
An Expeditious Total Synthesis of 5′-Deoxy-toyocamycin and 5′-Deoxysangivamycin


Key Laboratory of Organic Chemistry in Jiangxi Province, Institute of Organic Chemistry, Jiangxi Science & Technology Normal University, Nanchang 330013, China
*
Authors to whom correspondence should be addressed.
Molecules 2019, 24(4), 737; https://doi.org/10.3390/molecules24040737
Submission received: 4 February 2019
/
Revised: 15 February 2019
/
Accepted: 16 February 2019
/
Published: 19 February 2019
(This article belongs to the Special Issue Discovery, Design, Synthesis, and Application of Nucleoside/Nucleotides)
Abstract
:In present paper, an expeditious total synthesis of naturally occurring 5′-deoxytoyocamycin and 5′-deoxysangivamycin was accomplished. Because of the introduction of a benzoyl group at N-6 of 4-amino-5-cyano-6-bromo-pyrrolo[2,3-d]pyrimidine, a Vorbrüggen glycosylation with 1,2,3-tri-O-acetyl-5-deoxy-β-D-ribofuranose afforded a completely regioselective N-9 glycosylation product, which is unambiguously confirmed by X-ray diffraction analysis. All of the involved intermediates were well characterized by various spectra.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
The synthesis and biological evaluation of nucleoside/tide analogs as agents to treat cancers, viruses, and parasites, as well as fungal and bacterial infections, spans the past half century [1,2,3]. The first steps in this field can be traced back to the emergence of cytarabine to treat for acute myeloid leukemia and edoxudine for herpes simplex virus, which were both approved by the FDA in 1969. This strategy has become very successful and there are now more than 30 nucleoside/tide analogues approved for clinic use, with many more ongoing in preclinical and clinical trials [4,5,6].
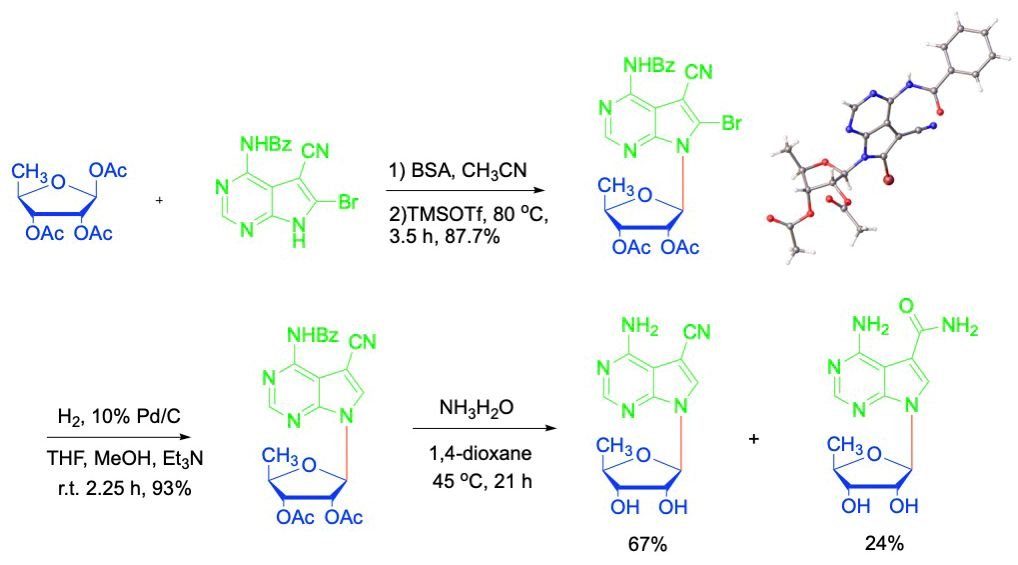
As important analogues of biogenic purine nucleosides, 7-deazapurine nucleosides have shown diverse biological activities [7,8,9,10]. Replacement of the purine’s N-7 atom with a carbon atom results a more electron rich pyrrole ring more prone to cation–π or π–π interactions, which subsequently leads to increased nucleobase-pairing in DNA or RNA and possibly better binding to proteins [11,12]. Many 7-deazapurine nucleosides were isolated as naturally occurring products, such as toyocamycin (1) and sangivamycin (2) from Streptomyces cultures (Figure 1). They showed potent cytotoxicity against various cancer cell lines with different modes of action. Moreover, toyocamycin (1) was identified recently as a potent inhibitor of induced XBP1 mRNA cleavage, which is a key component of the endoplasmic reticulum stress response [13]. On the other hand, the cytotoxic effect of sangivamycin (2) is mainly due to selective inhibition of protein kinase C through inhibition of Erk and Akt signaling pathway [14,15].
In the course of our continued research interest in the synthesis of 7-deazapurine nucleosides and their biological activities, 5′-deoxytoyocamycin (3) and 5′-deoxysangivamycin (4) (Figure 1) were needed as a control to compare their biological action mechanism with toyocamycin (1) and sangivamycin (2), respectively. In the published literature, 5′-deoxytoyocamycin (3) and 5′-deoxysangivamycin (4) were originally synthesized in moderate yields from toyocamycin and sangivamycin by radical reduction (AIBN and tributyltin hydride in dry THF) of the corresponding 5′-chloro-5′-deoxynucleosides [16,17]. This approach was inefficient, cumbersome and low yielding and the the starting materials very expensive. Later on, 5′-deoxytoyocamycin (3) was isolated from the fermentation broth of a Streptomyces sp. from a soil sample collected from Kepong, Malaysia. Although a manufacturing process for 5’-deoxytoyocamycin using the Streptomyces sp. was patented soon after its discovery, it was not practical to perform it in our laboratory. Thus, we turned our endeavor to total syntheses of 5′-deoxytoyocamycin (3) and 5′-deoxysangivamycin (4), which could provide enough material for further biological studies.
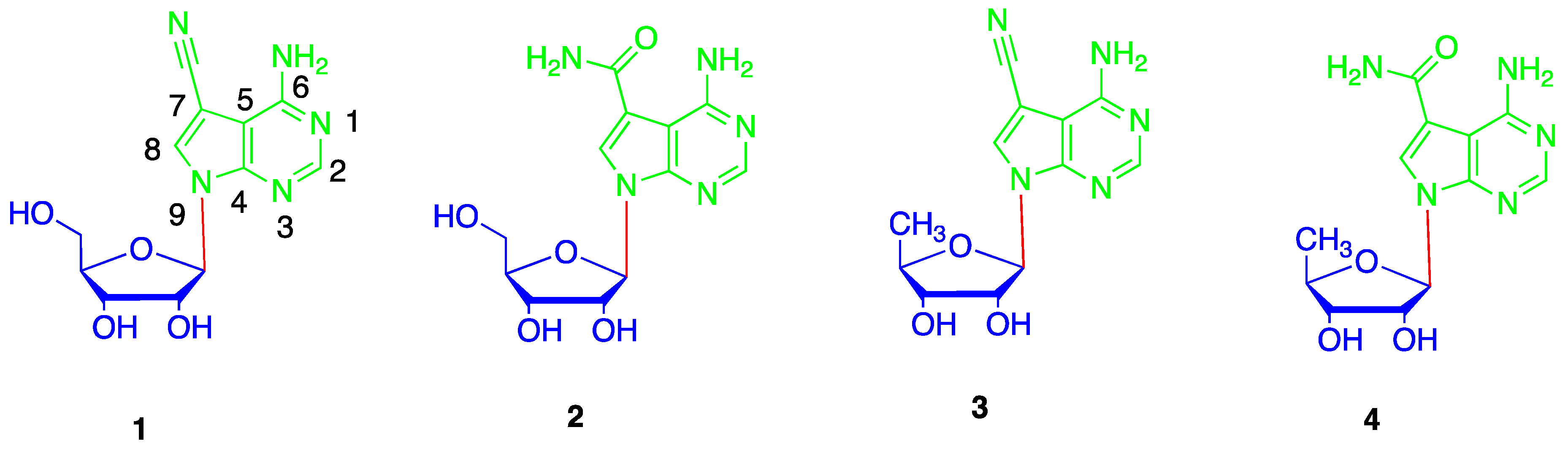
While various glycosylation approaches have been developed for the preparation of ribonucleosides, the most widely employed method is the silyl-Hilbert-Johnson reaction catalyzed by equimolar or excess amounts of Lewis acid catalysts (Vorbrüggen glycosylation), which has now been developed for more than fifty years [18]. However, the Vorbrüggen glycosylation when performed with 7-deazapurines was found to be much less efficient. Until now, Vorbrüggen glycosylation could only be used to prepare the corresponding ribonucleosides of 7-deazapurine nucleobases with an electron withdrawing substitute at C-7 [19]. Specifically, total synthesis of toyocamycin (1) by Vorbrüggen glycosylation of ribose 5 and nucleobase 6 was firstly reported by Bobek [20], and further improved by Townsend [21], but during our many repeats of this approach [22,23,24,25], it was found that the N-9 glycosylation product 7 and N-3 glycosylation product 8 were inevitably formed simultaneously (Figure 2). This phenomenon was also reported by several other groups [26,27,28]. The selectivity and equilibrium between N-9 glycosylation product 7 and N-3 glycosylation product 8 were subsequently studied in detail [27]. In order to resolve this problem, an improved synthetic approach is reported in the present paper, that was used successfully in the total synthesis of 5′-deoxytoyocamycin (3) and 5′-deoxysangivamycin (4).
2. Results and Discussion
According to the reaction mechanism of the Vorbrüggen glycosylation, we reasoned that N-3 product formation may result from the more electron rich pyrimidine in nucleobase 6. Thus, the formation of N-3 products, such as nucleoside 8, could be avoided by reducing the pyrimidine ring’s electron density. On the other hand, the solubility of nucleobase 6 is very low and its purification is really difficult. We speculated that nucleobase 12 with a benzoyl group at N-6 could fulfill our requirements. Although, the nucleobase 6 with an acetyl at N-6 was already reported, its use did not solve the above selectivity and solubility problems [20,29,30,31].
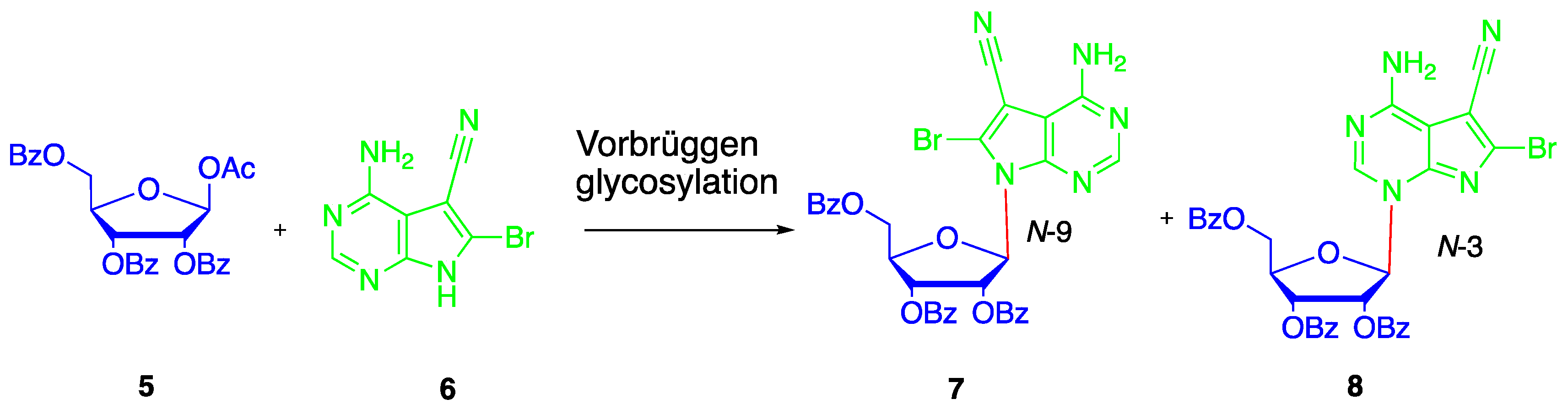
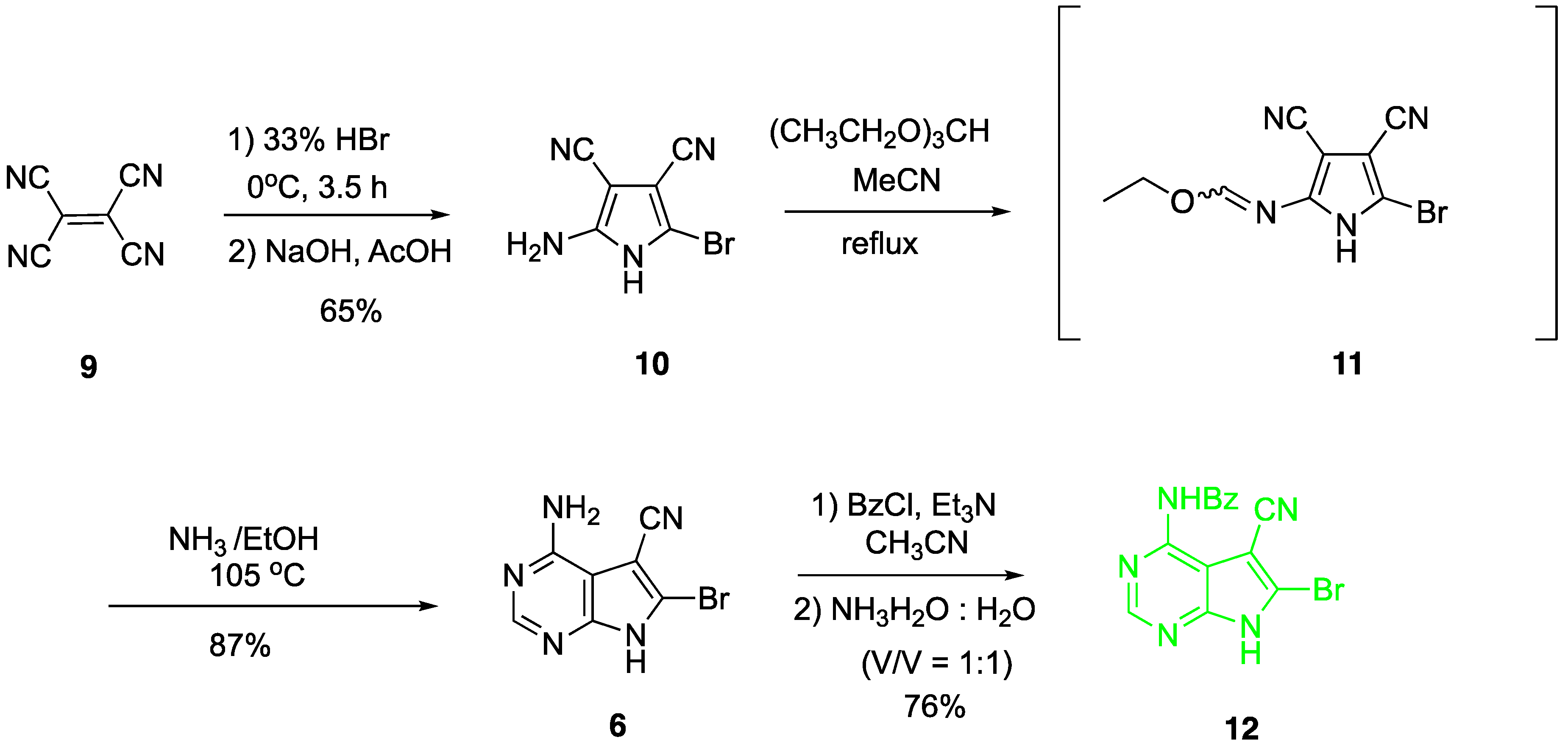
Preparation of nucleobase 6 from tetracyanoethylene 9 was reported in several references [32,33,34,35]. In order to synthesize it on a large scale, the corresponding synthetic procedure was improved in our following experiments (Scheme 1). Firstly, pyrrole 10 were synthesized from tetracyanoethylene 9 according to a modification of the protocol developed by Townsend [36]. It should be mentioned that the intermediate formed after addition of 33% HBr is very hydroscopic and not stable in air. After transformation to the free pyrrole 10, it could be stored on the bench for a long time. Then, pyrrole 10 was refluxed with ethyl orthoformate in CH3CN to give intermediate 11. After removing the volatile byproducts and without further purification, ring annulation to give nucleobase 6 in 87% yield was accomplished in freshly prepared saturated ethanolic ammonia at 105 °C in a closed stainless-steel container for 6 h.
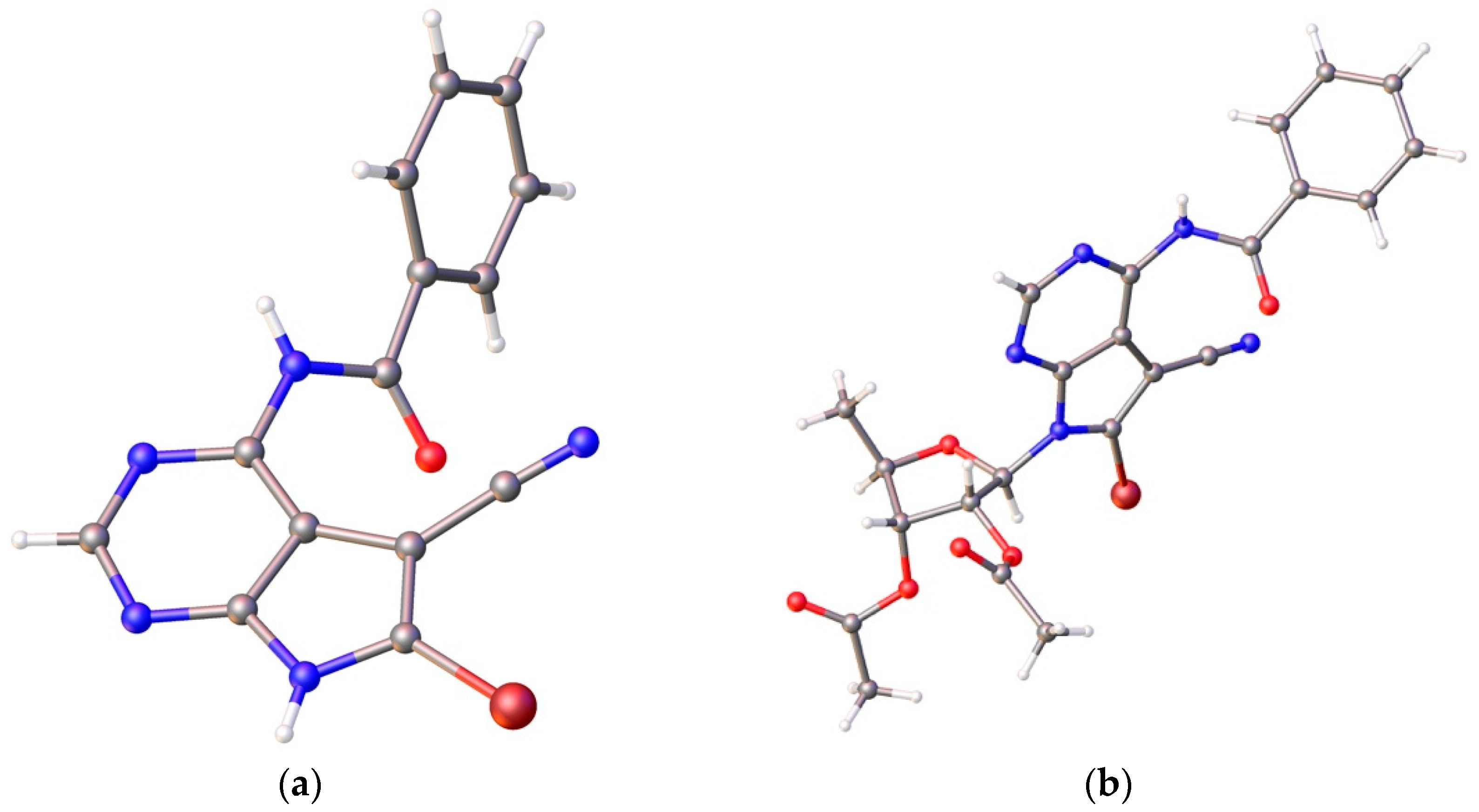
The subsequent monobenzoylation at N-6 to provide nucleobase 12 proved to be troublesome. Using pyridine as the base and solvent, at least four products are formed. Other attempts to directly synthesize the monobenzoylated product 12 also failed. After optimization, complete benzoylation of nucleobase 6 was conducted firstly to give the corresponding N6,N6,N9-tribenzoyl derivative of 6 using a large excess of benzoyl chloride (5 eq.) and Et3N (7.5 eq) in CH3CN. Then the tribenzoyl intermediate, without isolation, was stirred directly with concentrated ammonia and H2O (V/V = 1:1) to afford the desired nucleobase 12 in 76% yield. This modified protocol for the synthesis of nucleobase 12 from 9 was repeated several times at 50 g scale without requiring chromatographic purification. Furthermore, a single crystal of nucleobase 12 suitable for X-ray crystallographic analysis was obtained and the X-ray structure shown in Figure 3a was determined [37].
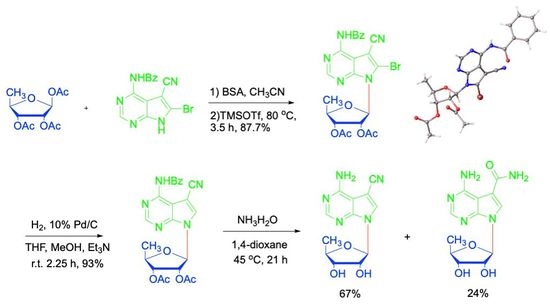
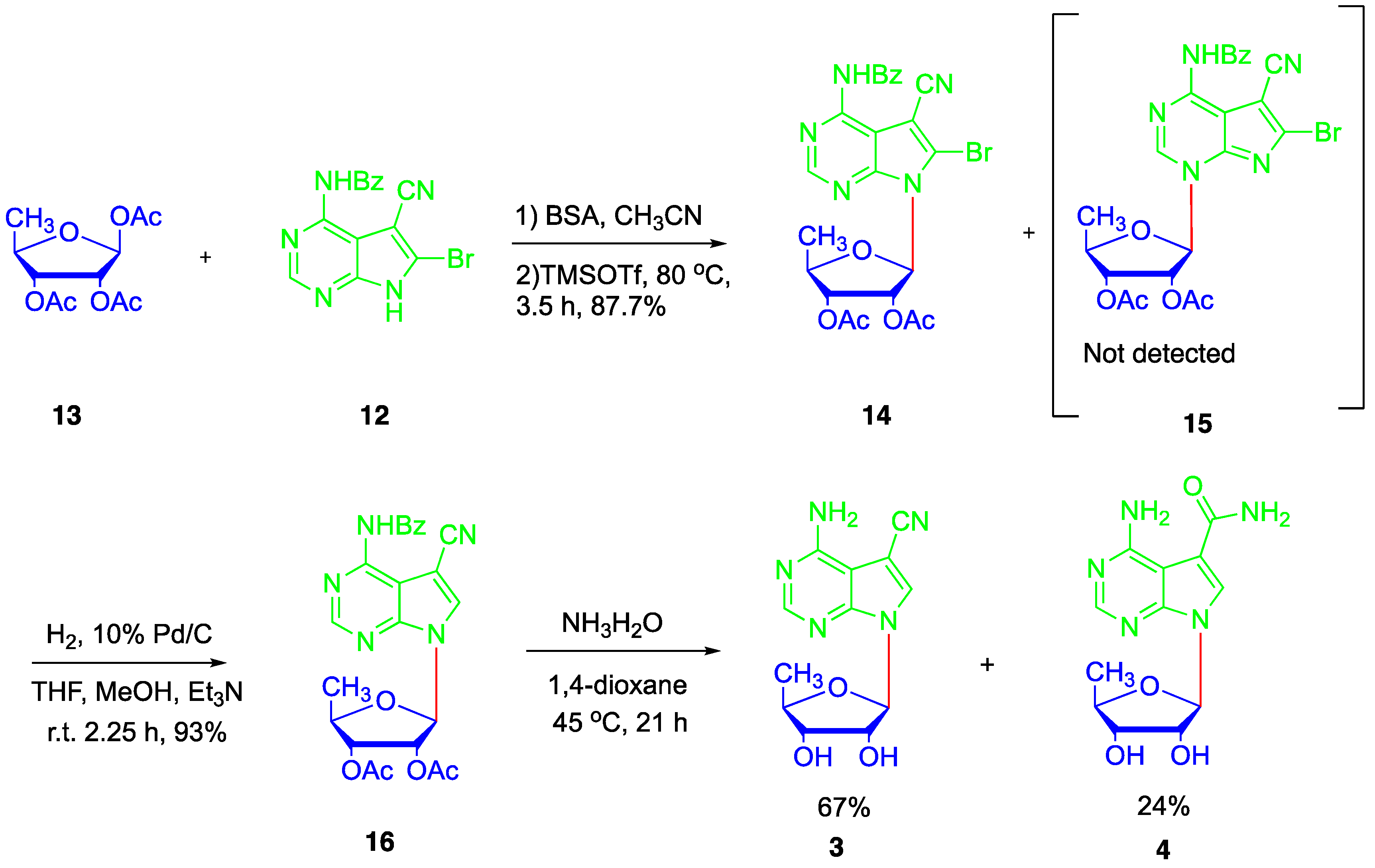
Next, the Vorbrüggen glycosylation between ribose 13 and nucleobase 12 was conducted (Scheme 2). After silylation of nucleobase 12 with N,O-Bis(trimethylsilyl)acetamide (BSA) in dry MeCN, 1,2,3-tri-O-acetyl-5-deoxy-β-D-ribofuranose (13) was added, followed by TMSOTf. The resulting mixture was heated at 80 °C for 3.5 h. After purification, 7-deazapurine nucleoside 14 was obtained in 87.7% yield. To our delight, the N-3 glycosylation product 15 was not detected. Thus, our introduction of a benzoyl group at N-6 successfully solved the regioselectivity problem of the glycosylation. At the same time, a single crystal of nucleoside 14 suitable for X-ray diffraction analysis was also obtained and its structure is unambiguously confirmed, which showed an anti-glycosylic bond conformation and an envelope C2′-endo (2E) sugar conformation (S-type) (Figure 3b) [36]. It is worth mentioning that X-ray structures of toyocamycin and its analogues have seldom been reported [38].
Subsequently, the bromide at C-8 was hydrogenated in a solvent mixture of methanol and THF using 10% Pd/C as catalyst to give nucleoside 16 in 93% yield. Deprotection of the remaining four benzoyl groups proved troublesome. When heated with saturated ammonium in methanol, 5′-deoxytoyocamycin (3) and 5′-deoxysangivamycin (4) were obtained at the same time along with other side products.
A variety of other conditions were tried with no improvement. Looking back the X-ray structures of nucleobase 12 and nucleoside 14, it was obvious that the carbonyl of the N-6 benzoyl was close and syn to the C-7 nitrile group (Figure 3) [37]. Therefore, the nitrile group of nucleoside 16 could be activated by the electron-withdrawing benzoyl group, which makes the nitrile group susceptible to attack by ammonia. Luckily, 5′-deoxytoyocamycin (3) and 5′-deoxysangivamycin (4) were both targets, and they could be conveniently separated by chromatography. In the end, 5′-deoxytoyocamycin (3) and 5′-deoxysangivamycin (4) were obtained in 67% and 24% yield, respectively, after removing all the benzoyl protecting groups with ammonia in dioxane. All the spectra of 5′-deoxytoyocamycin (3) were in accordance with those of the naturally occurring product. Because different solvents were used in the original NMR determination of naturally occurring 5′-deoxytoyocamycin (3) [39], the corresponding NMR data obtained in various solvents are summarized in this work (Table S1 in the Supplementary Information).
3. Conclusions
In conclusion, we have accomplished an improved total synthesis of 5′-deoxytoyocamycin (3) and 5′-deoxysangivamycin (4) using a Vorbrüggen glycosylation between 1,2,3-tri-O-acetyl-5-deoxy-β-D-ribofuranose (13) and N4-benzoyl-5-cyano-6-bromo-7H-pyrrolo[2,3-d]pyrimidine (12) as the key step. The structure of 7-deazapurine nucleoside 14 was unambiguously confirmed by X-ray diffraction analysis, which showed an anti glycosylic bond conformation and an envelope C2′-endo (2E) sugar conformation. It was demonstrated that the regioselectivity during the Vorbrüggen glycosylation completely favored the N-9 glycosylation product after introducing a benzoyl group at the N-6 position of 4-amino-5-cyano-6-bromo-7H-pyrrolo[2,3-d]pyrimidine. Thus, this newly developed nucleobase 12 could be used to synthesize other toyocamycin analogues while avoiding the formation of N-3 glycosylation side products.
4. Materials and Methods
4.1. Materials and Instruments
Unless otherwise specified, all the reagents were acquired from commercial sources and used directly. NMR spectra (400 MHz/100 MHz) were recorded on an Advance DPX spectrometer (Bruker, Billerica, MA, USA) at room temperature with DMSO-d6 as solvent. Tetramethylsilane (TMS) was used as an internal reference. Specific rotations were acquired on an Autopol IV polarimeter (Rudolph, Hackettstown, NJ, USA). High resolution mass spectrometry (HRMS) data were measured with an AB Sciex TOF 4600 instrument (Billerica, MA, USA). Melting points were measured on an X-4 digital melting point apparatus (Beijing Taike Corparation, Beijing, China). X-ray diffraction analysis was performed on a Bruker Smart Apex II system (Bruker, Billerica, MA, USA).
4.2. Synthesis of 2-amino-5-bromo-3,4-dicyano-1H-pyrrole (10)
Tetracyanoethylene (9, 35.6 g, 277.93 mmol) was dissolved in fresh dried acetone (200 mL) and ethyl acetate (420 mL). Under an Ar atmosphere, 33% HBr in acetic acid (200 mL) was added slowly while keeping the internal reaction temperature below 2 °C. After addition, the reaction mixture was stirred for another 1.5 h. The formed yellow solid was filtered and washed with cold ethyl acetate (100 mL). Subsequently, the crude product was suspended in ice cold water (100 mL) and 50% NaOH in H2O (60 mL) was added slowly until a homogenous solution was obtained. After filtration through Celite, the filtrate pH was adjusted carefully to 5 using acetic acid. The thus obtained solid was filtered and washed thoroughly with water until neutral. After drying under vacuum with P2O5, a grey solid of 10 was obtained (38.12 g, 65%). Rf = 0.6 (DCM/MeOH, V/V = 10:1); mp: 224–226 °C; 1H-NMR δ: 12.25 (s, 1H), 6.45 (s, 2H); 13C-NMR δ: 149.8, 115.1, 114.4, 103.0, 93.5, 71.4; HRMS: calcd. for C6H2BrN4 [M − H]− 208.9468; found: 208.9446.
4.3. Synthesis of 4-amino-5-cyano-6-bromo-7H-pyrrolo[2,3-d]pyrimidine (6)
2-Amino-5-bromo-3,4-dicyano-1H-pyrrole (10, 65.5 g, 310.40 mmol) was suspended in fresh dried CH3CN (1.5 L) and ethyl orthoformate (77.44 mL, 465.59 mmol). The suspension was heated to reflux until TLC showed the reaction was finished (about 3 h). The solvent was removed under vacuum to give a grey solid (82 g). Subsequently, the obtained solid was suspended in freshly prepared saturated ammonia in ethanol (1150 mL) in a closed stainless-steel container and heated at 105 °C for 6 h. Once cooled to room temperature, the reaction mixture was poured to a 5 L beaker with activated charcoal (22 g). After stirring for 30 min, the mixture was filtered through Celite and washed thoroughly with concentrated ammonia in ethanol (V/V = 1/5, 150 mL × 3). The filtration was adjusted to pH = 5 with acetic acid (about 160 mL). The obtained solid was filtered and washed with H2O. After dried under vacuum with P2O5, 6 was obtained as greenish solid (64.28 g, 87%). Rf = 0.6 (DCM/MeOH, V/V = 10:1); mp: 279–281 °C; 1H-NMR δ: 8.21 (s, 1H, H-2), 7.20 (s, 2H, NH2); 13C-NMR δ: 156.0(C-4), 150.1(C-7a), 149.6 (C-2), 124.9 (C-6), 115.9 (CN), 103.7 (C-4a), 84.4(C-5); HRMS: calcd. for C7H3BrN5 [M−H]− 235.9577; found: 235.9585.
4.4. Synthesis of N4-benzoyl-5-cyano-6-bromo-7H-pyrrolo[2,3-d]pyrimidine (12)
Deazapurine base 6 (50 g, 210.05 mmol, 1 eq.) was suspended in fresh dried CH3CN (450 mL) and Et3N (220 mL, 1.58 mol, 7.5 eq.). After the above suspension was stirred for 1 h at room temperature, benzoyl chloride (122 mL, 1.05 mol, 5 eq.) was added and the reaction mixture was stirred overnight. After TLC showed the 7-deaazapurine base had vanished, the reaction pH was adjusted by acetic acid to 6 and he reaction mixture was filtered. The filtrate was extracted with DCM (300 mL × 3). The combined extracts were washed with H2O, saturated NaCl, and dried with anhydrous MgSO4. After filtration, the solvent was evaporated under vacuum. The obtained crude product was stirred with concentrated ammonium and H2O (1 L, V/V = 1:1) for 5 h to give light yellow protected deazapurine 12 (54.63 g, yield: 76%). Rf = 0.3 (CH2Cl2/MeOH, V/V = 10:1); mp 235–237 °C; 1H-NMR δ: 14.23 (s, 1H, NH), 11.36 (s, 1H, NH), 8.75 (s, 1H, H-2), 8.06 (d, J = 6.3 Hz, 2H), 7.65 (t, J = 7.2 Hz, 1H), 7.56 (t, J = 7.5 Hz, 2H); 13C-NMR δ: 167.4 (C=O), 153.3 (C-4), 152.7 (C-2), 150.6 (C-7a), 133.5, 132.9, 128.9, 128.8, 124.1 (C-6), 114.8 (CN), 111.9 (C-4a), 88.8 (C-5); HRMS: calcd. for C14H7BrN5O [M − H]− 339.9839; found: 339.9852.
4.5. Synthesis of N4-benzoyl-5-cyano-6-bromo-7-(2′,3′-bi-O-acetyl-5′-deoxy-β-D-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidine (14)
N,O-Bis(trimethylsilyl)acetamide (BSA, 14.27 g, 70.14 mmol) was added to a suspension of N4-benzoyl-5-cyano-6-bromo-7H-pyrrolo[2,3-d]pyrimidine (12, 6.00 g, 17.53 mmol) in dry MeCN (60 mL) under an argon atmosphere. The reaction mixture was stirred at room temperature for 60 min. A solution of 1,2,3-tri-O-acetyl-5-deoxy-β-D-ribofuranose 13 (5.48 g, 21.04 mmol) in dry MeCN (50 mL) along with TMSOTf (15.59 g, 70.14 mmol) were added to the above reaction mixture under ice-bath condition. The solution was stirred for 30 min at room temperature before heating to 80 °C for 3.5 h. Then the mixture was cooled and poured into sat. NaHCO3 solution (150 mL). It was extracted with CH2Cl2 (150 mL × 3). The combined organic layer was washed with sat. NaHCO3 (100 mL × 2), brine (100 mL × 2), and dried over anhydrous MgSO4. After filtration, the filtrate was evaporated to dryness under reduced pressure. The residue was purified by silica gel column chromatography (CH2Cl2 /EtOAc 20:1, v:v) to afford 14 as yellow solid (8.34 g, 87.7%). Rf = 0.4 (CH2Cl2 /EtOAc, V/V = 20:1); m.p. 230–232 °C; −22.40 (c = 0.098, CH2Cl2); 1H-NMR δ 11.58 (s, 1H, NH), 8.89 (s, 1H, H-2), 8.06 (d, J = 7.5 Hz, 2H), 7.67 (t, J = 7.3 Hz, 1H), 7.57 (t, J = 7.6 Hz, 2H), 6.23 (d, J = 4.0 Hz, 1H, H-1′), 6.18–6.14 (m, 1H, H-2′), 5.50 (t, J = 6.7 Hz, 1H, H-3′), 4.33 – 4.25 (m, 1H, H-4′), 2.13 (s, 3H, OAc), 2.08 (s, 3H, OAc), 1.39 (d, J = 6.2 Hz, 3H, H-5’); 13C-NMR δ 170.1 (C=O, OAc), 170.0 (C=O, OAc), 167.5 (C=O, Bz), 153.1 (C-2), 152.0 (C-4), 151.4 (C-7a), 133.1, 129.0, 128.9, 126.6 (C-6), 114.2 (CN), 111.8 (C-4a), 91.4 (C-5), 89.1 (C-1′), 77.8 (C-4′), 74.0 (C-3′), 72.5 (C-2′), 20.8(OAc), 20.7 (OAc), 18.2 (C-5′); HRMS: calcd for C23H21BrN5O6 [M+H]+ 542.0670; found 542.0679.
4.6. Synthesis of N4-benzoyl-5-cyano-7-(2′,3′-di-O-acetyl-5′-deoxy-β-D-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidine (16)
10% Pd/C (0.75 g, 0.70 mmol) and Et3N (0.94 g, 9.22 mmol) were added to a solution of nucleoside 14 (2.50 g, 4.61 mmol) in the mixture of THF (65 mL) and MeOH (35 mL). After stirring at room temperature for 2.25 h under H2 atmosphere (1 atm), the reaction mixture was filtered and concentrated to dryness under reduced pressure. The residue was purified by silica gel column chromatography (CH2Cl2/EtOAc = 20:1, v:v) to afford 16 as light yellow solid (1.99 g, 93%). Rf = 0.3 (CH2Cl2 /EtOAc, V/V = 20:1); m.p. 92–94 °C; −45.80 (c = 0.109, CH2Cl2); 1H-NMR δ 11.50 (s, 1H, NH), 8.88 (s, 2H, H-2, H-6), 8.07 (d, J = 7.5 Hz, 2H), 7.66 (t, J = 7.2 Hz, 1H), 7.57 (t, J = 7.4 Hz, 2H), 6.40 (d, J = 5.4 Hz, 1H, H-1′), 5.90 (t, J = 5.6 Hz, 1H, H-2′), 5.32 (t, J = 5.2 Hz, 1H, H-3′), 4.32–4.26 (m, 1H, H-4′), 2.13 (s, 3H, OAc), 2.04 (s, 3H, OAc), 1.44 (d, J = 6.3 Hz, 3H, H-5′); 13C-NMR δ 170.0 (C=O, OAc), 169.8 (C=O, OAc), 167.5 (C=O, Bz), 153.2 (C-2), 152.5 (C-4), 152.1(C-7a), 137.1 (C-6), 133.3, 133.0, 128.9, 128.8, 114.9 (CN), 111.4 (C-4a), 87.5 (C-5), 86.6 (C-1′), 78.6 (C-4′), 74.3 (C-3′,), 73.2 (C-2′), 20.8 (OAc), 20.6 (OAc), 18.8 (C-5′); HRMS: calcd for C23H22N5O6 [M + H]+ 464.1565; found 464.1579.
4.7. Synthesis of 5′-deoxytoyocamycin (3) and 5′-deoxysangivamycin (4)
Ammonium hydroxide (45 mL) was added to a solution of 5 (1.80 g, 3.88 mmol) in 1,4-dioxane (18 mL). The reaction mixture was sealed and heated to 45 °C for 21 h. Then the reaction mixture was evaporated to dryness under reduced pressure and the residue was purified by silica gel column chromatography (CH2Cl2/MeOH 20:1, v:v) to afford compounds 3 (716 mg, 67%) and 4 (274 mg, 24%), respectively.
4.8. 5′-Deoxytoyocamycin (3)
Rf = 0.2 (CH2Cl2/MeOH, V/V = 20:1); m.p. 190–192 °C (literature: 187–188 °C [16]); 87.88 (c = 0.099, DMSO); 1H-NMR δ 8.40 (s, 1H, H-8), 8.23 (s, 1H, H-2), 6.90 (s, 2H, NH2), 6.04 (d, J = 4.7 Hz, 1H, H-1′), 5.48 (d, J = 5.6 Hz, 1H, 2′-OH), 5.20 (d, J = 5.3 Hz, 1H, 3′-OH), 4.44–4.37 (m, 1H, H-2′), 4.00–3.91 (m, 1H, H-4′), 3.91–3.84 (m, 1H, H-3′), 1.30 (d, J = 6.3 Hz, 3H, CH3); 13C-NMR δ 157.5 (C-6), 154.1 (C-2), 150.8 (C-4), 132.9 (C-8), 115.8 (CN), 101.7 (C-5), 88.4 (C-1′), 83.8 (C-7), 80.2 (C-4′), 75.0 (C-3′), 74.2 (C-2′), 19.4 (CH3); HRMS: calcd for C12H14N5O3 [M + H]+ 276.1091; found 276.1099.
4.9. 5′-Deoxysangivamycin (4)
Rf = 0.15 (CH2Cl2/MeOH, V/V = 10:1); m.p. 257–259 °C (literature value 260–262 °C [16]); −77.05 (c = 0.122, DMSO); 1H-NMR δ 8.09 (s, 2H, H-2, H-8), 7.99 (s, 1H, NH), 7.39 (s, 1H, NH), 6.08 (d, J = 4.2 Hz, 1H, H-1′), 5.50 (d, J = 5.5 Hz, 1H, 2′-OH), 5.20 (d, J = 5.7 Hz, 1H, 3′-OH), 4.24–4.19 (m, 1H, H-2′), 4.00–3.91 (m, 1H, H-4′), 3.91–3.84 (m, 1H, H-3′), 1.32 (d, J = 6.3 Hz, 3H, CH3); 13C-NMR δ 166.8 (C=O), 158.5 (C-6), 153.4 (C-2), 151.4 (C-4), 125.4 (C-8), 111.7 (C-7), 101.3 (C-5), 87.7 (C-1′), 79.5 (C-4′), 75.2 (C-3′), 74.8 (C-2′), 19.4 (CH3); HRMS: calcd for C12H16N5O4 [M + H]+ 294.1197; found 294.1208.
Supplementary Materials
The NMR spectra of compound 3–4, 6, 9, 11–12, 14, 16 are available online at https://www.mdpi.com/1420-3049/24/4/737/s1.
Author Contributions
Q.X. and H.D. conceived and designed the experiments; X.D., J.T. and C.H. performed the experiments; C.H. and J.B. analyzed the data; Q.X. and H.D. wrote the paper.
Funding
This research was funded by the National Natural Science Foundation of China (no. 21676131 and no. 21462019), the Science Foundation of Jiangxi Province (20143ACB20012), and Jiangxi Science & Technology Normal University (Doctor Startup Fund for Dr. Haixin Ding) for financial support.
Conflicts of Interest
The authors declare no conflict of interest.
References and Note
- Yates, M.K.; Seley-Radtke, K.L. The evolution of antiviral nucleoside analogues: A review for chemists and non-chemists. Part II: Complex modifications to the nucleoside scaffold. Antivir. Res. 2019, 162, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Seley-Radtke, K.L.; Yates, M.K. The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold. Antivir. Res. 2018, 154, 66–86. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Wnuk, S.F. Modification of Purine and Pyrimidine Nucleosides by Direct C-H Bond Activation. Molecules 2015, 20, 4874–4901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.X.; Littler, E. Nucleoside analogs as anti-HBV agents. Curr. Top. Med. Chem. 2006, 6, 851–865. [Google Scholar] [CrossRef]
- De Clercq, E.; Li, G.D. Approved Antiviral Drugs over the Past 50 Years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perlikova, P.; Hocek, M. Pyrrolo[2,3-d]pyrimidine (7-deazapurine) as a privileged scaffold in design of antitumor and antiviral nucleosides. Med. Res. Rev. 2017, 37, 1429–1460. [Google Scholar] [CrossRef]
- Townsend, L.B.; Drach, J.C.; Wotring, L.L.; Vittori, S.; Pudlo, J.S.; Swayze, E.E.; Gupta, P.; Maruyama, T.; Saxena, N.; Coleman, L.A. Design, synthesis, and studies on the structure-activity relationships of certain pyrrolo[2,3-d]pyrimidine nucleosides and structurally related analogs as potential antineoplastic and antiviral agents. Farmaco 1991, 46, 113–139. [Google Scholar] [CrossRef]
- Dholakia, S.P.; Patel, M.M.; Patel, J.S. Role of pyrrolopyrimidine derivatives as anticancer agent: Mini review. Indo Am. J. Pharm. Res. 2015, 5, 858–867. [Google Scholar]
- De Coen, L.M.; Heugebaert, T.S.A.; Garcia, D.; Stevens, C.V. Synthetic Entries to and Biological Activity of Pyrrolopyrimidines. Chem. Rev. 2016, 116, 80–139. [Google Scholar] [CrossRef]
- Cahova, H.; Panattoni, A.; Kielkowski, P.; Fanfrlik, J.; Hocek, M. 5-Substituted Pyrimidine and 7-Substituted 7-Deazapurine dNTPs as Substrates for DNA Polymerases in Competitive Primer Extension in the Presence of Natural dNTPs. ACS Chem. Biol. 2016, 11, 3165–3171. [Google Scholar] [CrossRef] [PubMed]
- Seela, F.; Sirivolu, V.R. DNA containing side chains with terminal triple bonds: Base-pair stability and functionalization of alkynylated pyrimidines and 7-deazapurines. Chem. Biodivers. 2006, 3, 509–514. [Google Scholar] [CrossRef]
- Ri, M.; Tashiro, E.; Oikawa, D.; Shinjo, S.; Tokuda, M.; Yokouchi, Y.; Narita, T.; Masaki, A.; Ito, A.; Ding, J.; et al. Identification of Toyocamycin, an agent cytotoxic for multiple myeloma cells, as a potent inhibitor of ER stress-induced XBP1 mRNA splicing. Blood Cancer J. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, F.; Sanna, M.; Grossi, G.; Brullo, C.; Fallacara, A.L.; Schenone, S. Pyrrolo[2,3-d]Pyrimidines as Kinase Inhibitors. Curr. Med. Chem. 2017, 24, 2059–2085. [Google Scholar] [CrossRef]
- Musumeci, F.; Sanna, M.; Greco, C.; Giacchello, I.; Fallacara, A.L.; Amato, R.; Schenone, S. Pyrrolo[2,3-d]pyrimidines active as Btk inhibitors. Expert Opin. Ther. Pat. 2017, 27, 1305–1318. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hogenkamp, H.P.C.; Long, R.A.; Revankar, G.R.; Robins, R.K. A convenient synthesis of 5’-deoxyribonucleosides. Carbohydr. Res. 1977, 59, 449–457. [Google Scholar] [CrossRef]
- Porcari, A.R.; Ptak, R.G.; Borysko, K.Z.; Breitenbach, J.M.; Vittori, S.; Wotring, L.L.; Drach, J.C.; Townsend, L.B. Deoxy Sugar Analogues of Triciribine: Correlation of Antiviral and Antiproliferative Activity with Intracellular Phosphorylation. J. Med. Chem. 2000, 43, 2438–2448. [Google Scholar] [CrossRef] [PubMed]
- Vorbrueggen, H.; Ruh-Pohlenz, C. Synthesis of nucleosides. Org. React. (Hobokennju. S.) 2000, 55. [Google Scholar] [CrossRef]
- Seela, F.; Peng, X. Progress in 7-deazapurine-pyrrolo[2,3-d]pyrimidine-ribonucleoside synthesis. Curr. Top. Med. Chem. 2006, 6, 867–892. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Bloch, A.; Bobek, M. A practical synthesis of the antibiotic toyocamycin. Nucleosides Nucleotides 1993, 12, 643–648. [Google Scholar] [CrossRef]
- Porcari, A.R.; Townsend, L.B. Total synthesis of the naturally occurring antibiotic toyocamycin using new and improved synthetic procedures. Nucleosides Nucleotides 1999, 18, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Z.; Gong, Z.; Xiao, Q.; Yang, R. An improved total synthesis of 5’-deoxytoyocamycin. Adv. Mater. Res. 2014, 997, 200–202. [Google Scholar] [CrossRef]
- Dou, Y.-H.; Ding, H.-X.; Yang, R.-C.; Li, W.; Xiao, Q. A total synthesis of mycalisine A. Chin. Chem. Lett. 2013, 24, 379–382. [Google Scholar] [CrossRef]
- Hu, C.; Ruan, Z.Z.; Ding, H.X.; Zhou, Y.R.; Xiao, Q. An Expedient Total Synthesis of Triciribine. Molecules 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.Y.; Dou, Y.H.; Ding, H.X.; Yang, R.C.; Sun, Q.; Xiao, Q. First Total Synthesis of a Naturally Occurring Iodinated 5 ‘-Deoxyxylofuranosyl Marine Nucleoside. Mar. Drugs 2012, 10, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Leonard, P.; Ingale, S.A.; Ding, P.; Ming, X.; Seela, F. Studies on the glycosylation of pyrrolo[2,3-d]pyrimidines with 1-O-acetyl-2,3,5-tri-O-benzoyl-β-D-ribofuranose: The formation of regioisomers during toyocamycin and 7-deazainosine syntheses. Nucleosidesnucleotides Nucleic Acids 2009, 28, 678–694. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-J.; Kwon, S.H.; Bae, I.H.; Kim, B.M. Selectivity between N-1 and N-7 nucleosides: Regioselective synthesis of BMK-Y101, a potent cdk7 and 9 inhibitor. Tetrahedron Lett. 2013, 54, 5484–5488. [Google Scholar] [CrossRef]
- Zhou, L.; Amblard, F.; Zhang, H.; McBrayer, T.R.; Detorio, M.A.; Whitaker, T.; Coats, S.J.; Schinazi, R.F. Synthesis and evaluation of Janus type nucleosides as potential HCV NS5B polymerase inhibitors. Bioorganic Med. Chem. Lett. 2013, 23, 3385–3388. [Google Scholar] [CrossRef] [Green Version]
- Tolman, R.L.; Robins, R.K.; Townsend, L.B. Pyrrolopyrimidine nucleosides. III. Total synthesis of toyocamycin, sangivamycin, tubercidin, and related derivatives. J. Am. Chem. Soc. 1969, 91, 2102–2108. [Google Scholar] [CrossRef]
- Tolman, R.L.; Robins, R.K.; Townsend, L.B. Pyrrolo[2,3-d]pyrimidine nucleoside antibiotics. Total synthesis and structure of toyocamycin, unamycin B, vengicide, antibiotic E-212, and sangivamycin (BA-90912). J. Am. Chem. Soc. 1968, 90, 524–526. [Google Scholar] [CrossRef]
- Xiao, C.; Sun, C.; Han, W.W.; Pan, F.; Dan, Z.; Li, Y.; Song, Z.G.; Jin, Y.H. Synthesis of 6-(het) ary Xylocydine analogues and evaluating their inhibitory activities of CDK1 and CDK2 in vitro. Bioorganic Med. Chem. 2011, 19, 7100–7110. [Google Scholar] [CrossRef] [PubMed]
- Porcari, A.R.; Townsend, L.B. An improved synthesis of the versatile heterocycle, 4-amino-6-bromo-5-cyanopyrrolo[2,3-d]pyrimidine. Synth. Commun. 1998, 28, 3835–3843. [Google Scholar] [CrossRef]
- Suh, H.; Choi, K.-W.; Lee, J.; Ryou, C.; Rhee, H.; Lee, C.-H. Effects of a novel carbocyclic analog of pyrrolo[2,3-d]pyrimidine nucleoside on pleiotropic induction of cell death in prostate cancer cells with different androgen responsiveness. Bioorganic Med. Chem. Lett. 2016, 26, 1130–1135. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Seo, H.; Yoon, S.; Choi, K.; Lee, C.-H.; Rhee, H. The synthesis and evaluation of new carbocyclic pyrrolo[2,3-d]pyrimidine nucleoside analogs. Heterocycles 2014, 89, 1606–1619. [Google Scholar] [CrossRef]
- Wilding, B.; Winkler, M.; Petschacher, B.; Kratzer, R.; Glieder, A.; Klempier, N. Nitrile Reductase from Geobacillus kaustophilus: A Potential Catalyst for a New Nitrile Biotransformation Reaction. Adv. Synth. Catal. 2012, 354, 2191–2198. [Google Scholar] [CrossRef]
- Zemlicka, J.; Owens, J. Nucleic Acid Chemistry, Improved and New Synthetic Procedures, Methods and Techniques, Part 4; Wiley-Interscience: New York, NY, USA, 1991; pp. 16–18. [Google Scholar]
- Crystallographic data for nucleobase 12: C14H8BrN5O, M = 324.16, crystal dimensions 0.08 × 0.12 × 0.20 mm, colorless block, space group, C2/c (No. 15). CCDC 1525337 contains the supplementary crystallographic data. Crystallographic data for nucleoside 14: C23H20BrN5O6, M = 542.35, crystal dimensions 0.07 × 0.20 × 0.22 mm, monoclinic, space group, P21 (No. 4). CCDC 1895393 contains the supplementary crystallographic data. These data can be obtained free of charge from The Cambridge Crystallographic Date Centre via www.ccdc.cam.ac.uk/date_request/cif.
- Prusiner, P.; Sundaralingam, M. The crystal and molecular structure of toyocamycin monohydrate, a nucleoside antibiotic. Acta Crystallogr. Sect. B 1978, B34, 517–523. [Google Scholar] [CrossRef]
- Isaac, B.G.; Ayer, S.W.; Letendre, L.J.; Stonard, R.J. Herbicidal nucleosides from microbial sources. J. Antibiot. 1991, 44, 729–732. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 3–16 are available from Prof. Dr. Qiang Xiao. |
Figure 1.
Examples of naturally occurring 7-deazapurine nucleosides.

Figure 2.
Synthesis of 7-deazapurine nucleosides by Vorbrüggen glycosylation.

Scheme 1.
Synthesis of N4-benzoyl-5-cyano-6-bromo-7H-pyrrolo[2,3-d]pyrimidine.

Scheme 2.
Total synthesis of 5′-deoxytoyocamycin and 5′-deoxysangivamycin.

Figure 3.
X-Ray structure of nucleobase 12 (a) and nucleoside 14 (b).

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Dong, X.; Tang, J.; Hu, C.; Bai, J.; Ding, H.; Xiao, Q. An Expeditious Total Synthesis of 5′-Deoxy-toyocamycin and 5′-Deoxysangivamycin. Molecules 2019, 24, 737. https://doi.org/10.3390/molecules24040737
AMA Style
Dong X, Tang J, Hu C, Bai J, Ding H, Xiao Q. An Expeditious Total Synthesis of 5′-Deoxy-toyocamycin and 5′-Deoxysangivamycin. Molecules. 2019; 24(4):737. https://doi.org/10.3390/molecules24040737
Chicago/Turabian StyleDong, Xiangyou, Jie Tang, Chen Hu, Jiang Bai, Haixin Ding, and Qiang Xiao. 2019. "An Expeditious Total Synthesis of 5′-Deoxy-toyocamycin and 5′-Deoxysangivamycin" Molecules 24, no. 4: 737. https://doi.org/10.3390/molecules24040737