
Indole-Containing Phytoalexin-Based Bioisosteres as Antifungals: In Vitro and In Silico Evaluation against Fusarium oxysporum




Abstract
:
1. Introduction
2. Results and Discussion
2.1. Test Indole-Containing Phytoalexin Analogs and Docking Protocol Validation
2.2. Vina Scores-Related Trends
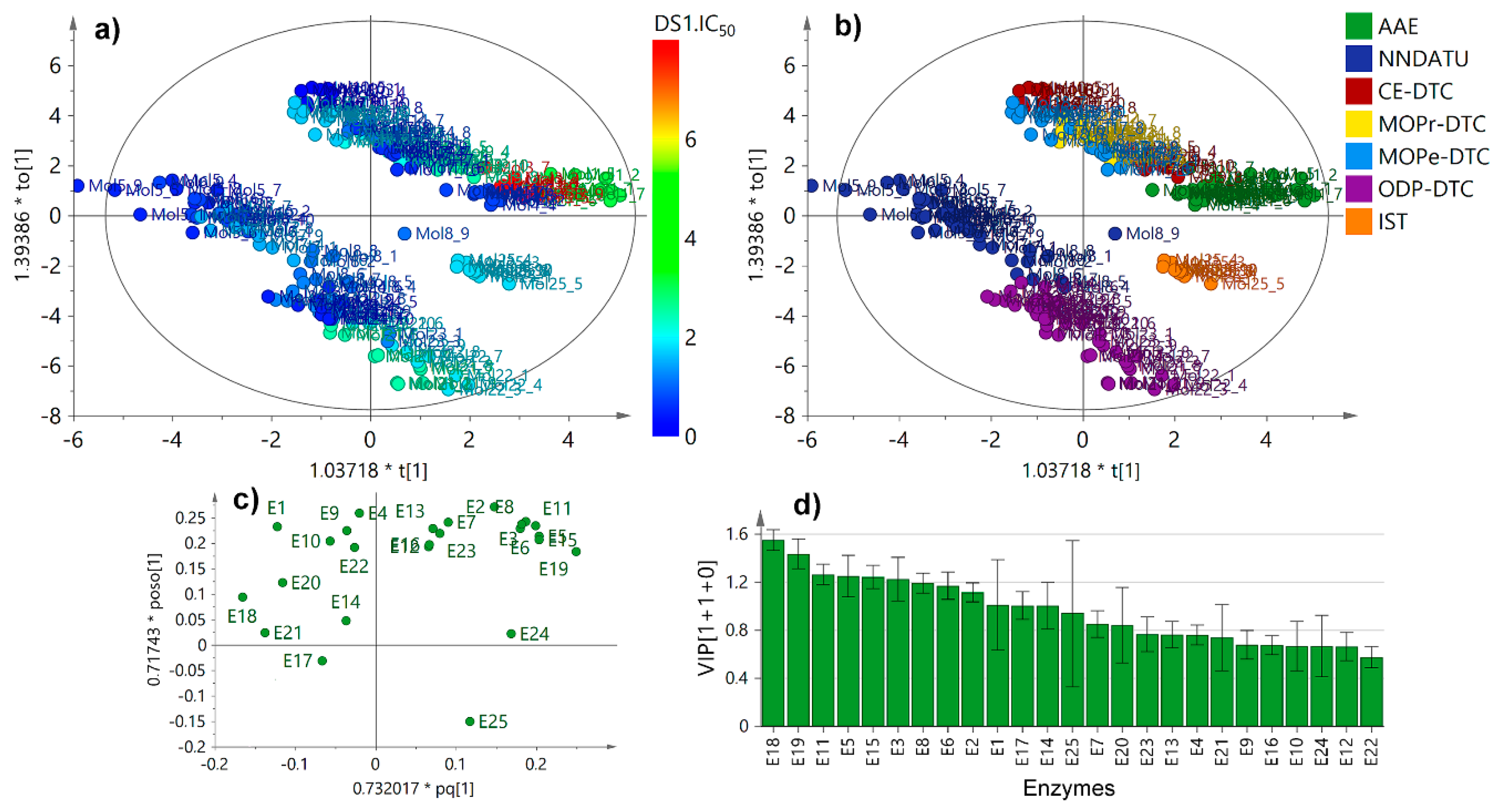
2.3. Unsupervised and Supervised Multivariate Statistics
2.4. Comparative Molecular Field Analysis (CoMFA)
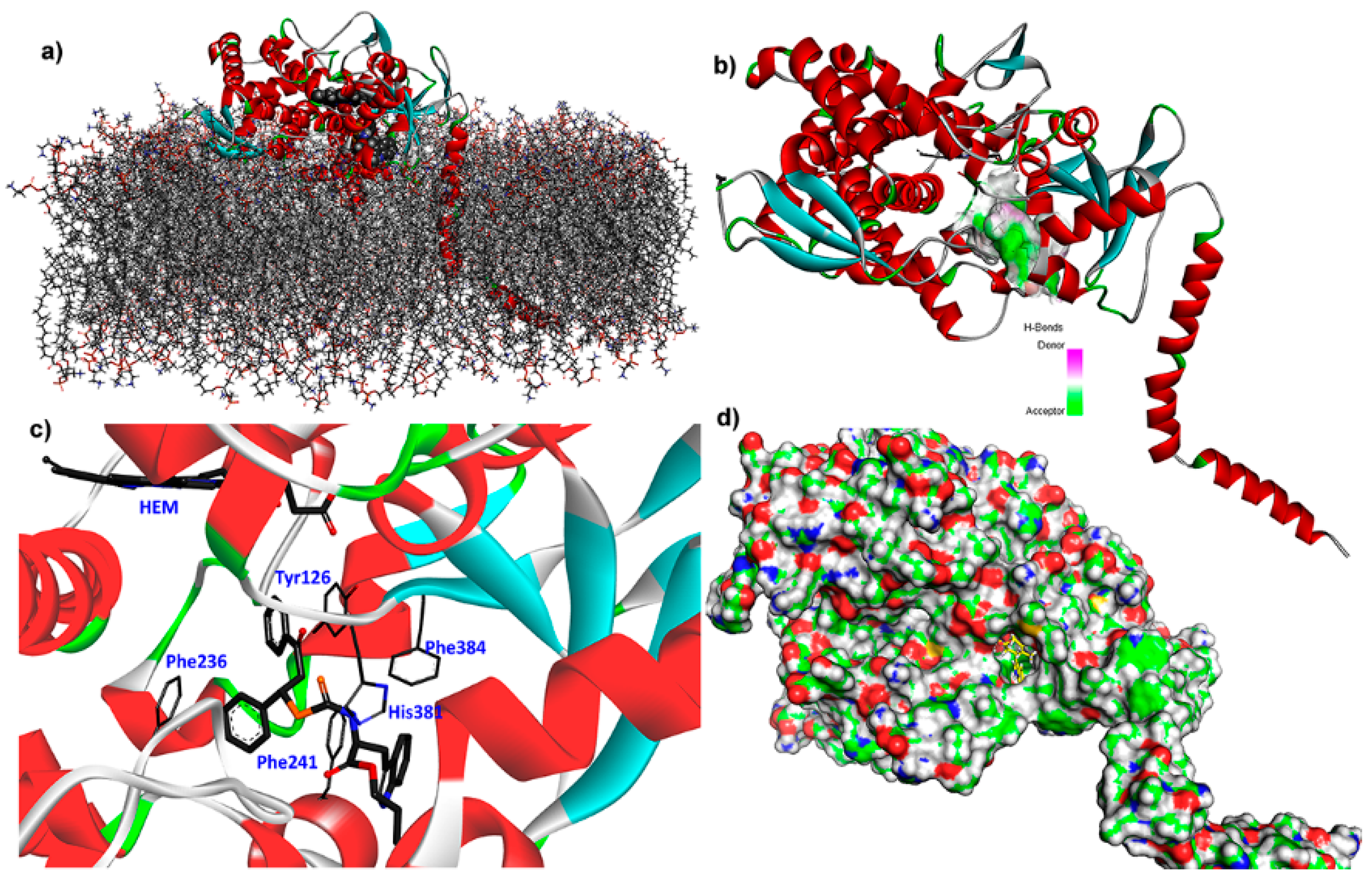
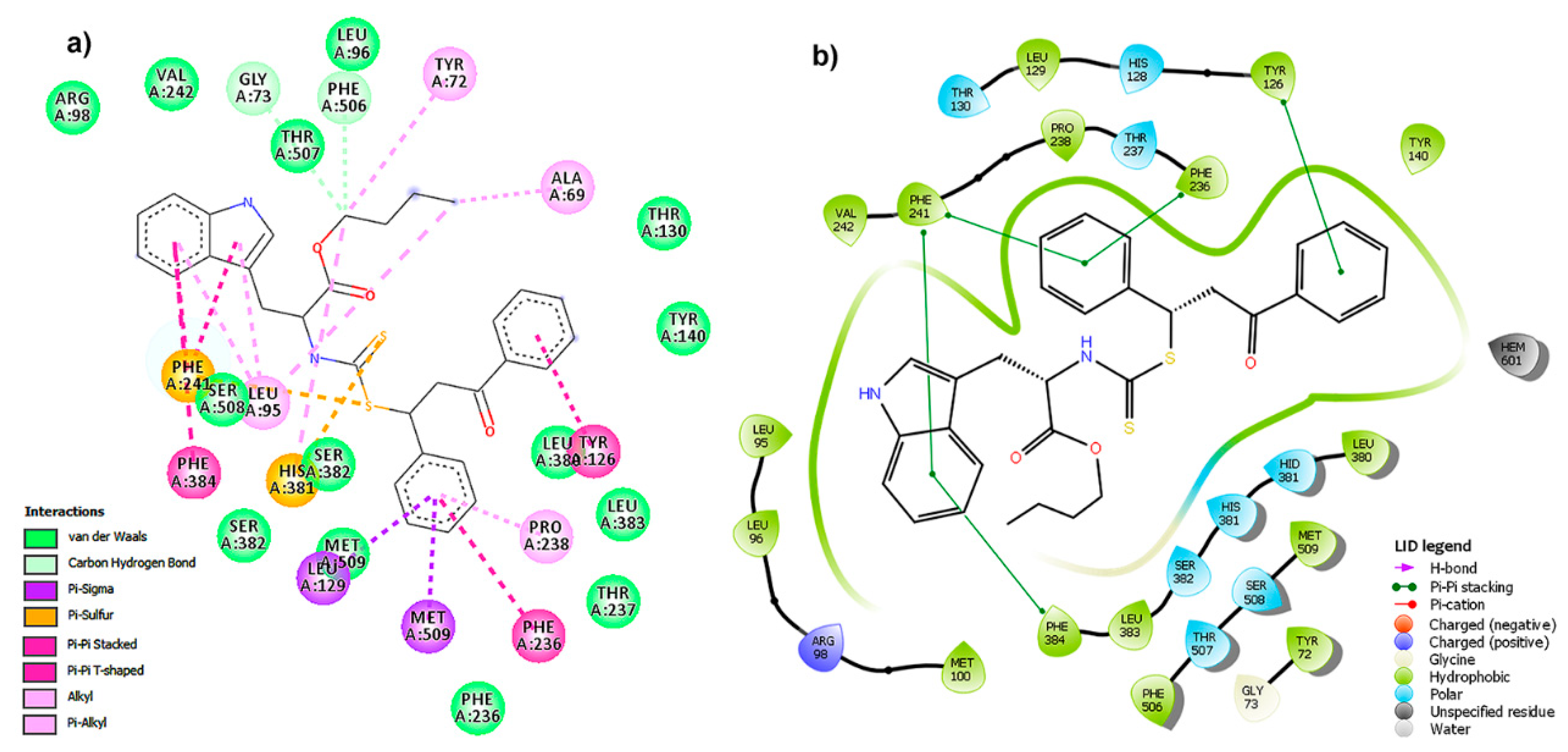
2.5. Binding Mode and Residual Interactions
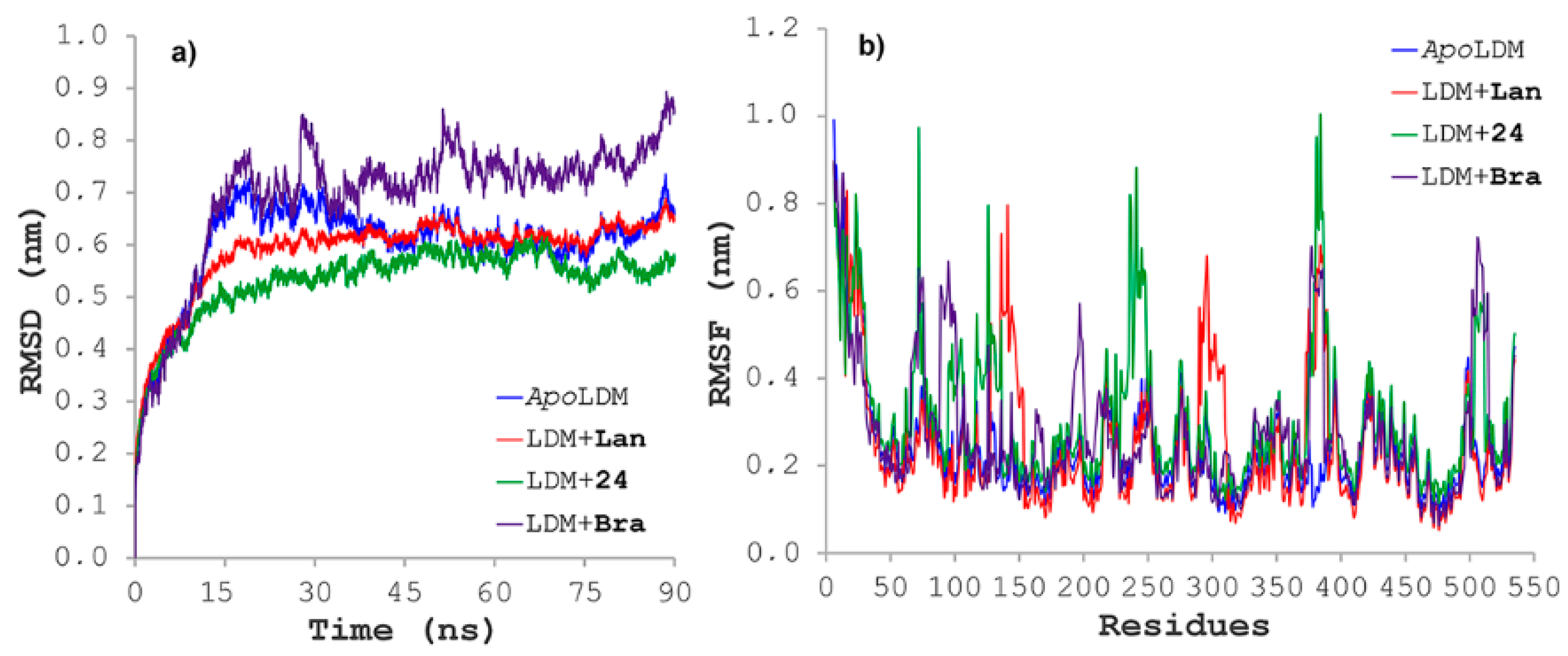
2.6. Molecular Dynamics Simulations
2.7. Binding Free Energy
3. Materials and Methods
3.1. Design and Synthesis of Indole-Containing Phytoalexin Analogs
3.2. Enzymes
3.3. Molecular Docking
3.4. Statistical Analysis
3.5. Molecular Dynamics Simulations of F. Cerealis Lanosterol 14α-demethylase (FcLDM)
3.6. Binding Free Energy Analyses
3.7. Antifungal Assay
3.8. Comparative Molecular Field Analysis (CoMFA)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Enzyme (Type) | PDB Code | Source |
|---|---|---|---|
| E1 | endoglucanase I (glycosyl hydrolase) [NAG cavity] a | 1OVW | F. oxysporum |
| E2 | endoglucanase I (glycosyl hydrolase) [native binding site cavity] a | 1OVW | F. oxysporum |
| E3 | endoglucanase I (glycosyl hydrolase) | 4OVW | F. oxysporum |
| E4 | endoglucanase I (cellobiohydrolase) | 3OVW | F. oxysporum |
| E5 | acetyltransferase (transferase) | 2ZBA | F. sporotrichioides |
| E6 | thrichodiene synthase (synthase) | 2PS8 | F. sporotrichioides |
| E7 | endoglucanase I (cellobiohydrolase) | 2OVW | F. oxysporum |
| E8 | trichodiene synthase (synthase) | 1YYQ | F. sporotrichioides |
| E9 | serine esterase (cutinase) | 1XZM | F. solani subsp. pisi |
| E10 | serine esterase (hydrolase-cutinase) | 1XZL | F. solani subsp. pisi |
| E11 | serine esterase (cutinase) | 1OXM | F. solani subsp. pisi |
| E12 | isocitrate lyase (lyase) | 5E9G | F. sporotrichioides |
| E13 | trichothecene 15-O-acetyltransferase (transferase) | 3FP0 | F. sporotrichioides |
| E14 | isocitrate lyase (lyase) | 5E9H | F. graminearum |
| E15 | trichodiene synthase (synthase) | 1JFG | F. oxysporum |
| E16 | trichothecene 3-O-acetyltransferase (transferase) | 3B30 | F. graminearum |
| E17 | nitroalkane oxidase (NAO) (oxidoreductase) | 2REH | F. oxysporum |
| E18 | nitroalkane oxidase (NAO) (oxidoreductase) | 2C0U | F. oxysporum |
| E19 | flavoenzyme nitroalkane oxidase (oxidoreductase) | 3D9E | F. oxysporum |
| E20 | nitric oxide reductase cytochrome (oxidoreductase) | 1ULW | F. oxysporum |
| E21 | NAD(P)-dependent dehydrogenase (dehydrogenase) | 1U3T b | F. vascular (A0A0D2YG03 c) |
| E22 | nitroreductase [NADPH] (oxidoreductase) | 2BII b | F. vascular wilt (P39863 c) |
| E23 | aspartate kinase (kinase) | 2CDQ b | F. verticilloides (W7MS01 c) |
| E24 | FMN-dependent dehydrogenase (oxidoreductase) | 1GOX b | F. verticilloides (W7NCP1 c) |
| E25 | 14-lanosterol demethylase (CYP51) (oxidoreductase) | 4LXJ b | F. cerealis (I6ZLS0 c) |
References
- Munkvold, G.P. Fusarium species and their associated mycotoxins. In Methods in Molecular Biology; Springer: New York, NY, USA, 2016; pp. 51–106. ISBN 1064-3745. [Google Scholar]
- Ma, L.-J.; Geiser, D.M.; Proctor, R.H.; Rooney, A.P.; O’Donnell, K.; Trail, F.; Gardiner, D.M.; Manners, J.M.; Kazan, K. Fusarium pathogenomics. Annu. Rev. Microbiol. 2013, 67, 399–416. [Google Scholar] [CrossRef] [Green Version]
- Bashir, M.R.; Atiq, M.; Sajid, M.; Mohsan, M.; Abbas, W.; Alam, M.W.; Bashair, M. Antifungal exploitation of fungicides against Fusarium oxysporum f. sp. capsici causing Fusarium wilt of chilli pepper in Pakistan. Environ. Sci. Pollut. Res. 2017, 25, 6797–6801. [Google Scholar] [CrossRef]
- Khomutov, R.M.; Khurs, E.N.; Osipova, T.I.; Zhemchuzhina, N.S.; Mikituk, O.D.; Nazarova, T.A.; Shcherbakova, L.A.; Dzhavakhiya, V.G. Chemical control of adaptive function of plant pathogenic fungi. Dokl. Biochem. Biophys. 2015, 461, 69–71. [Google Scholar] [CrossRef]
- Ahuja, I.; Kissen, R.; Bones, A.M. Phytoalexins in defense against pathogens. Trends Plant Sci. 2012, 17, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Pedras, M.S.C.; Minic, Z.; Jha, M. Brassinin oxidase, a fungal detoxifying enzyme to overcome a plant defense - purification, characterization and inhibition. FEBS J. 2008, 275, 3691–3705. [Google Scholar] [CrossRef] [PubMed]
- Lima, L.; Barreiro, E. Bioisosterism: A useful strategy for molecular modification and drug design. Curr. Med. Chem. 2005, 12, 23–49. [Google Scholar] [CrossRef] [PubMed]
- Quiroga, D.; Becerra, L.; Sadat-Bernal, J.; Vargas, N.; Coy-Barrera, E. Synthesis and antifungal activity against Fusarium oxysporum of some Brassinin analogs derived from L-tryptophan: A DFT/B3LYP study on the reaction mechanism. Molecules 2016, 21, 1349. [Google Scholar] [CrossRef] [PubMed]
- Quiroga, D.; Becerra, L.D.; Coy-Barrera, E. Ultrasound-assisted synthesis, antifungal activity against Fusarium oxysporum, and three-dimensional quantitative structure–activity relationship of N,S-dialkyl dithiocarbamates derived from 2-amino acids. ACS Omega 2019, 4, 13710–13720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W., Jr. Computational methods in drug discovery. Pharmacol. Rev. 2013, 66, 334–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Lim-Wilby, M. Molecular Docking BT—Molecular Modeling of Proteins. In Molecular Modeling of Proteins; Kukol, A., Ed.; Humana Press: Totowa, NJ, USA, 2008; pp. 365–382. ISBN 978-1-59745-177-2. [Google Scholar]
- Chen, Y.-C. Beware of docking! Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef]
- Westermaier, Y.; Barril, X.; Scapozza, L. Virtual screening: An in silico tool for interlacing the chemical universe with the proteome. Methods 2015, 71, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Bandos, A.I.; Gur, D. On the use of partial area under the ROC curve for comparison of two diagnostic tests. Biom. J. 2015, 57, 304–320. [Google Scholar] [CrossRef] [PubMed]
- Zhan, D.; Wang, D.; Min, W.; Han, W. Exploring the molecular basis for selective binding of homoserine dehydrogenase from Mycobacterium leprae TN toward inhibitors: A virtual screening study. Int. J. Mol. Sci. 2014, 15, 1826–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garvey, G.S.; Rayment, I.; McCormick, S.P.; Alexander, N.J. Structural and functional characterization of TRI3 trichothecene 15-O-acetyltransferase from Fusarium sporotrichioides. Protein Sci. 2009, 18, 747–761. [Google Scholar]
- González, G.; Yutronic, N.; Jara, M. Conformational equilibria in N,N-dialkyltthioureas. Spectrochim. Acta Part A Mol. Spectrosc. 1990, 46, 1729–1736. [Google Scholar] [CrossRef]
- Veignie, E.; Ceballos, C.; Len, C.; Rafin, C. Design of new antifungal dithiocarbamic esters having bio-based acrylate moiety. ACS Omega 2019, 4, 4779–4784. [Google Scholar] [CrossRef] [Green Version]
- Russell, P.E. A century of fungicide evolution. J. Agric. Sci. 2005, 143, 11–25. [Google Scholar] [CrossRef]
- Foster, A.J.; Thines, E. Identification of fungicide targets in pathogenic fungi. In Physiology and Genetics; Springer: Berlin/Heidelberg, Germany, 2009; pp. 233–245. ISBN 9783642002854. [Google Scholar]
- Roy, K.; Kar, S.; Das, R.N. Introduction to 3D-QSAR. In Understanding the Basics of QSAR for Applications in Pharmaceutical Sciences and Risk Assessment; Elsevier: London, UK, 2015; pp. 291–317. ISBN 9780128015056. [Google Scholar]
- Doweyko, A.M. Three-dimensional quantitative structure–activity relationship: The state of the art. In Comprehensive Medicinal Chemistry II; Elsevier: London, UK, 2007; pp. 575–595. ISBN 9780080450445. [Google Scholar]
- Clark, R.D.; Fox, P.C. Statistical variation in progressive scrambling. J. Comput. Aided Mol. Des. 2004, 18, 563–576. [Google Scholar] [CrossRef]
- Sheng, C.; Miao, Z.; Ji, H.; Yao, J.; Wang, W.; Che, X.; Dong, G.; Lu, J.; Guo, W.; Zhang, W. Three-dimensional model of lanosterol 14-demethylase from Cryptococcus neoformans: Active-site characterization and insights into azole binding. Antimicrob. Agents Chemother. 2009, 53, 3487–3495. [Google Scholar] [CrossRef] [Green Version]
- Warrilow, A.G.; Parker, J.E.; Kelly, D.E.; Kelly, S.L. Azole affinity of sterol 14α-demethylase (CYP51) enzymes from Candida albicans and Homo sapiens. Antimicrob. Agents Chemother. 2012, 57, 1352–1360. [Google Scholar] [CrossRef] [Green Version]
- Land, H.; Humble, M.S. YASARA: A tool to obtain structural guidance in biocatalytic investigations. In Methods in Molecular Biology; Springer: New York, NY, USA, 2017; pp. 43–67. ISBN 1064-3745. [Google Scholar]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef] [PubMed]
- Kim, S. Getting the most out of PubChem for virtual screening. Expert Opin. Drug Discov. 2016, 11, 843–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Empereur-Mot, C.; Zagury, J.-F.; Montes, M. Screening Explorer–an interactive tool for the analysis of screening results. J. Chem. Inf. Model. 2016, 56, 2281–2286. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Perilla, C.; Bernal, F.A.; Coy-Barrera, E. Insights into the interaction and binding mode of a set of antifungal azoles as inhibitors of potential fungal enzyme-based targets. Mol. Divers. 2018, 22, 929–942. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Li, Y.; Tian, S.; Xu, L.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys. Chem. Chem. Phys. 2014, 16, 16719–16729. [Google Scholar] [CrossRef]
- Sun, H.; Li, Y.; Shen, M.; Tian, S.; Xu, L.; Pan, P.; Guan, Y.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 5. Improved docking performance using high solute dielectric constant MM/GBSA and MM/PBSA rescoring. Phys. Chem. Chem. Phys. 2014, 16, 22035–22045. [Google Scholar] [CrossRef]
- Tosco, P.; Balle, T. Open3DQSAR: A new open-source software aimed at high-throughput chemometric analysis of molecular interaction fields. J. Mol. Model. 2010, 17, 201–208. [Google Scholar] [CrossRef]
Sample Availability: Samples of the all test compounds are available from the authors. |









| CC a | C1 | C2 | C3 | C4 | C5 | C6 | C7 |
|---|---|---|---|---|---|---|---|
| C1 | 1.00 | ||||||
| C2 | 0.699 | 1.00 | |||||
| C3 | 0.872 | 0.832 | 1.00 | ||||
| C4 | 0.926 | 0.731 | 0.901 | 1.00 | |||
| C5 | 0.921 | 0.747 | 0.886 | 0.985 | 1.00 | ||
| C6 | 0.647 | 0.626 | 0.627 | 0.678 | 0.724 | 1.00 | |
| C7 | 0.928 | 0.704 | 0.886 | 0.912 | 0.888 | 0.636 | 1.00 |
| C8 | 0.728 | 0.444 | 0.670 | 0.693 | 0.671 | 0.446 | 0.694 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angarita-Rodríguez, A.; Quiroga, D.; Coy-Barrera, E. Indole-Containing Phytoalexin-Based Bioisosteres as Antifungals: In Vitro and In Silico Evaluation against Fusarium oxysporum. Molecules 2020, 25, 45. https://doi.org/10.3390/molecules25010045
Angarita-Rodríguez A, Quiroga D, Coy-Barrera E. Indole-Containing Phytoalexin-Based Bioisosteres as Antifungals: In Vitro and In Silico Evaluation against Fusarium oxysporum. Molecules. 2020; 25(1):45. https://doi.org/10.3390/molecules25010045
Chicago/Turabian StyleAngarita-Rodríguez, Andrea, Diego Quiroga, and Ericsson Coy-Barrera. 2020. "Indole-Containing Phytoalexin-Based Bioisosteres as Antifungals: In Vitro and In Silico Evaluation against Fusarium oxysporum" Molecules 25, no. 1: 45. https://doi.org/10.3390/molecules25010045