16516–16519. [Google Scholar] [CrossRef]
- Vijaykumar, G.; Pariyar, A.; Ahmed, J.; Shaw, B.K.; Adhikari, D.; Mandal, S.K. Tuning the redox non-innocence of a phenalenyl ligand toward efficient nickel-assisted catalytic hydrosilylation. Chem. Sci. 2018, 9, 2817–2825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ai, X.; Evans, E.W.; Dong, S.; Gillett, A.J.; Guo, H.; Chen, Y.; Hele, T.J.H.; Friend, R.H.; Li, F. Efficient radical-based light-emitting diodes with doublet emission. Nature 2018, 563, 536–540. [Google Scholar] [CrossRef]
- Mas-Torrent, M.; Crivillers, N.; Rovira, C.; Veciana, J. Attaching Persistent Organic Free Radicals to Surfaces: How and Why. Chem. Rev. 2012, 112, 2506–2527. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |
Figure 1.
Resonance structure and singly occupied molecular orbital (SOMO) of phenalenyl radical (2).
Figure 1.
Resonance structure and singly occupied molecular orbital (SOMO) of phenalenyl radical (2).

Figure 2.
Dimerization mode of phenalenyl radicals.

Figure 3.
Dimerization mode of (a) 4 and (b) 5.

Figure 4.
Various dimer forms of 6. ORTEP (Oak Ridge Thermal-Ellipsoid Plot Program) drawing of (a) 62-σ-chiral and (b) 62-π. (c) Three structural isomers of the dimer of 6.
Figure 4.
Various dimer forms of 6. ORTEP (Oak Ridge Thermal-Ellipsoid Plot Program) drawing of (a) 62-σ-chiral and (b) 62-π. (c) Three structural isomers of the dimer of 6.

Figure 5.
Attractive interactions between radical units of (a) 42 and (b) 52.

Figure 6.
One-dimensional stack of 4, (a) Top view and (b) side view. (c) Side view of alternating one-dimensional stack of 4 and 5.
Figure 6.
One-dimensional stack of 4, (a) Top view and (b) side view. (c) Side view of alternating one-dimensional stack of 4 and 5.

Figure 7.
(a) Fluorenyl radical (7). (b) Spin density of 7 calculated by a BLYP/6-31G** method. (c) Anthryl-protected dibenzo-fluorenyl radicals (8, 9, 10).
Figure 7.
(a) Fluorenyl radical (7). (b) Spin density of 7 calculated by a BLYP/6-31G** method. (c) Anthryl-protected dibenzo-fluorenyl radicals (8, 9, 10).

Figure 8.
σ-Dimer structures of (a) 8 and (b) 10 determined by X-ray crystallographic analysis.

Figure 9.
Ring condensation modes for 8, 9, and 10.

Figure 10.
Manner of the delocalization of an unpaired electron based on the spin density calculation (UBLYP/6-31G**//UB3LYP/6-31G**).
Figure 10.
Manner of the delocalization of an unpaired electron based on the spin density calculation (UBLYP/6-31G**//UB3LYP/6-31G**).

Figure 11.
Energy diagram for twisted 10 to 102 (σ-dimer) calculated with the UB3LYP/6-31G** method.
Figure 11.
Energy diagram for twisted 10 to 102 (σ-dimer) calculated with the UB3LYP/6-31G** method.
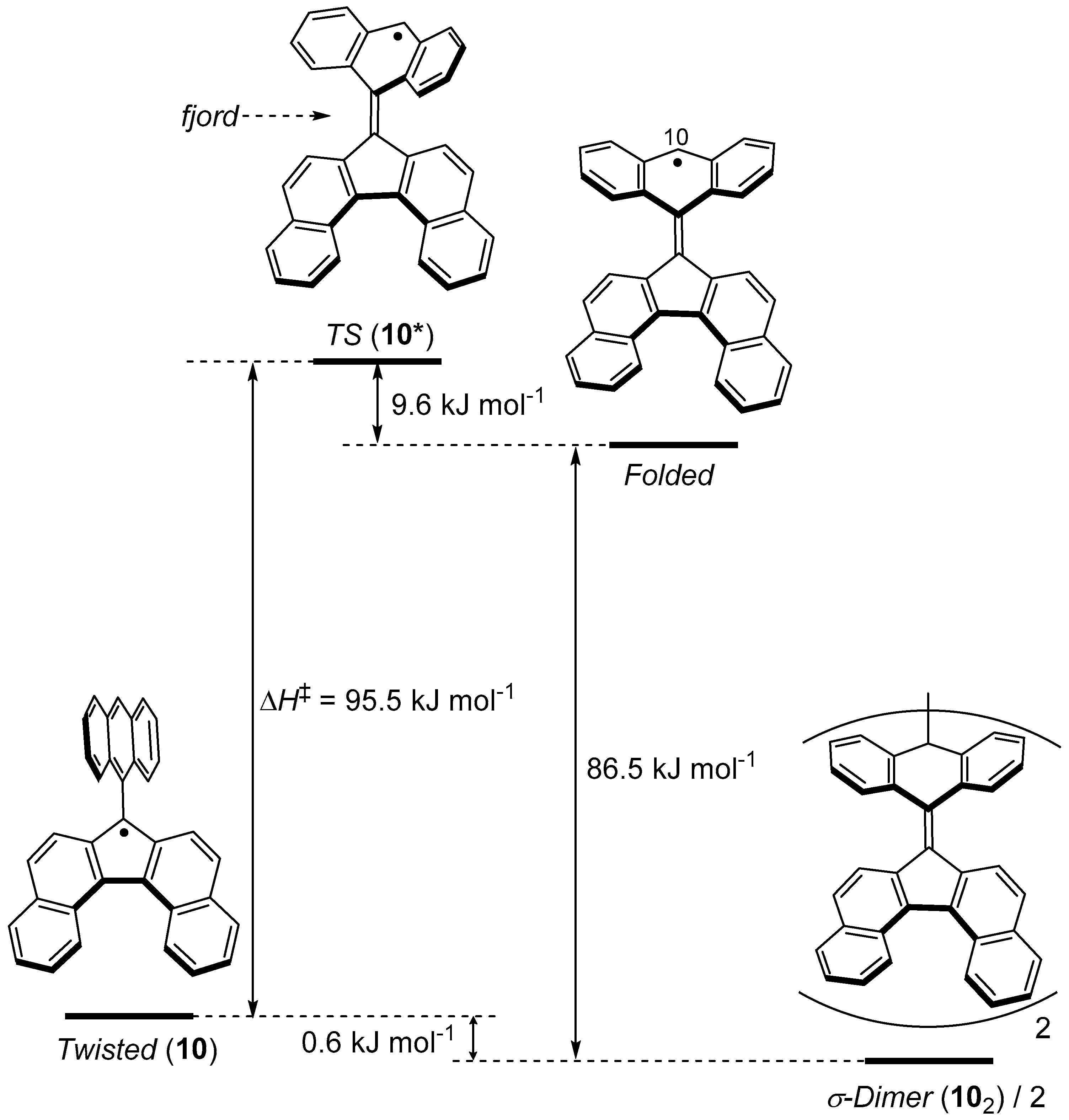
Figure 12.
Anthryl-substituted tricyclic radicals (11 (a), 12 (b), and 13 (c)). Spin density maps calculated by UBLYP/6-31G**//UωB97X-D/6-31G** are also shown.
Figure 12.
Anthryl-substituted tricyclic radicals (11 (a), 12 (b), and 13 (c)). Spin density maps calculated by UBLYP/6-31G**//UωB97X-D/6-31G** are also shown.

Figure 13.
Structures of the transition state of (a) 11 and (b) 13 calculated at the UBLYP-D3/6-31G** level of theory.
Figure 13.
Structures of the transition state of (a) 11 and (b) 13 calculated at the UBLYP-D3/6-31G** level of theory.

Figure 14.
(a) Trianthrylmethyl radicals (14, 15, and 16). Space-filling models of (b) 1 and (c) 14.
Figure 14.
(a) Trianthrylmethyl radicals (14, 15, and 16). Space-filling models of (b) 1 and (c) 14.

Figure 15.
Dimerization reaction of 16. Mes = mesityl.

Figure 16.
Relative energies for propeller flipping motion of 14 calculated at the UωB97X-D/6-31G** level of theory.
Figure 16.
Relative energies for propeller flipping motion of 14 calculated at the UωB97X-D/6-31G** level of theory.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Radical stabilization energies (RSEs) of selected carbon-centered radicals.
| Radical | RSE/kJ mol−1 |
|---|---|
| Methyl (•CH3) | 0 (as standard) |
| tert-Butyl | −28.3 |
| Allyl | −77.5 |
| Benzyl | −50.4 |
| Cyclopentadienyl | −98.3 |
| Cycloheptatrienyl | −134.1 |
| Triphenylmethyl (1) | −103.4 |
| Phenalenyl (2) | −201.6 |
| 9-Fluorenyl (7) | −90.7 |
Table 2.
Redox potentials (V vs Fc/Fc+) of 11, 12, and 13.
| Compounds | Eox/V | Ered/V |
|---|---|---|
| 11 | +0.28 | −1.11 |
| 12 | −0.10 | −1.76 |
| 13 | −0.30 | −1.78 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kubo, T. Synthesis, Physical Properties, and Reactivity of Stable, π-Conjugated, Carbon-Centered Radicals. Molecules 2019, 24, 665. https://doi.org/10.3390/molecules24040665
AMA Style
Kubo T. Synthesis, Physical Properties, and Reactivity of Stable, π-Conjugated, Carbon-Centered Radicals. Molecules. 2019; 24(4):665. https://doi.org/10.3390/molecules24040665
Chicago/Turabian StyleKubo, Takashi. 2019. "Synthesis, Physical Properties, and Reactivity of Stable, π-Conjugated, Carbon-Centered Radicals" Molecules 24, no. 4: 665. https://doi.org/10.3390/molecules24040665