
Synthesis and Antiproliferative Activities of Conjugates of Paclitaxel and Camptothecin with a Cyclic Cell-Penetrating Peptide

 , and
, and 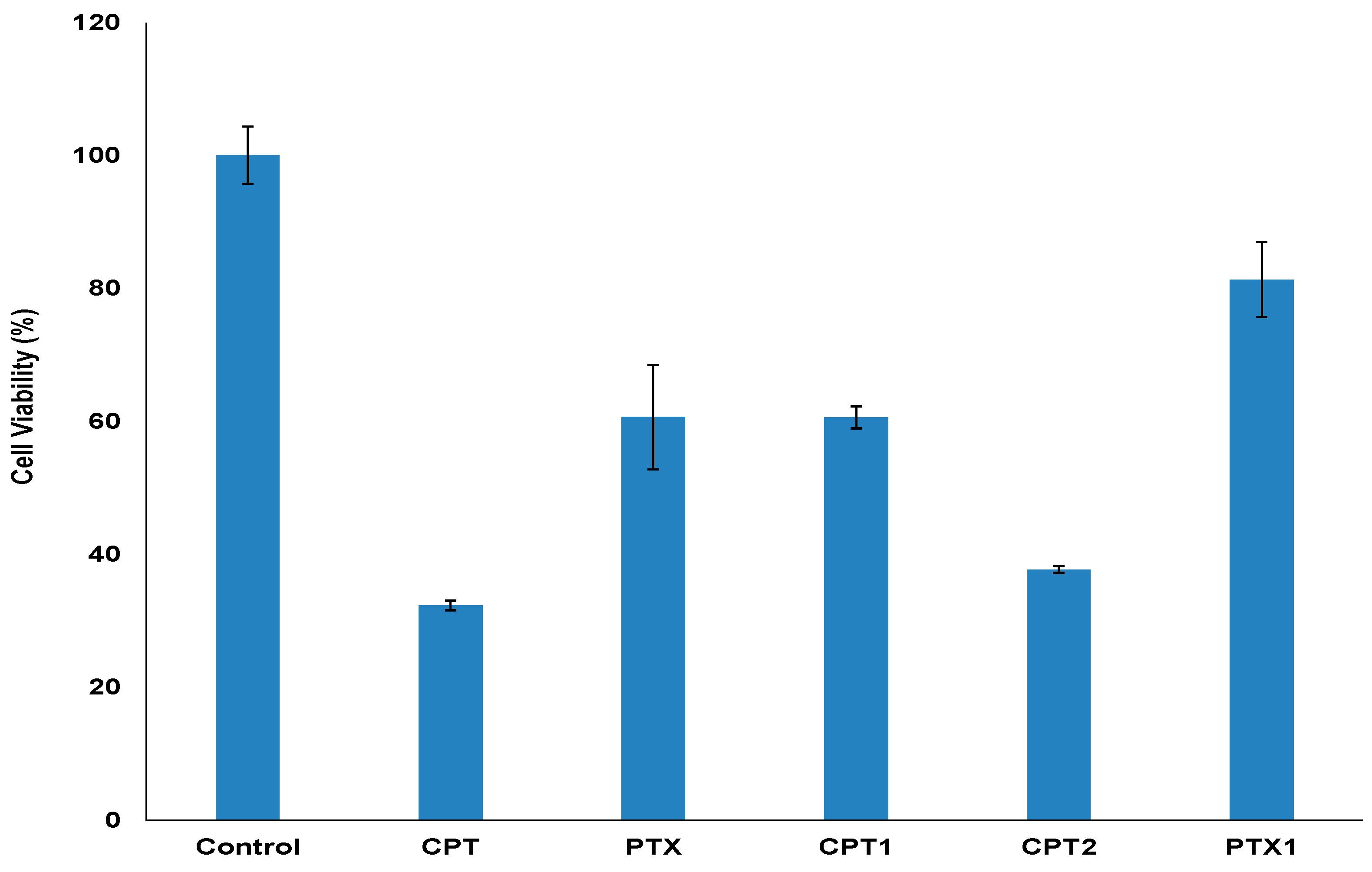{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
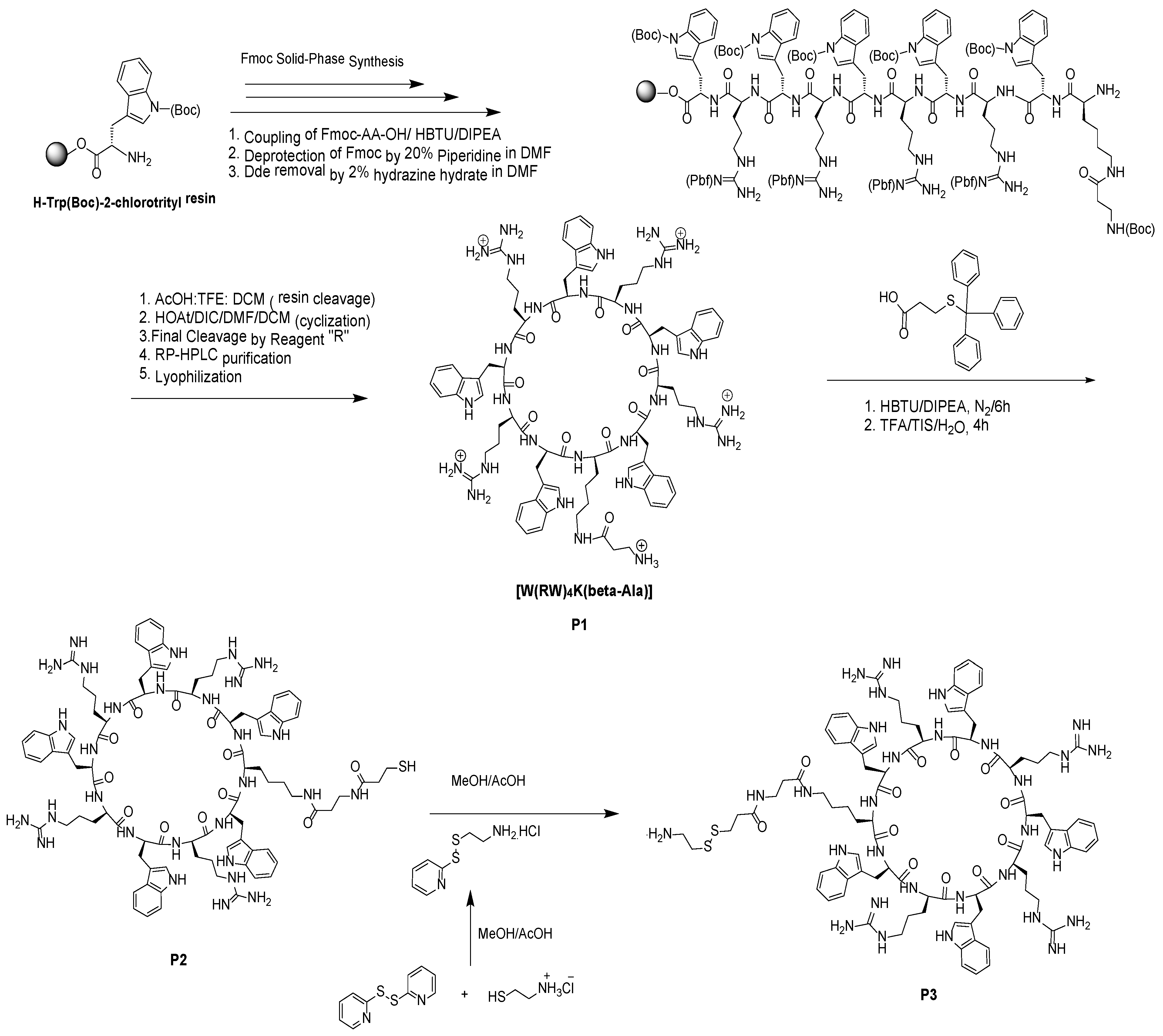
2.1. Synthesis of Cyclic Peptide [(WR)4K(βAla)] and Its Functionalization
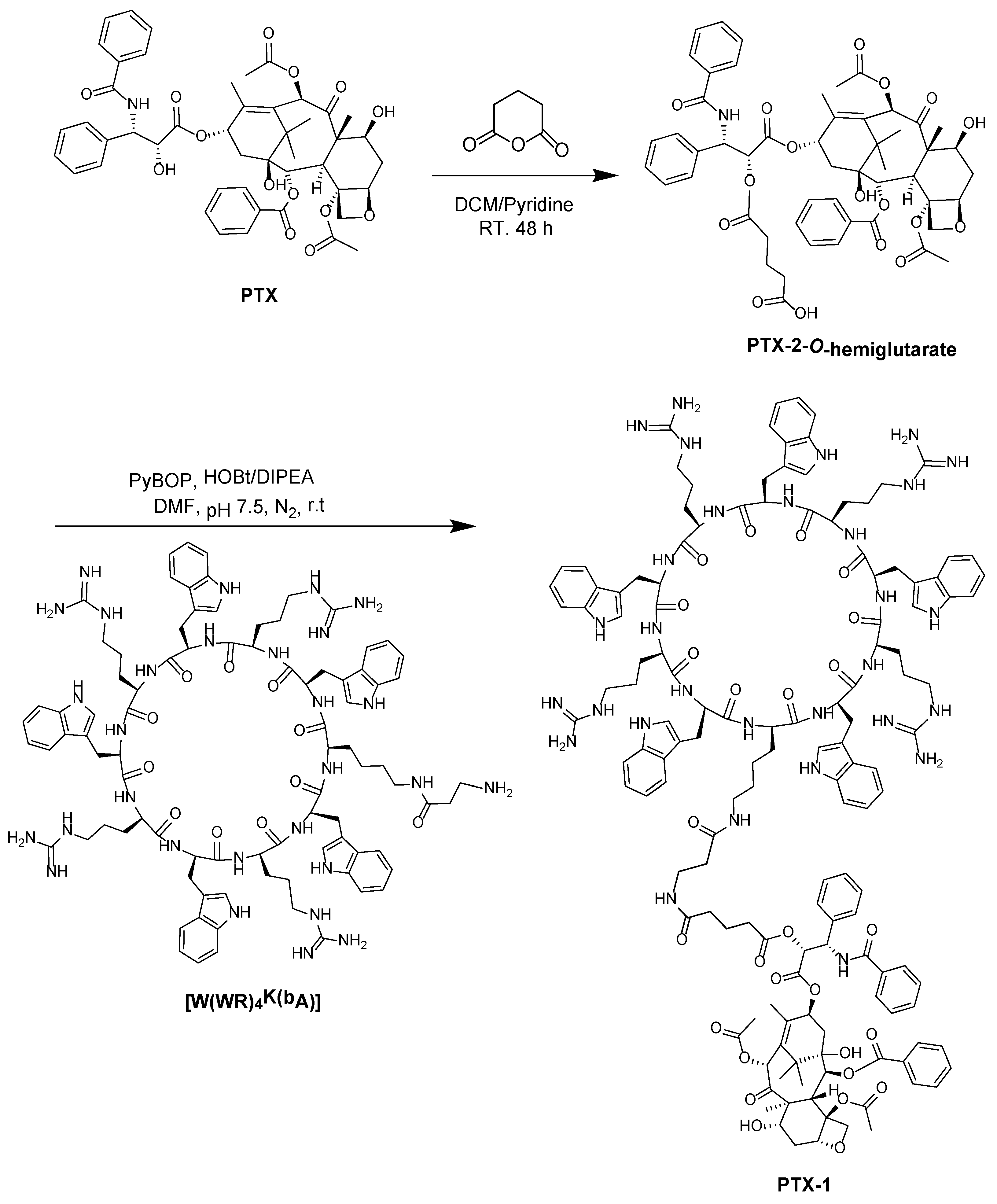
2.2. Coupling Hydrophobic Drugs to [W(WR)4K(βAla)]
Coupling of PTX to [W(WR)4K(βAla)]
2.3. Biological Activity
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Synthesis of Cyclic Peptide [W(RW)4K(βAla)] (P1)
3.2.2. Synthesis of [(WR)4WK(βAla-Thiopropionic acid)] (P2) and [(WR)4WK(βAla-Thiopropionyl-ethaneamine] (P3)
3.2.3. Synthesis of Paclitaxel-2-O-Hemiglutarate
3.2.4. General Procedure for the Synthesis of PTX-1 from Coupling of [W(WR)4K(βAla)] Peptide with PTX-2-O-Hemiglutarate
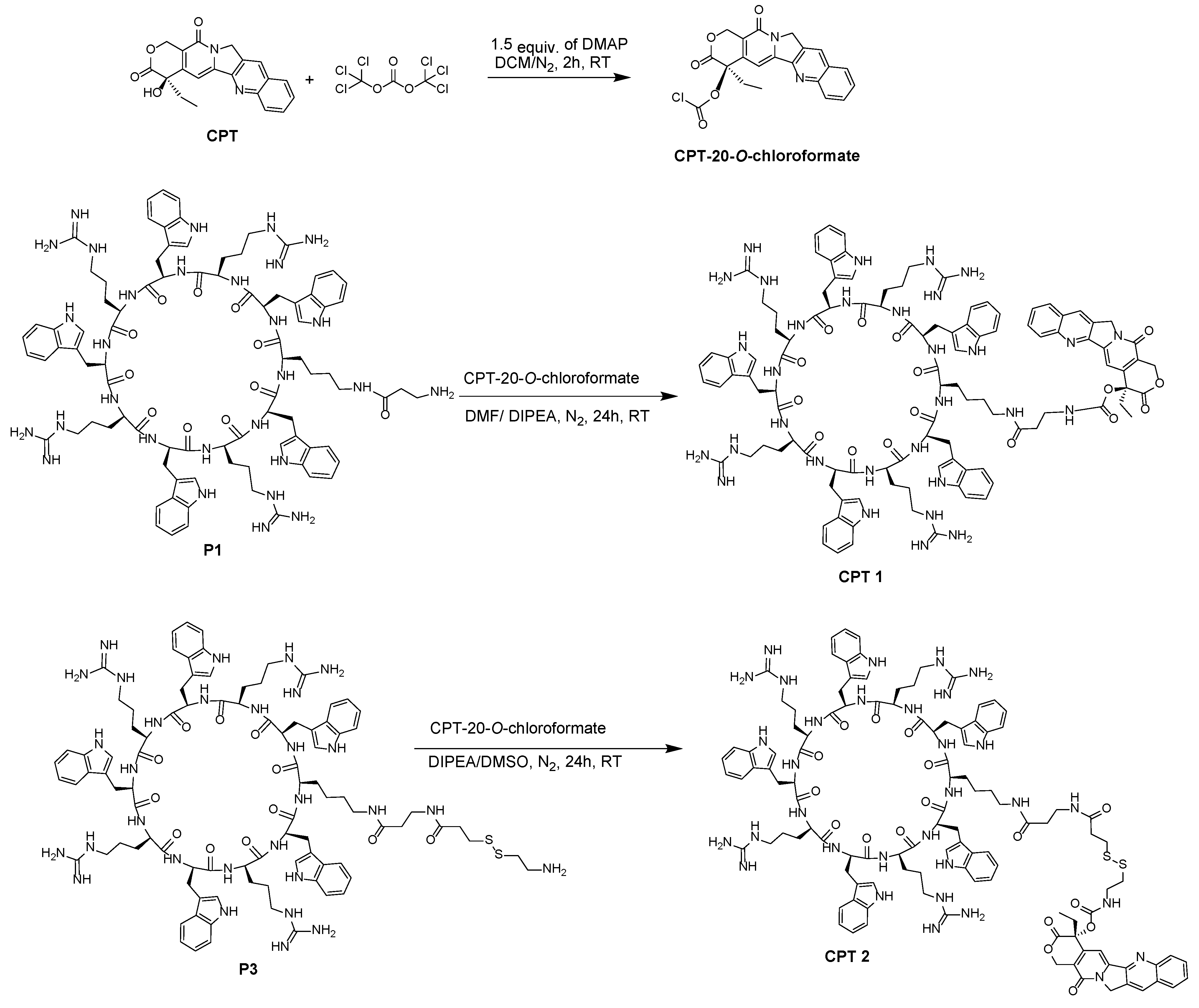
3.2.5. Synthesis of Camptothecin-20-O-Chloroformate
3.2.6. Synthesis of CPT1 from Coupling of [W(WR)4K(βAla)] Peptide with CPT-20-O-Chloroformate
3.2.7. Synthesis of CPT2 from Coupling of [(WR)4WK(βAla-thiopropionyl-S-S-ethaneamine)] with CPT-20-O-chloroformate
3.2.8. Cell Culture
3.2.9. Antiproliferative Assay
4. Conclusion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Bae, Y.H. Drug targeting and tumor heterogeneity. J. Control. Release 2009, 133, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Gullotti, E.; Yeo, Y. Extracellularly activated nanocarriers: A new paradigm of tumor targeted drug delivery. Mol. Pharm. 2009, 6, 1041–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, F.; Hashida, M. Pharmacokinetic considerations for targeted drug delivery. Adv. Drug Deliv. Rev. 2013, 65, 139–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danhier, F.; Feron, O.; Préat, V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J. Control. Release 2010, 148, 135–146. [Google Scholar] [CrossRef]
- Muro, S. Challenges in design and characterization of ligand-targeted drug delivery systems. J. Control. Release 2012, 164, 125–137. [Google Scholar] [CrossRef] [Green Version]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 2018, 9, 1410. [Google Scholar] [CrossRef]
- Löwik, D.W.; Leunissen, E.H.P.; van den Heuvel, M.; Hansen, M.B.; van Hest, J.C. Stimulus responsive peptide-based materials. Chem. Soc. Rev. 2010, 39, 3394–3412. [Google Scholar] [CrossRef] [PubMed]
- Myrberg, H.; Zhang, L.; Mäe, M.; Langel, Ü. Design of a tumor-homing cell-penetrating peptide. Bioconjug. Chem 2007, 19, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Luque-Ortega, J.R.; Hof, W.V.T.; Veerman, E.C.; Saugar, J.M.; Rivas, L. Human antimicrobial peptide histatin 5 is a cell-penetrating peptide targeting mitochondrial ATP synthesis in Leishmania. FASEB J. 2008, 22, 1817–1828. [Google Scholar] [CrossRef] [Green Version]
- Derakhshankhah, H.; Jafari, S. Cell penetrating peptides: A concise review with emphasis on biomedical applications. Biomed. Pharmacother. 2018, 108, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Splith, K.; Neundorf, I. Antimicrobial peptides with cell-penetrating peptide properties and vice versa. Eur. Biophys. J. 2011, 40, 387–397. [Google Scholar] [CrossRef]
- Vasconcelos, L.; Pärn, K.; Langel, Ü. Therapeutic potential of cell-penetrating peptides. Therap. Deliv. 2013, 4, 573–591. [Google Scholar] [CrossRef]
- Reissmann, S. Cell penetration: Scope and limitations by the application of cell-penetrating peptides. J. Pept. Sci. 2014, 20, 760–784. [Google Scholar] [CrossRef]
- Dong, H.; Hartgerink, J.D. Role of hydrophobic clusters in the stability of α-helical coiled coils and their conversion to amyloid-like β-sheets. Biomacromolecules 2007, 8, 617–623. [Google Scholar] [CrossRef]
- Mart, R.J.; Osborne, R.D.; Stevens, M.M.; Ulijn, R.V. Peptide-based stimuli-responsive biomaterials. Soft Matter 2006, 2, 822–835. [Google Scholar] [CrossRef]
- El-Sayed, N.S.; Miyake, T.; Shirazi, A.; Park, S.; Clark, J.; Buchholz, S.; Parang, K.; Tiwari, R. Design, Synthesis, and Evaluation of Homochiral Peptides Containing Arginine and Histidine as Molecular Transporters. Molecules 2018, 23, 1590. [Google Scholar] [CrossRef]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-penetrating peptides: From basic research to clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef]
- Aroui, S.; Brahim, S.; Hamelin, J.; De Waard, M.; Bréard, J.; Kenani, A. Conjugation of doxorubicin to cell penetrating peptides sensitizes human breast MDA-MB 231 cancer cells to endogenous TRAIL-induced apoptosis. Apoptosis 2009, 14, 1352–1365. [Google Scholar] [CrossRef]
- Zhang, W.; Song, J.; Zhang, B.; Liu, L.; Wang, K.; Wang, R. Design of acid-activated cell penetrating peptide for delivery of active molecules into cancer cells. Bioconj. Chem. 2011, 22, 1410–1415. [Google Scholar] [CrossRef]
- Song, J.; Zhang, Y.; Zhang, W.; Chen, J.; Yang, X.; Ma, P.; Zhang, B.; Liu, B.; Ni, J.; Wang, R. Cell penetrating peptide TAT can kill cancer cells via membrane disruption after attachment of camptothecin. Peptides 2015, 63, 143–149. [Google Scholar] [CrossRef]
- Shirazi, A.N.; Salem El-Sayed, N.; Tiwari, R.K.; Tavakoli, K.; Parang, K. Cyclic peptide containing hydrophobic and positively charged residues as a drug delivery system for curcumin. Curr. Drug Deliv. 2016, 13, 409–417. [Google Scholar] [CrossRef]
- Sharma, M.; El-Sayed, N.S.; Do, H.; Parang, K.; Tiwari, R.K.; Aliabadi, H.M. Tumor-targeted delivery of siRNA using fatty acyl-CGKRK peptide conjugates. Sci. Rep. 2017, 7, 6093. [Google Scholar] [CrossRef] [Green Version]
- Jafarzade, B.S.; Bolhassani, A.; Sadat, S.M.; Yaghobi, R. Delivery of HIV-1 Nef protein in mammalian cells using cell penetrating peptides as a candidate therapeutic vaccine. Int. J. Pept. Res. Ther. 2017, 23, 145–153. [Google Scholar] [CrossRef]
- Mandal, D.; Shirazi, A.N.; Parang, K. Cell-penetrating homochiral cyclic peptides as nuclear-targeting molecular transporters. Angew. Chem. Int. Ed. 2011, 50, 9633–9637. [Google Scholar] [CrossRef]
- Shirazi, A.N.; Tiwari, R.K.; Oh, D.; Banerjee, A.; Yadav, A.; Parang, K. Efficient delivery of cell impermeable phosphopeptides by a cyclic peptide amphiphile containing tryptophan and arginine. Mol. Pharm. 2013, 10, 2008–2020. [Google Scholar] [CrossRef]
- Shirazi, A.N.; Tiwari, R.; Chhikara, B.S.; Mandal, D.; Parang, K. Design and biological evaluation of cell-penetrating peptide–doxorubicin conjugates as prodrugs. Mol. Pharm. 2013, 10, 488–499. [Google Scholar] [CrossRef]
- Weaver, B.A. How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Porter, M.; Konstantopoulos, A.; Zhang, P.; Cui, H. Preclinical development of drug delivery systems for paclitaxel-based cancer chemotherapy. J. Control Release 2017, 267, 100–118. [Google Scholar] [CrossRef]
- Wall, M.E.; Wani, M.C.; Cook, C.E.; Palmer, K.H.; McPhail, A.A.; Sim, G.A. Plant antitumor agents. I. The isolation and structure of camptothecin, a novel alkaloidal leukemia and tumor inhibitor from camptotheca acuminate 1,2. J. Am. Chem. Soc. 1966, 88, 3888–3890. [Google Scholar]
- Sparreboom, A.; Van Asperen, J.; Mayer, U.; Schinkel, A.H.; Smit, J.W.; Meijer, D.K.; Borst, P.; Nooijen, W.J.; Beijnen, J.H.; Tellingen, O.V. Limited oral bioavailability and active epithelial excretion of paclitaxel (Taxol) caused by P-glycoprotein in the intestine. Proc. Natl. Acad. Sci. USA 1997, 94, 2031–2035. [Google Scholar] [CrossRef] [Green Version]
- Svenson, S.; Wolfgang, M.; Hwang, J.; Ryan, J.; Eliasof, S. Preclinical to clinical development of the novel camptothecin nanopharmaceutical CRLX101. J. Control. Release 2011, 153, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Cai, H.; Jiang, L.; Hu, J.; Bains, A.; Hu, J.; Gong, Q.; Luo, K.; Gu, Z. Enzyme-sensitive and amphiphilic PEGylated dendrimer-paclitaxel prodrug-based nanoparticles for enhanced stability and anticancer efficacy. ACS Appl. Mater. Interfaces 2017, 9, 6865–6877. [Google Scholar] [CrossRef]
- Fox, M.E.; Guillaudeu, S.; Fréchet, J.M.; Jerger, K.; Macaraeg, N.; Szoka, F.C. Synthesis and in vivo antitumor efficacy of PEGylated poly (l-lysine) dendrimer− camptothecin conjugates. Mol. Pharm. 2009, 6, 1562–1572. [Google Scholar] [CrossRef]
- Martins, S.; Tho, I.; Reimold, I.; Fricker, G.; Souto, E.; Ferreira, D.; Brandl, M. Brain delivery of camptothecin by means of solid lipid nanoparticles: Formulation design, in vitro and in vivo studies. Int. J. Pharm. 2012, 439, 49–62. [Google Scholar] [CrossRef]
- He, Z.; Wan, X.; Schulz, A.; Bludau, H.; Dobrovolskaia, M.A.; Stern, S.T.; Montgomery, S.A.; Yuan, H.; Li, Z.; Alakhova, D.; Sokolsky, M. A high capacity polymeric micelle of paclitaxel: Implication of high dose drug therapy to safety and in vivo anti-cancer activity. Biomaterials 2016, 101, 296–309. [Google Scholar] [CrossRef]
- Du, X.; Khan, A.R.; Fu, M.; Yu, A.; Zhai, G. Current development in the formulations of non-injection administration of paclitaxel. Int. J. Pharm. 2018, 542, 242–252. [Google Scholar] [CrossRef]
- Zugates, G.T.; Anderson, D.G.; Little, S.R.; Lawhorn, I.E.; Langer, R. Synthesis of poly (β-amino ester)s with thiol-reactive side chains for DNA delivery. J. Am. Chem. Soc. 2006, 128, 12726–12734. [Google Scholar] [CrossRef]
- Sundaram, S.; Durairaj, C.; Kadam, R.; Kompella, U.B. Luteinizing hormone-releasing hormone receptor–targeted deslorelin-docetaxel conjugate enhances efficacy of docetaxel in prostate cancer therapy. Mol. Cancer Ther. 2009, 8, 1655–1665. [Google Scholar] [CrossRef]
- Henne, W.A.; Doorneweerd, D.D.; Hilgenbrink, A.R.; Kularatne, S.A.; Low, P.S. Synthesis and activity of a folate peptide camptothecin prodrug. Bioorg. Med. Chem. Lett. 2006, 16, 5350–5355. [Google Scholar] [CrossRef]
- Zhang, Q.; He, J.; Zhang, M.; Ni, P. A polyphosphoester-conjugated camptothecin prodrug with disulfide linkage for potent reduction-triggered drug delivery. J. Mat. Chem. B 2015, 3, 4922–4932. [Google Scholar] [CrossRef]
- Darwish, S.; Sadeghiani, N.; Fong, S.; Mozaffari, S.; Hamidi, P.; Withana, T.; Yang, S.; Tiwari, R.K.; Parang, K. Synthesis and antiproliferative activities of doxorubicin thiol conjugates and doxorubicin-SS-cyclic peptide. Eur. J. Med. Chem. 2019, 161, 594–606. [Google Scholar] [CrossRef]
- Risinger, A.L.; Dybdal-Hargreaves, N.F.; Mooberry, S.L. Breast Cancer Cell Lines Exhibit Differential Sensitivities to Microtubule-targeting Drugs Independent of Doubling Time. Anticancer Res. 2015, 35, 5845–5850. [Google Scholar]
- Thomas, C.J.; Rahier, N.J.; Hecht, S.M. Camptothecin: Current perspectives. Bioorg. Med. Chem. 2004, 12, 1585–1604. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available for short time from the authors. |





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Sayed, N.S.; Shirazi, A.N.; Sajid, M.I.; Park, S.E.; Parang, K.; Tiwari, R.K. Synthesis and Antiproliferative Activities of Conjugates of Paclitaxel and Camptothecin with a Cyclic Cell-Penetrating Peptide. Molecules 2019, 24, 1427. https://doi.org/10.3390/molecules24071427
El-Sayed NS, Shirazi AN, Sajid MI, Park SE, Parang K, Tiwari RK. Synthesis and Antiproliferative Activities of Conjugates of Paclitaxel and Camptothecin with a Cyclic Cell-Penetrating Peptide. Molecules. 2019; 24(7):1427. https://doi.org/10.3390/molecules24071427
Chicago/Turabian StyleEl-Sayed, Naglaa Salem, Amir Nasrolahi Shirazi, Muhammad Imran Sajid, Shang Eun Park, Keykavous Parang, and Rakesh Kumar Tiwari. 2019. "Synthesis and Antiproliferative Activities of Conjugates of Paclitaxel and Camptothecin with a Cyclic Cell-Penetrating Peptide" Molecules 24, no. 7: 1427. https://doi.org/10.3390/molecules24071427