3.1. General
NMR spectra were recorded in CDCl3 (internal Me4Si) with a Varian AM-600 (600 MHz 1H, 150 MHz 13C) spectrometer (Sugar Land, TX, USA) at rt. Chemical shifts (δ) are reported in ppm relative to Me4Si (δ 0.00) for 1H and residual chloroform (δ 77.00) for 13C. All significant resonances (carbon skeleton) were assigned by COSY (1H-1H), HSQC (1H-13C), and HMBC (1H-13C) correlations. The relative configuration of the stereogenic centers was assigned on the basis of 1D-NOESY spectra. Mass spectra (ESI) were recorded with an Applied Biosystems 4000 Q-TRAP (low resolution) (Toronto, ON, Canada) and Waters AutoSpec Premier (Waters, Milford, MA, USA) or Waters MALDISynapt G2-S HDMS (high resolution) spectrometers (Manchester, UK). HPLC analyses were conducted on Merck-Hitachi apparatus (Darmstadt, Germany) equipped with Merck LiChrospher 100 RP-18 (250 × 0.4 mm, 5 μm) column and UV detector. Column chromatography was performed on silica gel 60 (70–230 mesh, Merck. Flash chromatography was performed on Buchi glass columns packed with silica gel 60 (230–400 mesh, Merck), using Knauer Smartline system with a Buchi fraction collector. Reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA), Alfa Aesar (Tianjin, China) or ABCR (Karlsruhe, Germany), and used without purification. Thin-layer chromatography was carried out on silica gel 60 F254 (Merck). The organic solutions were dried over MgSO4.
3.1.1. 2,3,4,5-Tetra-O-benzyl-6-O-tert-butyl-diphenylsilyl-D-glucose 17
To a cooled to 0 °C solution of 2,3,4,5-tetra-
O-benzyl-D-glucose propane-1,3-diyl dithioacetal [
20] (
16; 21.89 g; 34.7 mmol) and imidazole (4.74 g, 69.4 g, 2.0 eq) in methylene chloride (300 mL),
tert-butyl-diphenylsilyl chloride (9.6 mL, 37.5 mmol, 1.1 equiv.) was added dropwise within 1 h and the mixture was stirred overnight at room temperature. Water (150 mL) was added, the layers were separated and the organic solution was washed with 1M H
2SO
4 (100 mL) and water (100 mL), dried, concentrated, and the crude product was isolated by column chromatography (hexanes-ethyl acetate 7:1→3:1) to afford the 6-
O-protected derivative (27.47 g; 91%) as an oil.
1H NMR δ: 7.69−7.52 (m, 4H), 7.48−7.15 (m, 26H), 4.88 (d, J = 11.5 Hz, 1H), 4.76−4.57 (m, 5H), 4.49 (d, J = 11.2 Hz, 1H), 4.38 (d, J = 11.5 Hz, 1H), 4.23 (d, J = 4.8 Hz, 1H), 4.07 (t, J = 5.2 Hz, 1H), 3.99−3.91 (m, 2H), 3.85−3.81 (m, 1H), 3.69 (dd, J = 6.2, 4.6 Hz, 1H), 3.54 (dt, J = 5.6, 4.3 Hz, 1H), 2.87−2.74 (m, 1H), 2.77−2.53 (m, 3H), 1.97−1.91 (m, 1H), 1.91−1.81 (m, 1H), 1.10 (s, 9H);
13C NMR δ: 138.9, 138.7, 2 × 138.4, 135.9–127.0, 81.9, 79.8, 79.9, 78.4, 75.2, 74.7, 73.8, 72.5, 62.4, 49.5, 30.0, 29.5, 27.0, 26.2, 19.3.
HR-MS: m/z = 868.3647; calcd for C53H60O5S2SiNa [M + Na]+: 868.3651.
To a solution of the above prepared compound (4.36 g; 5.02 mmol) in MeCN:CH
2Cl
2:H
2O (8:1:1; 50 mL), Dess-Martin reagent (4.47 g, 10.0 mmol, 2.0 eq) was added, the mixture was stirred at room temperature overnight, and concentrated. The residue was purified by column chromatography (hexanes-ethyl acetate 7:1→3:1) affording the title product
17 (3.28 g; 84%) (
Figure 2), which was used immediately in the next step.
LR-MS: m/z = 833 [C51H58O7SiNa (M + MeOH+Na)+].
3.1.2. Olefin 19
Generation of the titanium reagent 18: To a cooled to −78 °C solution of allyltrimethylsilane (3.7 g, 32.4 mmol, 10.0 eq) in dry THF (32 mL), a solution of 2.5 M BuLi in hexane (12 mL, 29.1 mmol, 9.0 eq.) was added within 1h with a syringe pump; after another 30 min. the mixture became yellowish. Then, a solution of 1.0 M ClTi(OiPr)3 in methylene chloride (29 mL) was added within 30 min (syringe pump) and the red mixture was stirred for another 30 min.
To such prepared reagent
18, a solution of
17 (2.10 g, 3.19 mmol) in dry THF (6.4 mL) was added within 1h, the mixture was stirred overnight at −78 °C (using immersion cooler with temperature control Huber TC100E, temperature range −100 to +40 °C) and partitioned between water (70 mL) and ether (70 mL). The white precipitate was filtered off and discarded; the organic layer was separated, washed with brine, and concentrated to afford a crude mixture of (anticipated) anti-isomers, pure enough to be used in the next steps (
Figure 3).
LR-MS: m/z = 915 [C56H68O6Si2Na (M + Na)+].
3.1.3. Dienoalcohol 21
To a solution of crude
19 obtained above in THF (16 mL) a solution of 1.0 M TBAF in THF (16 mL, 16.0 mmol, 5.0 eq.) was added and the mixture was stirred at room temperature overnight. Then it was concentrated and the residue was subjected to column chromatography (hexane-ethyl acetate, 92:8→36:64) to afford the title product
21 (1.11 g, 73% over two steps) as an oil (
Figure 4).
1H NMR δ: 7.39–7.17 (m, 20H, arom.), 6.51 (ddd, J = 16.6, 11.0 and 10.1 Hz, 1H, H-2), 6.29 (t, J = 11.2 Hz, 1H, H-3), 5.46 (t, J = 10.3 Hz, 1H, H-4), 5.28 (d, J = 16.7 Hz, 1H, H-1), 5.11 (d, J = 10.1 Hz, 1H, H-1′), 4.86 (d, J = 11.4 Hz, 1H, OCH2Ph), 4.73 (d, J = 11.4 Hz, 1H, OCH2Ph), 4.68 (d, J = 11.5 Hz, 1H, OCH2Ph), 4.63–4.57 (m, 3H, H-5 and 2×OCH2Ph), 4.49 (d, J = 11.6 Hz, 1H, OCH2Ph), 4.37 (d, J = 11.6 Hz, 1H, OCH2Ph), 4.33 (d, J = 11.7 Hz, 1H, OCH2Ph), 3.90 (t, J = 4.9 Hz, 1H, H-8), 3.85 (ddd, J = 12.0, 5.4, and 4.0 Hz, 1H, H-9), 3.77 (ddd, J = 11.7, 6.9, 4.0 Hz, 1H, H-9′), 3.73 (dd, J = 6.1, 4.4 Hz, 1H, H-6), 3.66 (dt, J = 5.5 and 4.0 Hz, 1H, H-7), 2.15 (dd, J = 6.9 and 5.5 Hz, 1H, OH);
13C NMR δ: 138.8, 138.7 and 2 × 138.3(quat. benzyl), 134.2 (C-3), 131.9 (C-2), 128.6−127.6 (arom. and C-4), 120.4 (C-1), 81.9 (C-6), 79.7 (C-7), 79.3 (C-8), 75.6 (C-5), 75.2, 74.5, 71.7 and 70.5 (4×OCH2Ph), 60.8 (C-9).
HR-MS: m/z = 587.2770; calcd for C37H40O5Na [M + Na]+: 587.2773.
3.1.4. Dienoaldehyde 22
To a cooled to 0 °C solution of alcohol
21 (350 mg, 0.62 mmol) and TEMPO (1.0 mg; 6.0 μmol) in dry CH
2Cl
2 (8 mL), trichloroisocyanuric acid (155 mg; 0.66 mmol; 1.1 eq) was added and the mixture was stirred for 15 min. Then it was filtered through short pad of Celite, the filtrate was diluted with Et
2O (6.0 mL) and washed with 5% Na
2S
2O
3 (2.0 mL), 1M NaOH (5 mL), 1M H
2SO
4 (5 mL), and water (3.0 mL). The organic phase was dried and concentrated, and the crude aldehyde was used immediately in the next step (
Figure 5).
LR-MS: m/z = 617 [C38H42NO6Na (M + MeOH + Na)+].
3.1.5. Triene 24
To a solution of aldehyde
22 (170 mg, 0.3 mmol) and phosphonate
23 [
21] (120 mg, 1.5 eq.) in dry CH
3CN (3 mL), LiBr (40.5 mg, 0.466 mmol, 1.5 eq) was added followed by di-isopropylethylamine (80 μL, 0.466 mmol, 1.5 eq) and the mixture was stirred for 20 h. It was then concentrated and the crude product was purified by column chromatography (hexane 100% then: hexane-ethyl acetate 92:8→36:64) (
Figure 6).
1H NMR δ: 7.46 (d, J = 15.6, 1H, H-10), 7.35–7.16 (m, 21H, arom. and H-9), 6.51 (ddd, J = 16.7, 11.3 and 10.1 Hz, 1H, H-2), 6.27 (t, J = 11.2 Hz, 1H, H-3), 5.45 (dd, J = 11.1 and 9.7 Hz, 1H, H-4), 5.25 (d, J = 16.7 Hz, 1H, H-1), 5.09 (d, J = 10.1 Hz, 1H, H-1′), 4.84 (d, J = 11.5 Hz, 1H, OCH2Ph), 4.67–4.59 (m, 3H, 2×OCH2Ph and H-5), 4.56 (d, J = 11.7 Hz, 1H, OCH2Ph), 4.55–4.47 (m, 2H, 2×OCH2Ph), 4.38–4.30 (m, 3H, OCH2Ph and 2×H-13), 4.24 (dd, J = 6.8 and 5.6 Hz, 1H, H-8), 4.14 (d, J = 11.4 Hz, 1H, OCH2Ph), 3.97 (qdd, J = 11.1, 8.9 and 7.1 Hz, 2H, 2×H-14), 3.81 (dd, J = 5.6 and 4.2 Hz, 1H, H-7), 3.74 (dd, J = 6.3 and 4.2 Hz, 1H, H-6);
13C NMR δ: 164.5 (C-12), 153.3 (C-11), 147.6 (C-9), 138.9, 138.7, 138.4 and 137.9 (quat. benzyl), 134.1 (C-3), 132.1 (C-2), 128.6−127.3 (arom. and C-4), 122.3 (C-10), 120.2 (C-1), 82.0 (C-7), 81.6 (C-6), 79.2 (C-8), 75.5 (C-5), 75.3, 74.2, 71.4, 70.4 (4×OCH2Ph), 62.1 (C-13), 42.7 (C-14).
HR-MS: m/z = 696.2922; calcd for C42H43NO7Na [M + Na]+: 696.2937.
3.1.6. Triene 25
Aldehyde
22 (45 mg) and Ph
3P=CHCO
2Me (54 mg; 2.0 eq.) were dissolved in dry benzene and stirred at room temperature for 16 h. The mixture was concentrated and the product was isolated by column chromatography (hexanes-ethyl acetate, 3:1→2:1) as a colorless oil in 74% yield (36.5 mg) (
Figure 7).
1H NMR δ: 7.43–7.18 (m, 20H), 7.01 (dd, J = 15.8, 6.6 Hz, 1H, H-9), 6.45 (ddd, J = 16.7, 11.2, 10.0 and 6.0 Hz, 1H, H-2), 6.26 (t, J = 11.2 Hz, 1H, H-3), 6.01 (d, J = 15.9 Hz, 1H, H-10), 5.45 (t, J = 10.3 Hz, 1H, H-4), 5.26 (d, J = 16.7 Hz, 1H, H-1), 5.09 (d, J = 10.0 Hz, 1H, H-1′), 4.82 (d, J = 11.5 Hz, 1H, OCH2Ph), 4.63 (m, 2H, 2×OCH2Ph), 4.59–4.53 (m, 2H, OCH2Ph and H-5), 4.51 (d, J = 11.3 Hz, 1H, OCH2Ph), 4.46 (d, J = 11.5 Hz, 1H, OCH2Ph), 4.32 (d, J = 11.7 Hz, 1H, OCH2Ph), 4.15 (m, 2H, OCH2Ph and H-8), 3.81 (s, 3H, OMe), 3.79 (dd, J = 5.6 and 4.4 Hz, 1H, H-7), 3.73 (m, 1H, H-6);
13C NMR δ: 166.3 (C-11), 146.0 (C-9), 138.7, 138.4, 138.1 and 137.8 (quat. benzyl), 133.9 (C-3), 131.8 (C-2), 128.5−127.3 (arom. and C-4), 123.3 (C-10), 120.2 (C-1), 81.6 and 81.5 (C-6 and C-7), 78.8 (C-8), 75.2 (OCH2Ph), 75.1 (C-5), 74.1, 71.1,and 70.3 (3×OCH2Ph), 51.7 (OMe).
HR-MS: m/z = 641.2886; calcd for C40H42O6Na [M + Na]+: 641.2879.
3.1.7. Triene 26
This triene was prepared analogously as
25 from aldehyde
22 (47 mg) and Ph
3P=CHCOMe (55 mg; 2.0 eq) as a colorless oil in 76% yield (36.5 mg) (
Figure 8).
1H NMR δ: 7.36–7.19 (m, 20H, arom.), 6.78 (dd, J = 16.3, 6.5 Hz, 1H, H-9), 6.46 (dt, J = 16.2, 10.6 Hz, 1H, H-2), 6.28 (t, J = 11.1 Hz, 1H, H-3), 6.18 (d, J = 16.2 Hz, H-10), 5.45 (t, J = 10.4 Hz, 1H, H-4), 5.27 (d, J = 16.7 Hz, 1H, H-1), 5.11 (dd, J = 10.0 and 1.7 Hz, 1H, H-1′), 4.85 (d, J = 11.4 Hz, 1H, OCH2Ph), 4.64–4.56 (m, 4H, 3×OCH2Ph and H-5), 4.53 (d, J = 11.4 Hz, 1H, OCH2Ph), 4.45 (d, J = 11.6 Hz, 1H, OCH2Ph), 4.32 (d, J = 11.7 Hz, 1H, OCH2Ph), 4.18 (d, J = 11.6 Hz, 1H, OCH2Ph), 4.15 (ddd, J = 6.5 and 5.3 Hz, 1H, H-8), 3.81 (t, J = 4.8 Hz, 1H, H-7), 3.70 (dd, J = 6.3 and 4.3 Hz, 1H, H-6), 2.11 (s, 3H, H-12);
13C NMR δ: 198.2 (C-11), 144.7 (C-9), 138.6, 138.3, 138.1 and 137.8 (quat. benzyl), 134.1 (C-3), 132.6 (C-10), 131.7 (C-2), 128.5−127.4 (arom. and C-4), 120.3 (C-1), 81.7 and 81.6 (C-6 and C-7), 79.3 (C-8), 2 × 75.3 (C-5 and OCH2Ph), 74.0, 71.3, 70.3 (3×OCH2Ph), 26.9 (C-12).
HR-MS: m/z = 625.2936; calcd for C40H42O5Na [M + Na]+: 625.2930.
3.1.8. Weinreb Amide 29
To a cooled to 0 °C suspension of MeNHOMe x HCl (5.85 g, 60.0 mmol, 3.0 eq) in dry CH2Cl2 (175 mL), a 2M solution of Me3Al in toluene (30 mL, 60 mmol, 2 eq.) was added dropwise during 30 min. by a syringe pump. The mixture was stirred for an additional 30 min., then lactone 27 (8.37 g, 20 mmol) in dry CH2Cl2 (25 mL) was added within 30 min. by a syringe pump, and the mixture was stirred at room temperature for 3 h. Aqueous H2SO4 (1M solution, 100 mL) was carefully added and the organic phase was separated, washed with water (100 mL), brine (100 mL), and dried.
Imidazole (4.08 g, 60 mmol, 3.0 eq) was added to this containing crude
28 and the resulting mixture was cooled to 0 °C. A solution of t
ert-butyldiphenylchlorosilane (7.8 mL, 30.0 mmol, 1.5 eq.) in CH
2Cl
2 (20 mL) was added dropwise within 1 h by a syringe pump and the mixture was stirred for additional 16 h. Aqueous H
2SO
4 (1M solution, 50 mL) was carefully added, the organic phase was separated, washed with water (100 mL), brine (100 mL), dried, and the crude product was purified by column chromatography (hexanes-ethyl acetate: 13:1→7:1) to give the title product
29 (12.35 g, 86% over two steps) as a colorless oil (
Figure 9).
1H NMR δ: 7.49–7.42 (m, 3H, arom.), 7.36–7.22 (m, 15H, arom.), 7.18–7.15 (m, 2H, arom.), 4.78–4.71 (m, 2H, 2×OCH2Ph), 4.60–4.51 (m, 4H, H-5 and 3×OCH2Ph), 4.35 (d, J = 11.4 Hz, 1H, OCH2Ph), 3.72–3.65 (m, 3H, H-2, H-3 and H-4), 3.62 (s, 3H, OCH3), 3.41 (m, 1H, H-4′), 3.15 (bs, 3H, NCH3), 1.07 [s, 9H, OSiPh2C(CH3)3];
13C NMR δ 170.4 (C-1), 138.5, 138.3 and 138.0 (quat. benzyl), 135.8, 133.3, 129.6 and 129.3−126.5 (arom.), 82.3 (C-3), 79.6 (C-4), 76.1 (C-2), 74.2, 73.3 and 71.6 (3×OCH2Ph), 61.4 (OCH3), 32.8 (NCH3), 26.8 [OSiPh2C(CH3)3], 18.8 [OSiPh2C(CH3)3].
HR-MS: m/z = 740. 3387; calcd for C44H51NO6SiNa [M + Na]+: 740.3383.
3.1.9. 2,3,4-Tri-O-benzyl-5-O-tert-butyl-diphenylsilyl-D-xylose 30
To a cooled to 0 °C solution of amide
29 (2.94 g, 4.10 mmol, 1.0 eq.) in dry Et
2O (41 mL), LiAlH
4 (0.19 g, 5.12 mmol, 1.25 eq.) was added in three portions within 15 min., and the mixture was stirred for 1 h. Aqueous H
2SO
4 (1M solution, 20 mL) was added, the organic phase was separated, washed with water (20 mL) and brine (20 mL), dried, and concentrated. The crude product was purified by column chromatography (hexanes→hexanes-ethyl acetate: 68:32) to give the aldehyde
30 (2.05 g, 76%) as a colorless oil (
Figure 10). This product was used immediately in the next step.
LR-MS: m/z = 713 [C43H50O6SiNa (M + MeOH + Na)+].
3.1.10. Silanol 31
This compound was prepared analogously as
19, from
30 (2.79 g, 4.23 mmol) as a colorless oil as a mixture of (anticipated) two anti-isomers (
Figure 11).
LR-MS: m/z = 795 [C48H60O5Si2Na (M + Na)+].
3.1.11. Dienoalcohol 32
This compound was prepared, analogously as
21, from
31. It was purified by column chromatography (hexanes→hexanes-ethyl acetate: 36:64) to give title dienalcohol
32 as a colorless oil (1.36 g, 72% over two steps) (
Figure 12).
1H NMR δ: 7.37–7.22 (m, 15H, arom.), 6.58 (ddd, J = 16.7, 11.2 and 10.1 Hz, 1H, H-2), 6.27 (t, J = 11.2 Hz, 1H, H-3), 5.53 (t, J = 10.3 Hz, 1H, H-4), 5.29 (d, J = 16.7 Hz, 1H, H-1), 5.16 (d, J = 10.2 Hz, 1H, H-1′), 4.78 (d, J = 11.4 Hz, 1H, OCH2Ph), 4.73 (d, J = 11.4 Hz, 1H, OCH2Ph), 4.64–4.55 (m, 4H, H-5 and 3×OCH2Ph), 4.35 (d, J = 11.7 Hz, 1H, OCH2Ph), 3.72 (ddd, J = 11.3, 6.6 and 4.2 Hz, 1H, H-8), 3.69–3.65 (m, 2H, H-6 and H-7), 3.55 (ddd, J = 11.4, 5.6 and 3.5 Hz, 1H, H-8′), 2.12 (t, J = 6.3 Hz, 1H, OH);
13C NMR δ: 138.6, 138.4 and 138.1 (quat. benzyl), 133.8 (C-3), 132.0 (C-2), 128.8−127.8 (arom. and C-4), 120.2 (C-1), 82.1 (C-6 or C-7), 79.7 (C-6 or C-7), 75.2 (OCH2Ph), 74.8 (C-5), 72.8 and 70.6 (2×OCH2Ph), 61.7 (C-8).
HR-MS: m/z = 467.2180; calcd for C29H32NO4Na [M + Na]+: 467.2188.
3.1.12. Triene 34
Alcohol 32 (244 mg, 0.55 mmol) was oxidized as described for 22 and the resulting product was purified by column chromatography (hexanes 100%→hexanes-ethyl acetate: 1:2) to afford the corresponding aldehyde 33 (204 mg, 84%) as an oil. LR-MS: m/z = 497 [C30H34O5Na (M + MeOH + Na)+].
This aldehyde (96 mg, 0.216 mmol) was reacted with phosphonate
23 analogously as for
22. The crude product was purified column chromatography (hexanes 100%→hexanes-ethyl acetate: 90:10→20:80) to give the title triene
34 as a colorless oil (87.3 mg, 73%) (
Figure 13).
1H NMR δ: 7.46 (d, J = 15.9, 1H, H-9), 7.35–7.16 (m, 16H, arom. and H-8), 6.51 (ddd, J = 16.7, 11.3 and 10.1 Hz, 1H, H-2), 6.27 (t, J = 11.2 Hz, 1H, H-3), 5.45 (dd, J = 11.1 and 9.7 Hz, 1H, H-4), 5.25 (d, J = 16.7, 1H, H-1), 5.09 (d, J = 10.1 Hz, 1H, H-1′), 4.74 (d, J = 11.5 Hz, 1H, OCH2Ph), 4.67 (d, J = 11.3 Hz, 1H, OCH2Ph), 4.58–4.30 (m, 6H, H-5, H-12, H-12′ and 4×OCH2Ph), 3.97 (m, 2H, H-13 and H-13′), 3.81−3.74 (m, 2H, H-6 and H-7);
13C NMR δ: 164.3 (C-10), 153.1 (C-11), 147.4 (C-8), 138.9, 138.7 and 138.1 (quat. benzyl), 134.0 (C-3), 132.1 (C-2), 128.5−127.2 (arom. and C-4), 122.5 (C-9), 120.1 (C-1), 82.0 and 79.8 (C-6 and C-7), 75.5 (×OCH2Ph), 75.0 (C-5), 72.4 and 70.4 (2×OCH2Ph), 62.1 (C-12), 42.7 (C-13).
HR-MS: m/z = 553.2456; calcd for C34H35NO6Na [M + Na]+: 553.2464.
3.1.13. Bicyclic Compound 35
To a cooled to −30 °C solution of
34 (39.6 mg, 7.86–10
−2 mmol) in dry CH
2Cl
2 (1.0 mL) a 1.0 M solution of Me
2AlCl (118 μL, 0.118 mmol, 1.5 eq.) was carefully added and the mixture was stirred for 5 h. Then the mixture was concentrated and the crude product was purified by preparative TLC (
n-heptane-MTBE: 6:4) to give the cyclic product
35 as colorless oil (26.9 mg, 68%) (
Figure 14).
1H NMR δ: 7.44–7.16 (m, 15H, arom.), 5.82–5.76 (m, 1H, H-4), 5.68–5.62 (m, 1H, H-5), 4.69–4.58 (m, 4H, 4×OCH2Ph), 4.59–4.51 (m, 2H, 2×OCH2Ph), 4.34 (td, J = 9.0 and 6.8 Hz, 1H, H-10), 4.28 (td, J = 9.1 and 7.0 Hz, 1H, H-10′), 4.03 (t, J = 5.0 Hz, 1H, H-2), 3.99–3.85 (m, 3H, H-7, H-11 and H-11′), 3.75 (t, J = 5.5 Hz, 1H, H-3), 3.60 (t, J = 5.6 Hz, 1H, H-1), 2.94 (brs, 1H, H-3a), 2.64 (q, J = 7.2 Hz, 1H, H-7a), 2.26 (dtd, J = 17.8, 4.2 and 2.0 Hz, 1H, H-6), 2.08 (dtd, J = 17.8, 3.8 and 1.9 Hz, 1H, H-6′);
13C NMR δ: 175.4 (C-8), 152.8 (C-9), 138.5, 138.4 and 138.3 (quat. benzyl), 128.4−127.5 (arom. and C-4), 124.3 (C-5), 90.3 (C-2), 88.5 (C-3), 83.4 (C-1), 72.0, 71.9 and 71.5 (3×OCH2Ph), 61.8 (C-10), 42.7 (C-11), 2 × 40.4 (C-3a and C-7a), 36.9 (C-7), 25.8 (C-6).
HR-MS: m/z = 553.2478; calcd for C34H35NO6Na [M + Na]+: 553.2464.
Configuration was established on the basis of 1D NOE (see
Figure 1 in the text).


 and
and 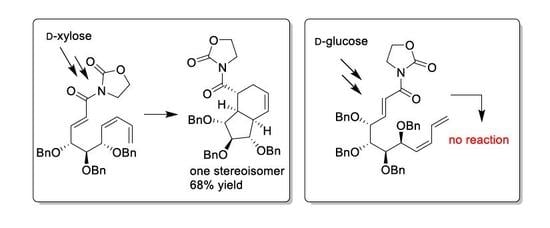




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
















