
Unravelling the Miscibility of Poly(2-oxazoline)s: A Novel Polymer Class for the Formulation of Amorphous Solid Dispersions

 , , , and
, , , and
Abstract
:1. Introduction
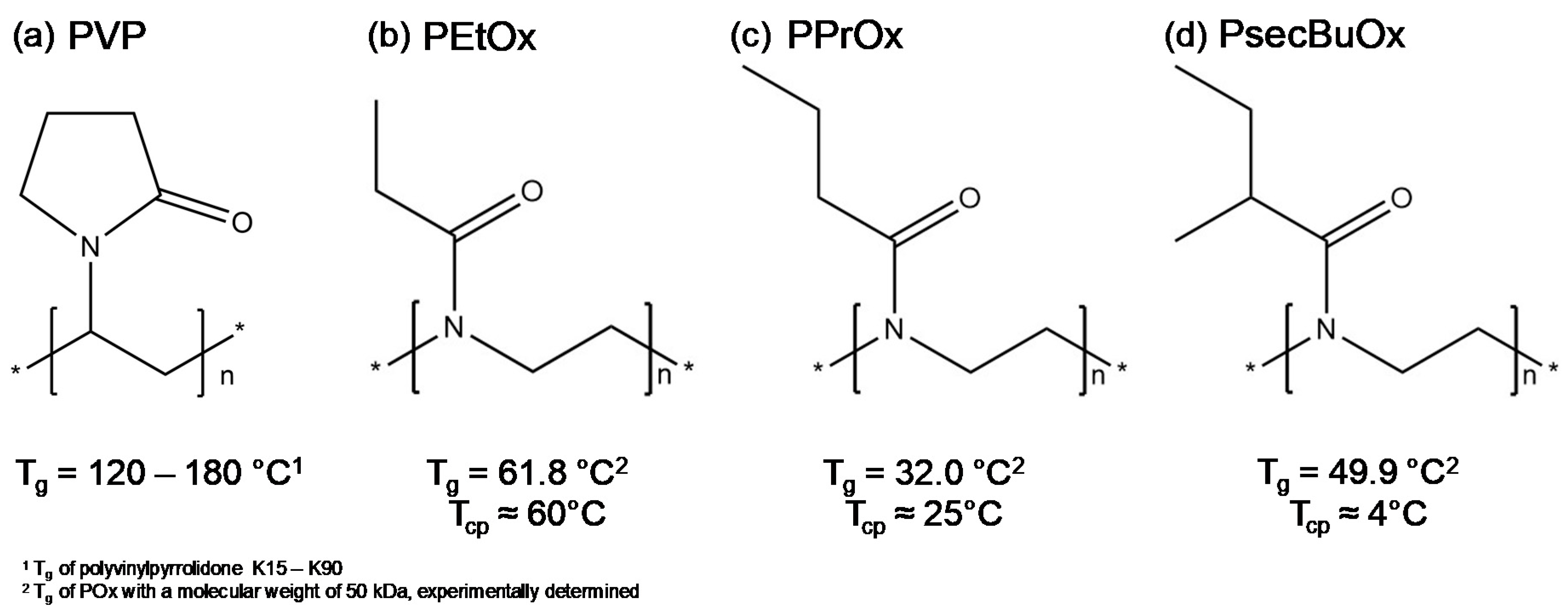
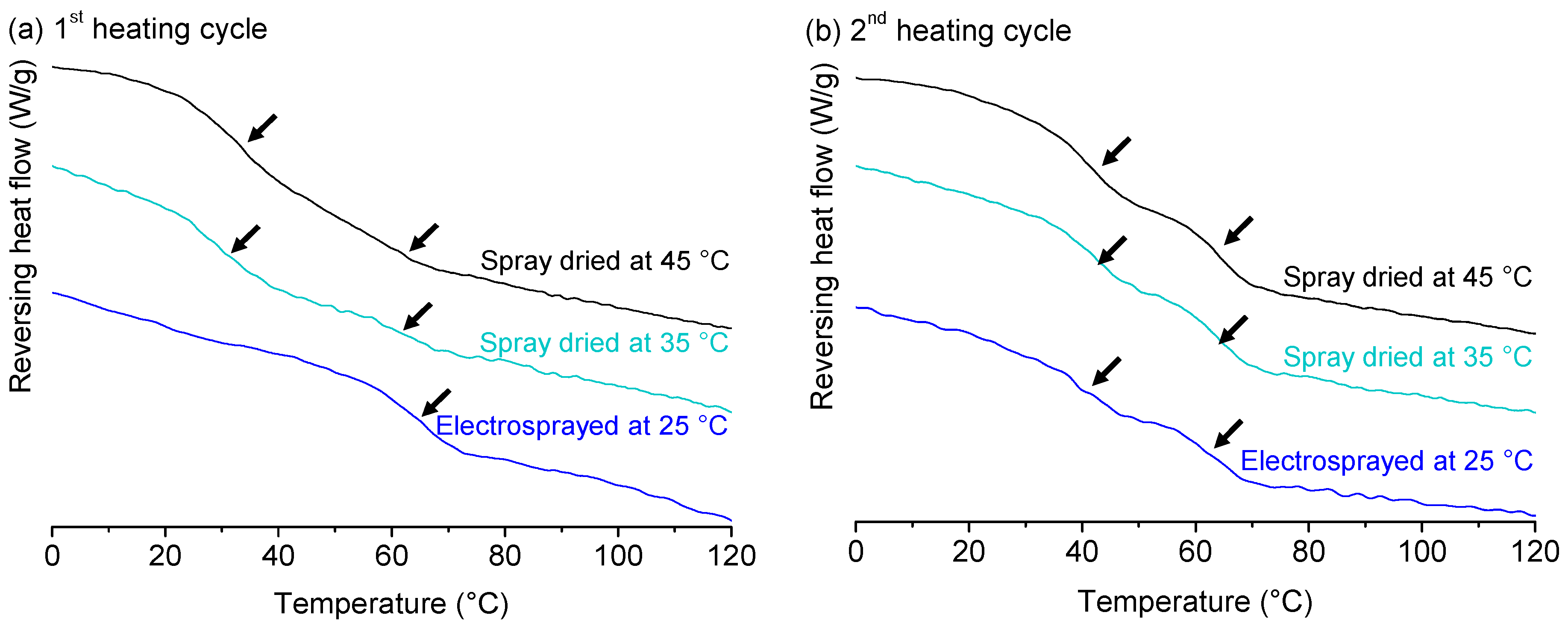
2. Results and Discussion
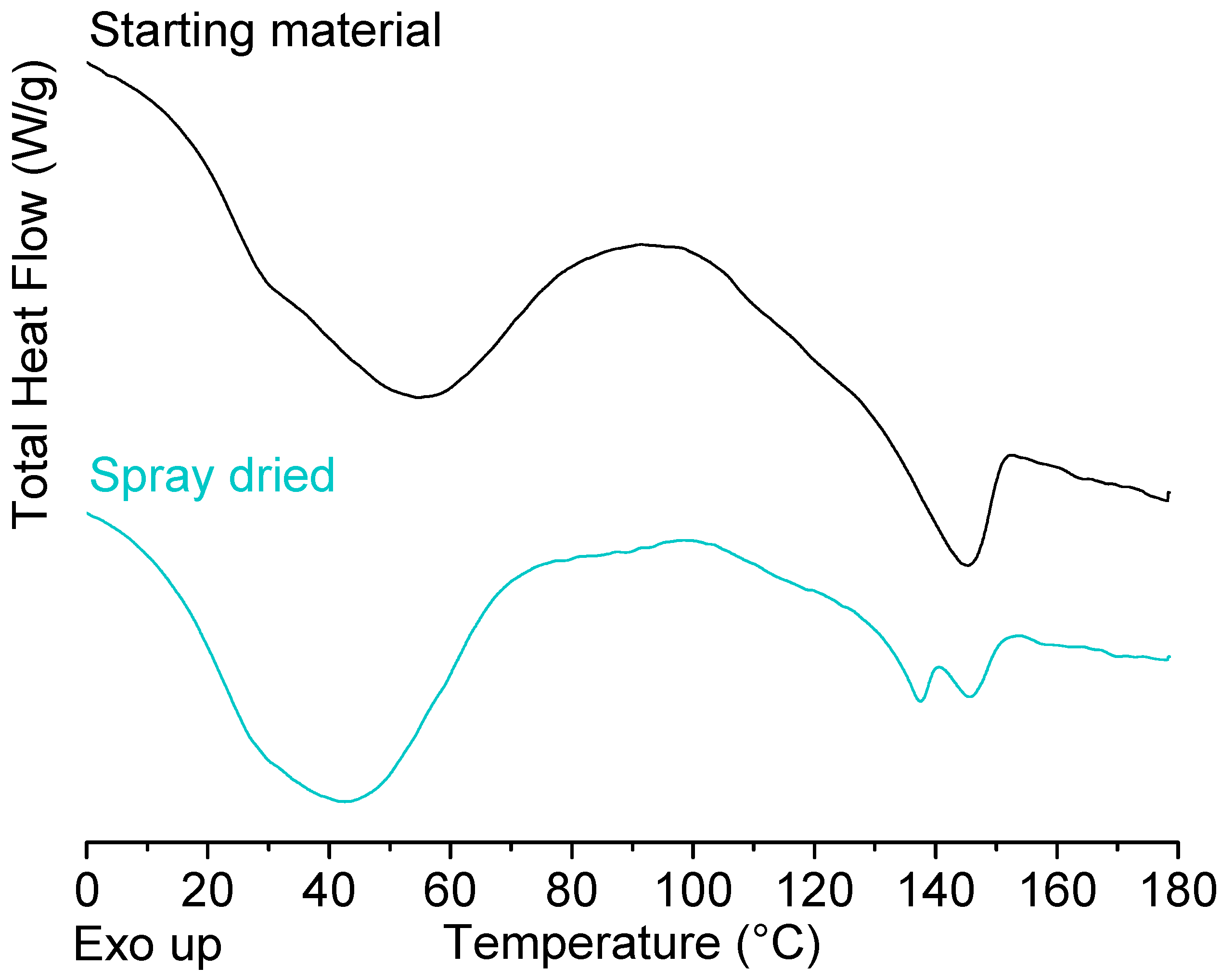
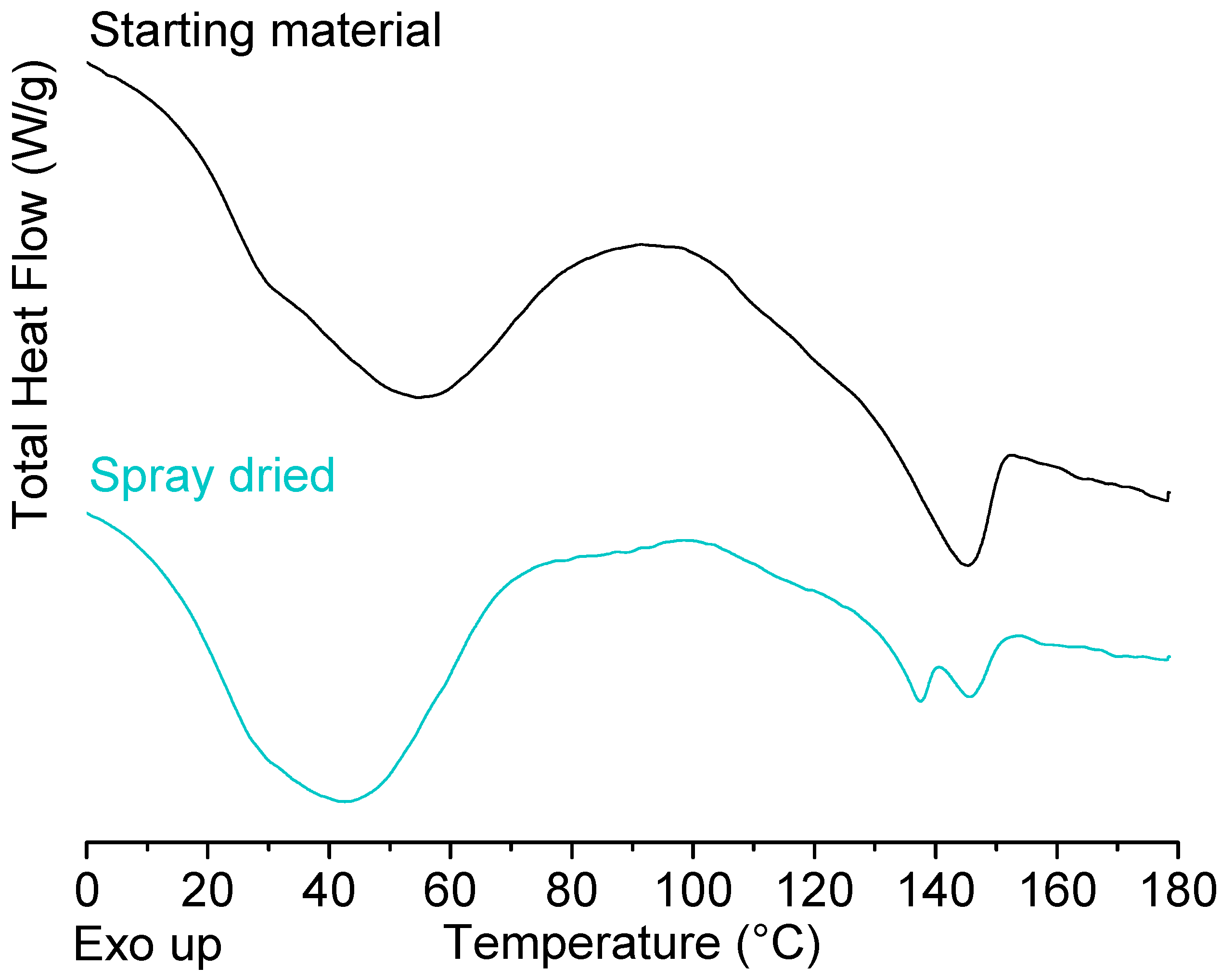
2.1. Solid State Analysis of Pure Polymers: Starting Material Versus Spray Dried Material
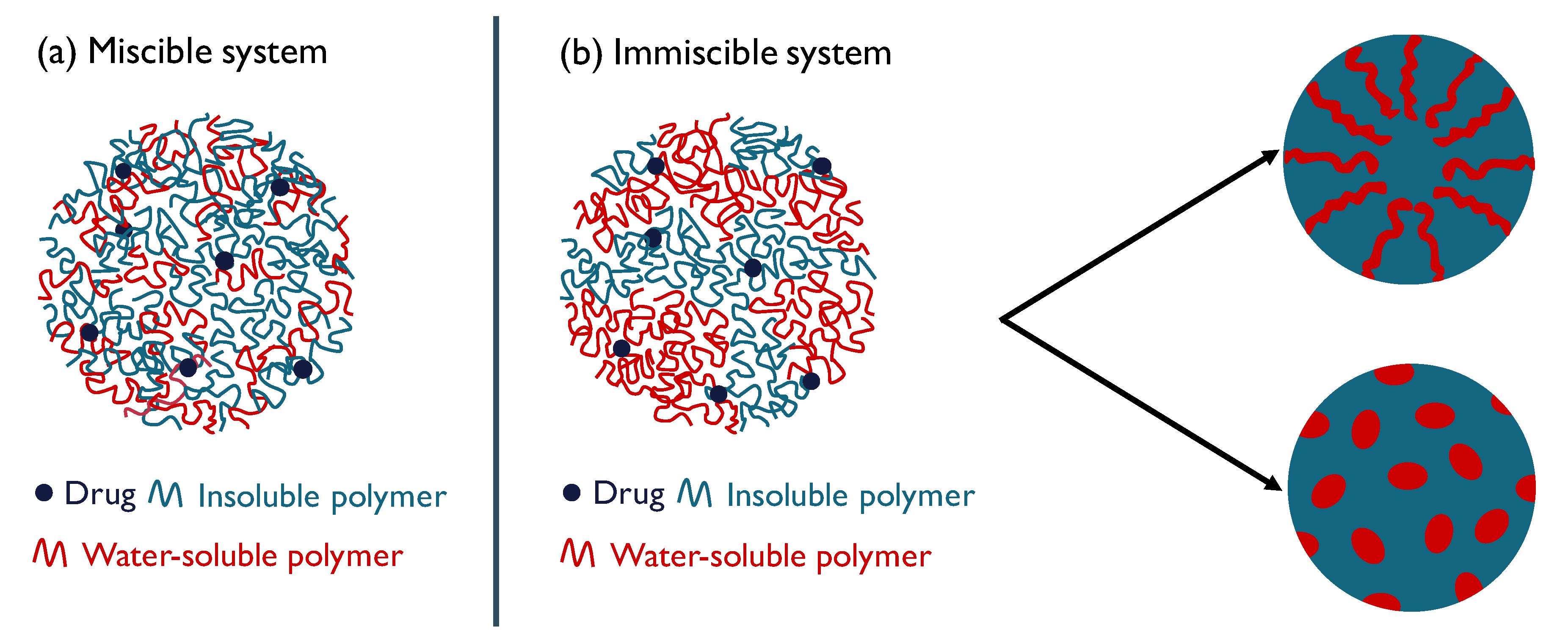
2.2. Solid State Analysis of Polymer Mixtures
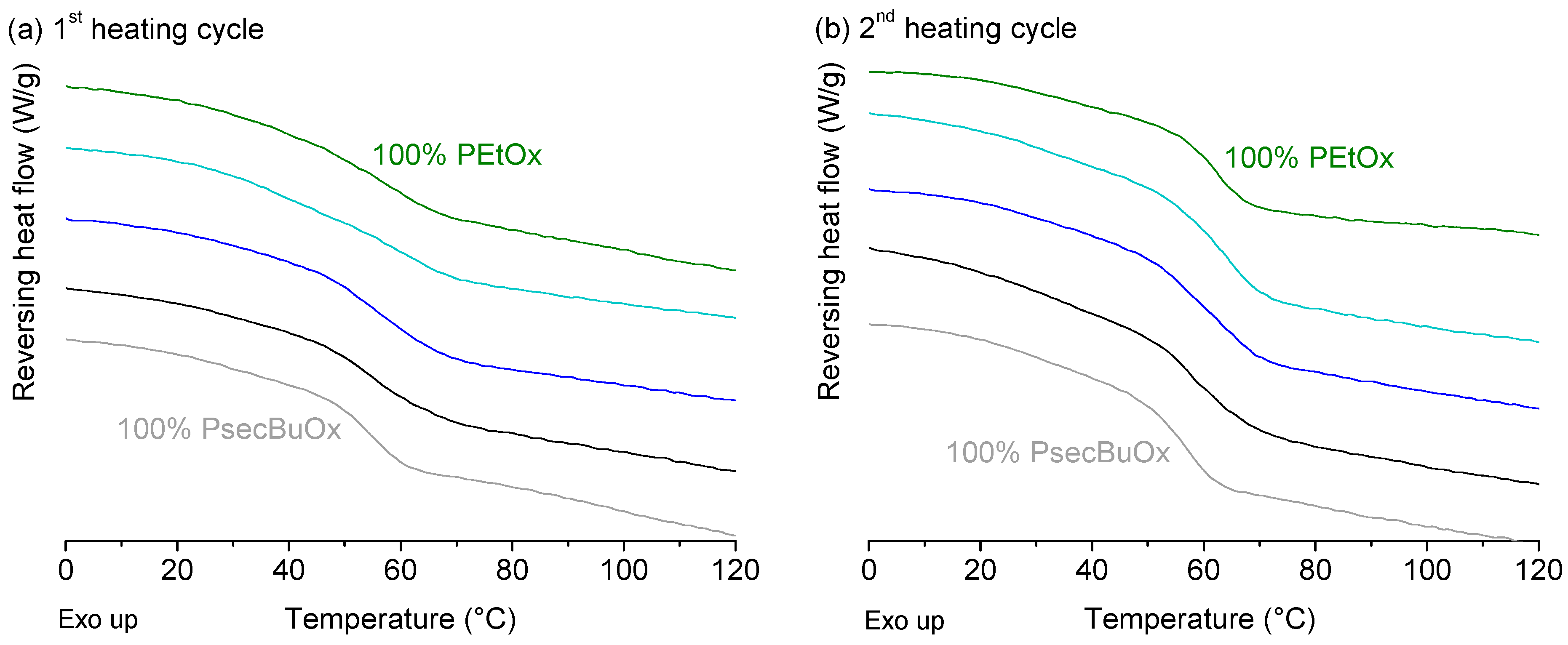
2.2.1. Poly(2-ethyl-2-oxazoline) and Poly(2-n-propyl-2-oxazoline)
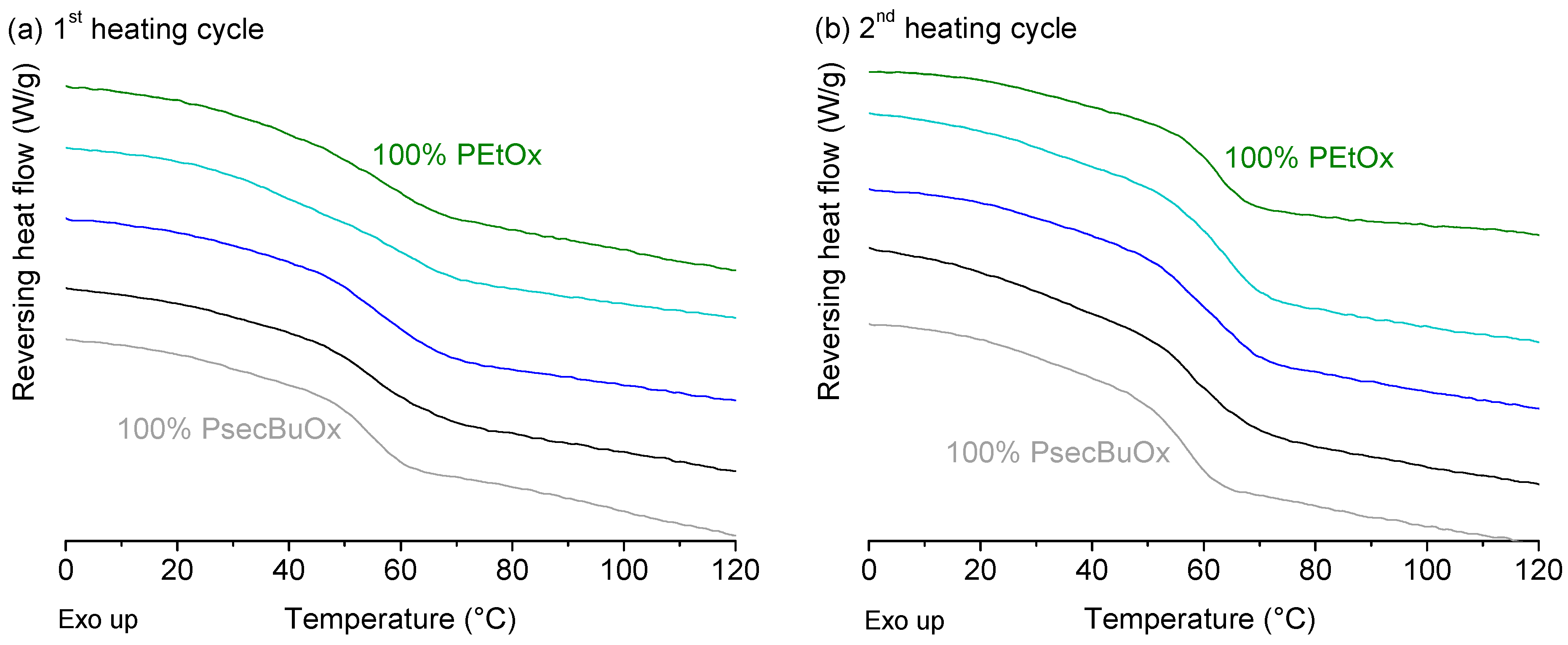
2.2.2. Poly(2-ethyl-2-oxazoline) and Poly(2-sec-butyl-2-oxazoline)
2.3. Solid State Analysis of Binary and Ternary Amorphous Solid Dispersions
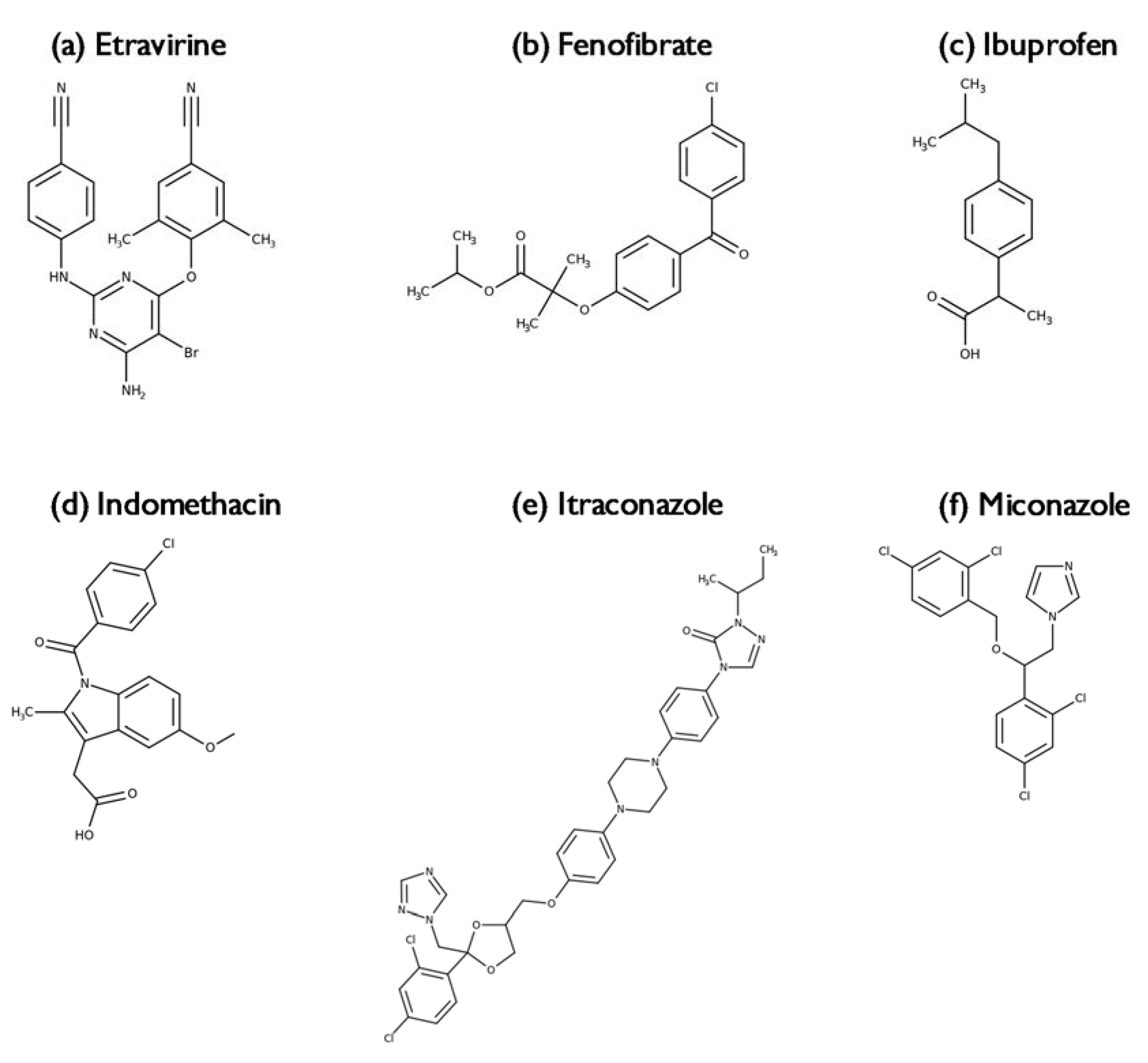
2.3.1. Indomethacin
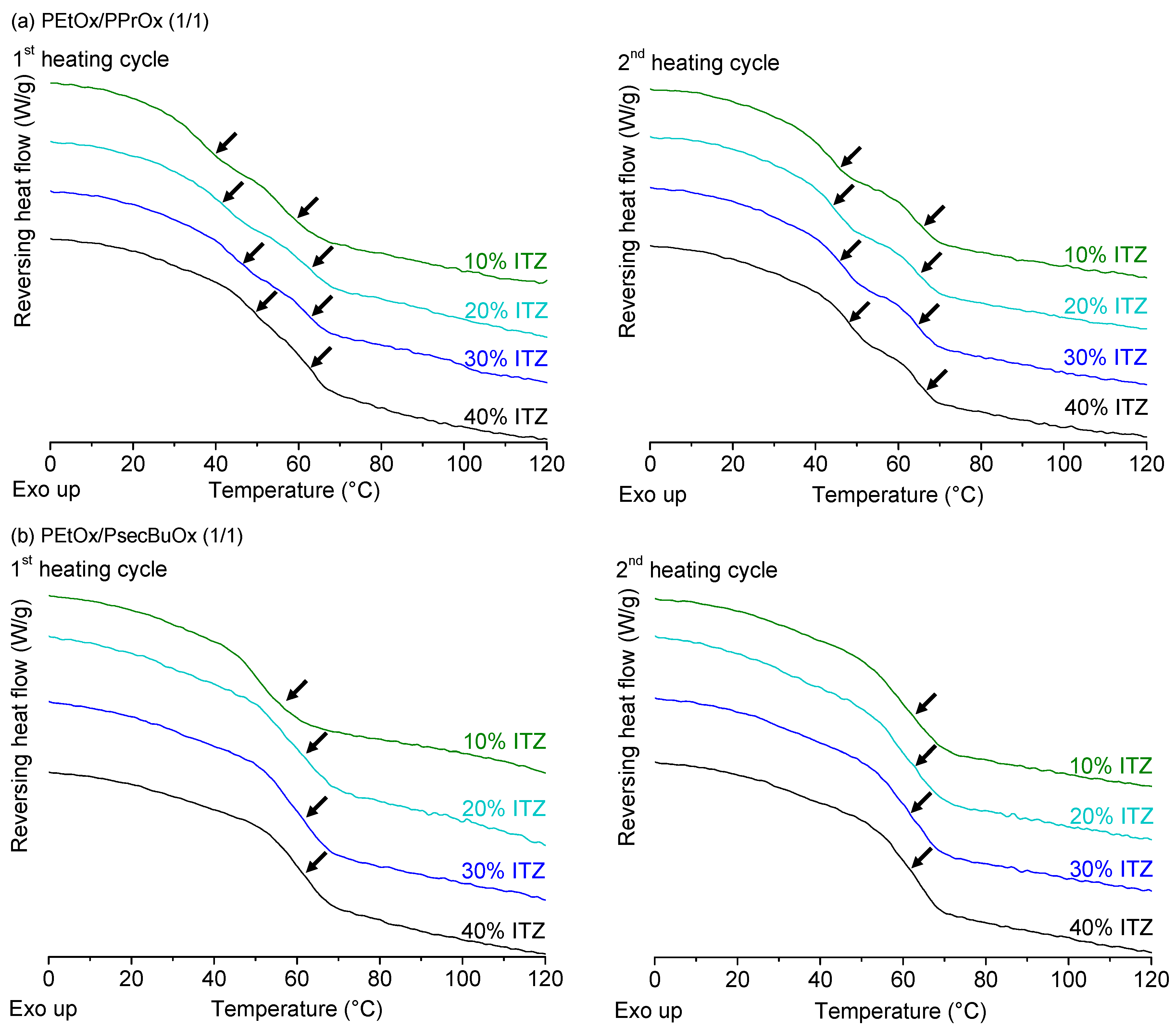
2.3.2. Itraconazole
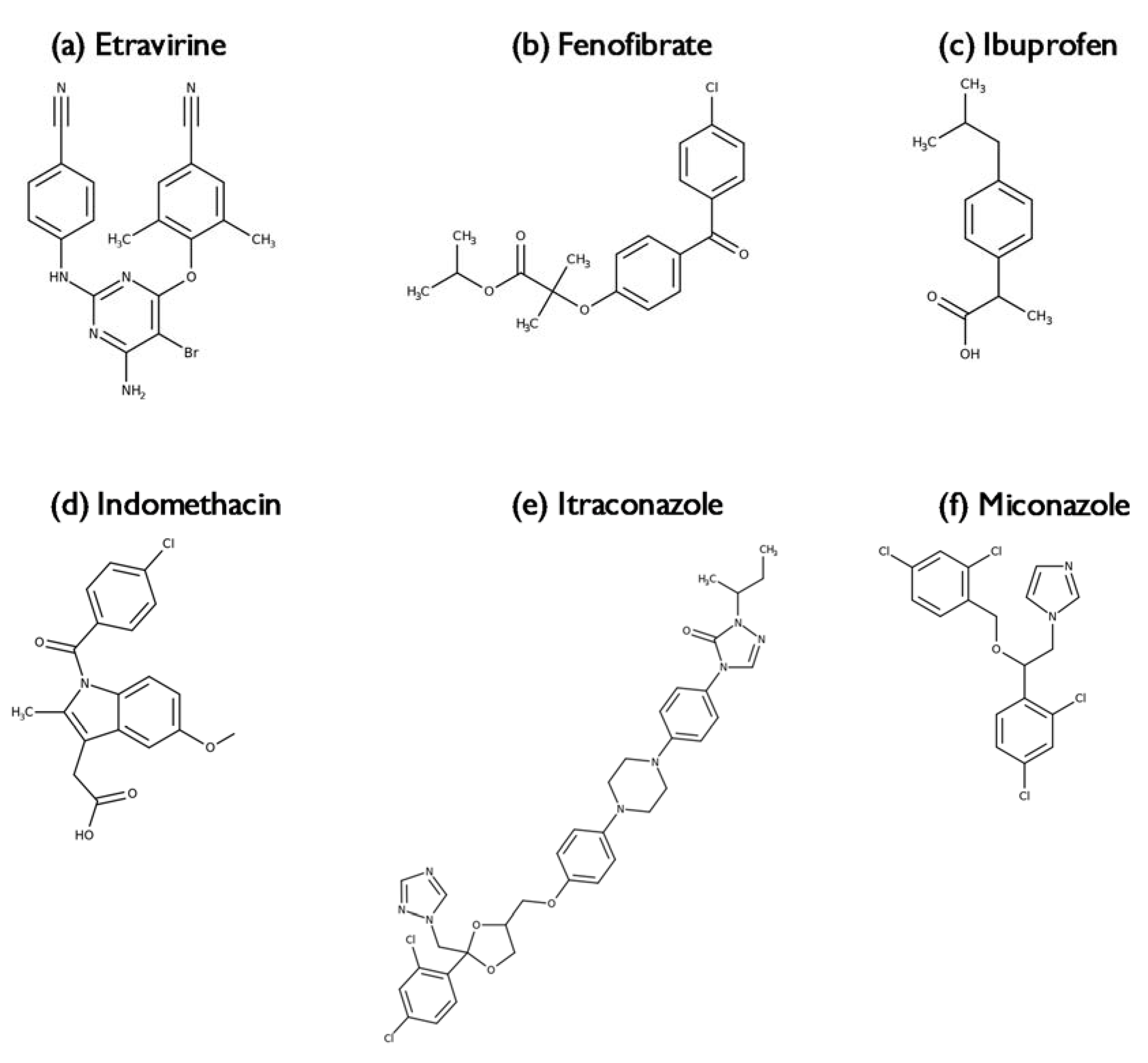
2.3.3. Miconazole, Fenofibrate, Ibuprofen and Etravirine
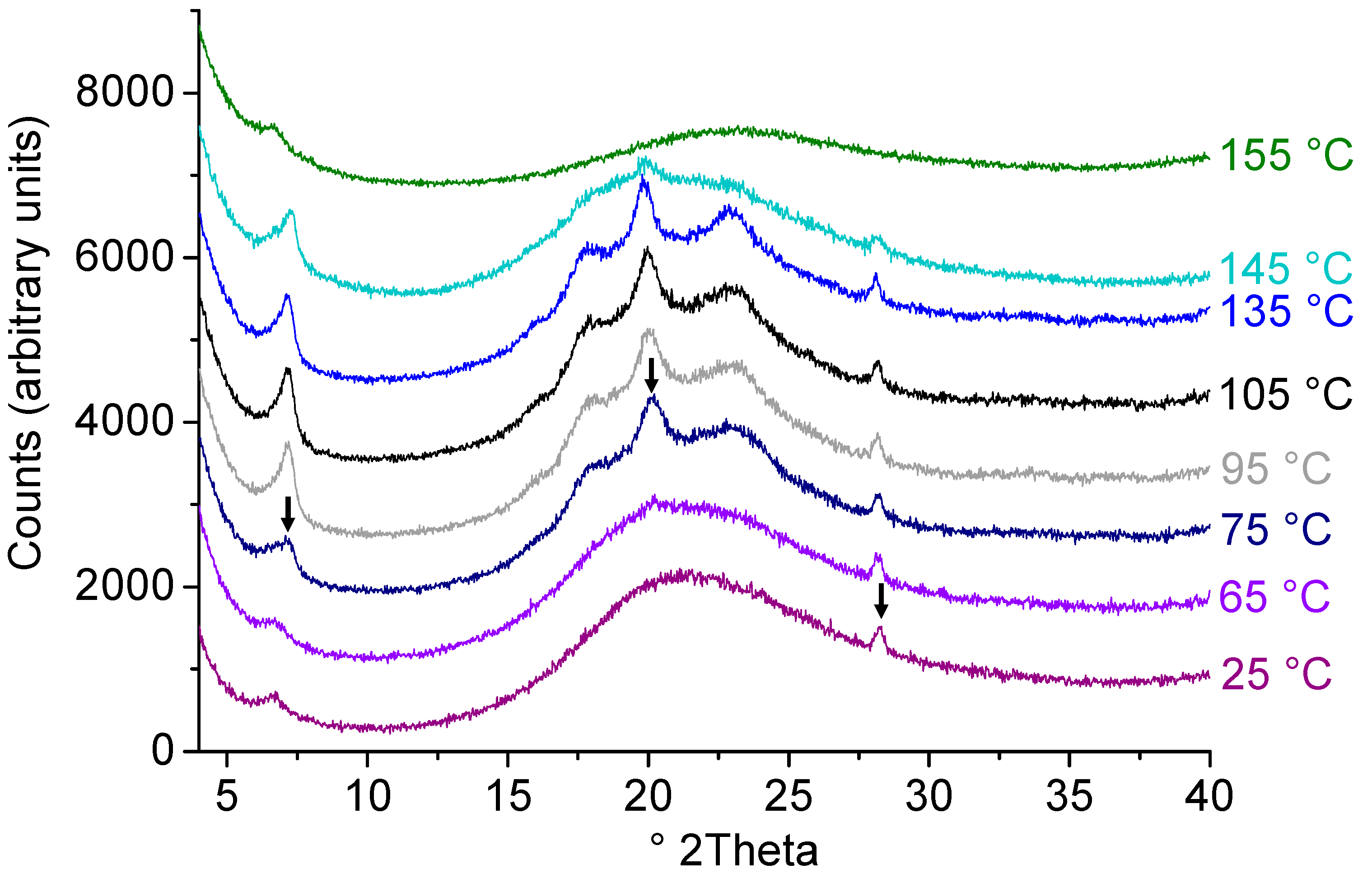
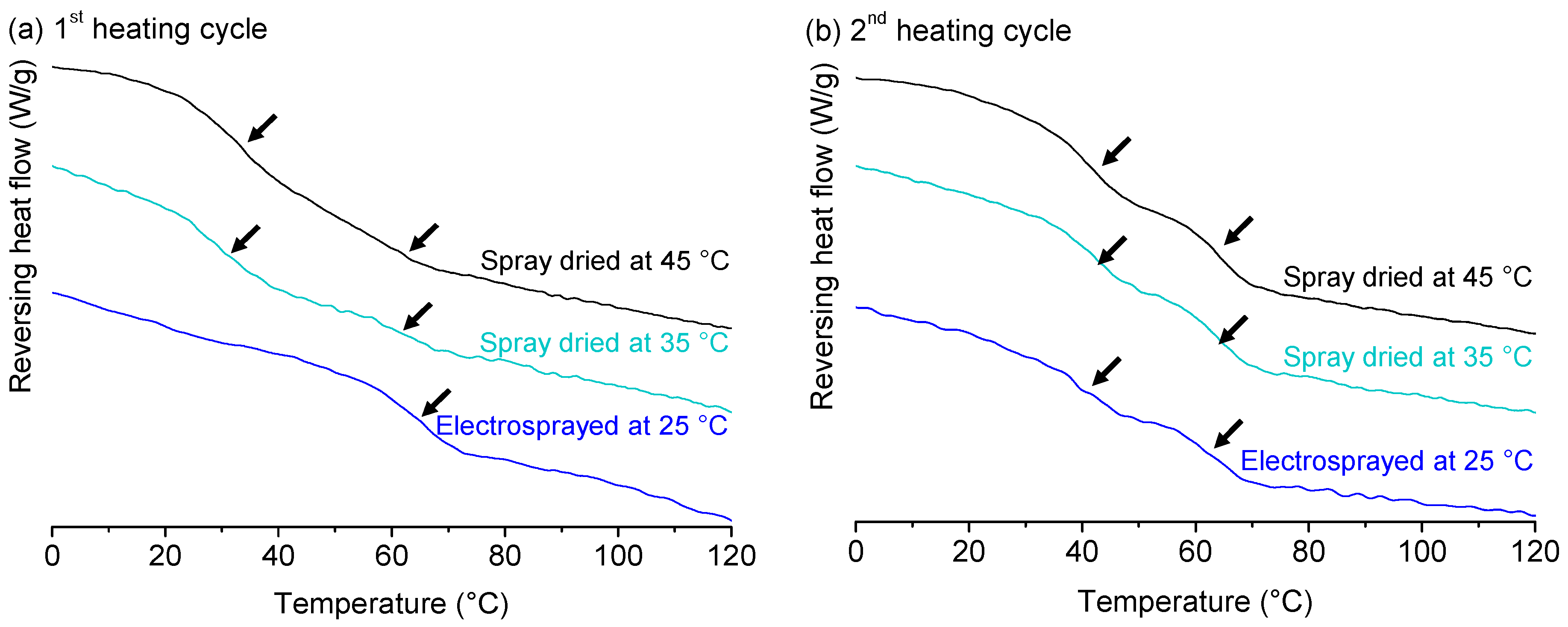
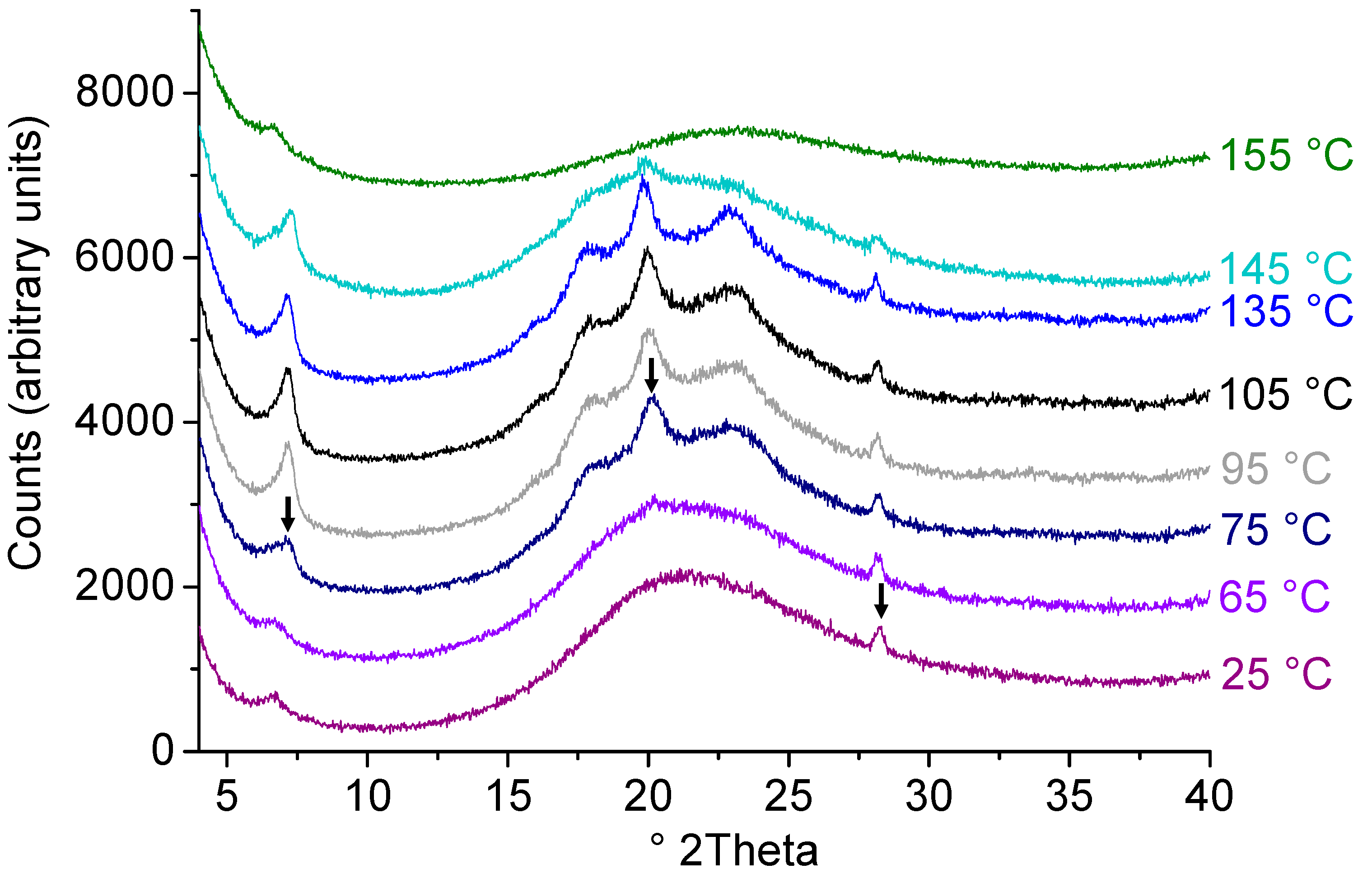
2.4. Effect of Process Conditions on Miscibility
3. Materials and Methods
3.1. Materials
3.2. Preparation of Polymers
- PEtOx:
- SEC with RI detector: Mn,RI = 61.4 kg/mol; ĐRI = 1.15 vs PMMA standards.SEC with MALS detector: Mn,MALS = 40.6 kg/mol; ĐRI = 1.07.
- PPrOx:
- SEC with RI detector: Mn,RI = 19.1 kg/mol; ĐRI = 1.16 vs PMMA standards.SEC with MALS detector: Mn,MALS = 49.6 kg/mol; ĐRI = 1.05.
- PsecBuOx:
- SEC with RI detector: Mn,RI = 26.9 kg/mol; ĐRI = 1.22 vs PMMA standards.SEC with MALS detector: Mn,MALS = 65.9 kg/mol; ĐRI = 1.11.
3.3. Preparation of Polymer Blends, Binary and Ternary Amorphous Solid Dispersions by Spray Drying
3.4. Electrospraying
3.5. Film Casting by Fast Evaporation of the Solvent
3.6. X-ray Powder Diffraction (XRPD)
3.7. Modulated Differential Scanning Calorimetry (mDSC)
3.8. Thermogravimetric Analysis (TGA)
3.9. Solid-State Nuclear Magnetic Resonance (ssNMR)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shah, N.; Sandhu, H.K.; Choi, D.S.; Chokshi, H.P.; Malick, A.W. Fundamentals of Amorphous Systems: Thermodynamic Aspects. In Amorphous Solid Dispersions; Shah, N., Sandhu, H.K., Choi, D.S., Chokshi, H.P., Malick, A.W., Eds.; Springer: New York, NY, USA, 2014; pp. 5–6. ISBN 9781493915989. [Google Scholar]
- Chiou, W.L.; Riegelman, S. Pharmaceutical applications of solid dispersion systems. J. Pharm. Sci. 1971, 60, 1281–1302. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Dai, W.-G. Fundamental aspects of solid dispersion technology for poorly soluble drugs. Acta Pharm. Sin. B 2014, 4, 18–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Mooter, G. The use of amorphous solid dispersions: A formulation strategy to overcome poor solubility and dissolution rate. Drug Discov. Today Technol. 2012, 9, e79–e85. [Google Scholar] [CrossRef] [PubMed]
- Van Speybroeck, M.; Mols, R.; Mellaerts, R.; Do Thi, T.; Martens, J.A.; Van Humbeeck, J.; Annaert, P.; Van den Mooter, G.; Augustijns, P. Combined use of ordered mesoporous silica and precipitation inhibitors for improved oral absorption of the poorly soluble weak base itraconazole. Eur. J. Pharm. Biopharm. 2010, 75, 354–365. [Google Scholar] [CrossRef]
- Warren, D.B.; Benameur, H.; Porter, C.J.H.; Pouton, C.W. Using polymeric precipitation inhibitors to improve the absorption of poorly water-soluble drugs: A mechanistic basis for utility. J. Drug Target. 2010, 18, 704–731. [Google Scholar] [CrossRef]
- Saboo, S.; Mugheirbi, N.A.; Zemlyanov, D.Y.; Kestur, U.S.; Taylor, L.S. Congruent release of drug and polymer: A “sweet spot” in the dissolution of amorphous solid dispersions. J. Control. Release 2019, 298, 68–82. [Google Scholar] [CrossRef]
- Sun, D.D.; Lee, P.I. Probing the mechanisms of drug release from amorphous solid dispersions in medium-soluble and medium-insoluble carriers. J. Control. Release 2015, 211, 85–93. [Google Scholar] [CrossRef]
- Everaerts, M.; Van den Mooter, G. Complex amorphous solid dispersions based on poly (2-hydroxyethyl methacrylate): Study of drug release from a hydrophilic insoluble polymeric carrier in the presence and absence of a porosity increasing agent. Int. J. Pharm. 2019, 566, 77–88. [Google Scholar] [CrossRef]
- Lugtu-Pe, J.A.; Ghaffari, A.; Chen, K.; Kane, A.; Yu Wu, X. Development of controlled release amorphous solid dispersions (CRASD) using polyvinyl acetate-based release retarding materials: Effect of dosage form design. Eur. J. Pharm. Sci. 2018, 124, 319–327. [Google Scholar] [CrossRef]
- Claeys, B.; Vervaeck, A.; Vervaet, C.; Remon, J.P.; Hoogenboom, R.; De Geest, B.G. Poly(2-ethyl-2-oxazoline) as matrix excipient for drug formulation by hot melt extrusion and injection molding. Macromol. Rapid Commun. 2012, 33, 1701–1707. [Google Scholar] [CrossRef]
- Policianova, O.; Brus, J.; Hruby, M.; Urbanova, M. In vitro dissolution study of acetylsalicylic acid solid dispersions. Tunable drug release allowed by the choice of polymer matrix. Pharm. Dev. Technol. 2015, 20, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Fael, H.; Ràfols, C.; Demirel, A.L. Poly(2-ethyl-2-oxazoline) as an alternative to poly(vinylpyrrolidone) in solid dispersions for solubility and dissolution rate enhancement of drugs. J. Pharm. Sci. 2018, 107, 2428–2438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Rubio, L.; Alonso, M.L.; Pérez-álvarez, L.; Alonso, R.M.; Vilas, J.L.; Khutoryanskiy, V.V. Formulation of Carbopol®/poly(2-ethyl-2-oxazoline)s mucoadhesive tablets for buccal delivery of hydrocortisone. Polymers 2018, 10, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abilova, G.K.; Kaldybekov, D.B.; Ozhmukhametova, E.K.; Saimova, A.Z.; Kazybayeva, D.S.; Irmukhametova, G.S.; Khutoryanskiy, V.V. Chitosan/poly(2-ethyl-2-oxazoline) films for ocular drug delivery: Formulation, miscibility, in vitro and in vivo studies. Eur. Polym. J. 2019, 116, 311–320. [Google Scholar] [CrossRef]
- Moustafine, R.I.; Viktorova, A.S.; Khutoryanskiy, V.V. Interpolymer complexes of carbopol® 971 and poly(2-ethyl-2-oxazoline): Physicochemical studies of complexation and formulations for oral drug delivery. Int. J. Pharm. 2019, 558, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Boel, E.; Smeets, A.; Vergaelen, M.; De la Rosa, V.R.; Hoogenboom, R.; Van den Mooter, G. Comparative study of the potential of poly (2-ethyl-2-oxazoline) as carrier in the formulation of amorphous solid dispersions of poorly soluble drugs. Eur. J. Pharm. Biopharm. 2019, 144, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Vergaelen, M. Poly(2-oxazoline)s as matrix excipient for oral drug formulations. Ph.D. Thesis, Ghent University, Ghent, Belgium, 2018. [Google Scholar]
- Glassner, M.; Vergaelen, M.; Hoogenboom, R. Poly(2-oxazoline)s: A comprehensive overview of polymer structures and their physical properties. Polym. Int. 2018, 67, 32–45. [Google Scholar] [CrossRef]
- Sedlacek, O.; Monnery, B.D.; Filippov, S.K.; Hoogenboom, R.; Hruby, M. Poly(2-oxazoline)s-Are they more advantageous for biomedical applications than other polymers? Macromol. Rapid Commun. 2012, 33, 1648–1662. [Google Scholar] [CrossRef]
- Turner, D.T.; Schwartz, A. The glass transition temperature of poly(N-vinyl pyrrolidone) by differential scanning calorimetry. Polymer (Guildf) 1985, 26, 757–762. [Google Scholar] [CrossRef]
- Bühler, V. Kollidon ® Polyvinylpyrrolidone Excipients for the Pharmaceutical Industry; 9th ed.; BASF SE Pharma Ingredients & Services: Ludwigshafen, Germany, 2008; p. 120. [Google Scholar]
- Schoolaert, E.; Merckx, R.; Becelaere, J.; Everaerts, M.; Van Guyse, J.F.R.; Sedlacek, O.; De Geest, B.; Van den Mooter, G.; D’hooge, D.R.; De Clerck, K.; et al. Immiscibility of chemically alike amorphous polymers: Phase separation of poly(2-ethyl-2-oxazoline) and poly(2-n-propyl-2-oxazoline). Macromolecules 2020, in press. [Google Scholar]
- Goh, S.H. Miscible Polymer Blends. In Polymer Blends Handbook; Utracki, L.A., Wilkie, C.A., Eds.; Springer: Dordrecht, The Netherlands, 2014; pp. 1915–2151. [Google Scholar]
- Boel, E.; Koekoekx, R.; Dedroog, S.; Babkin, I.; Vetrano, M.R.; Clasen, C.; Van den Mooter, G. Unraveling Particle Formation: From Single Droplet Drying to Spray Drying and Electrospraying. Pharmaceutics 2020, 12, 625. [Google Scholar] [CrossRef] [PubMed]
- Smeets, A.; Koekoekx, R.; Clasen, C.; Van den Mooter, G. Amorphous solid dispersions of darunavir: Comparison between spray drying and electrospraying. Eur. J. Pharm. Biopharm. 2018, 130, 96–107. [Google Scholar] [CrossRef]
- Demirel, A.L.; Tatar Güner, P.; Verbraeken, B.; Schlaad, H.; Schubert, U.S.; Hoogenboom, R. Revisiting the crystallization of poly(2-alkyl-2-oxazoline)s. J. Polym. Sci. Part. B Polym. Phys. 2016, 54, 721–729. [Google Scholar] [CrossRef]
- Litt, M.; Rahl, F.; Roldan, L.G. Polymerization of cyclic imino ethers. VI. X-ray study of some polyaziridines. J. Polym. Sci. Part. A-2 Polym. Phys. 1969, 7, 463–473. [Google Scholar] [CrossRef]
- Katsumoto, Y.; Tsuchiizu, A.; Qiu, X.; Winnik, F.M. Dissecting the mechanism of the heat-induced phase separation and crystallization of poly(2-isopropyl-2-oxazoline) in water through vibrational spectroscopy and molecular orbital calculations. Macromolecules 2012, 45, 3531–3541. [Google Scholar] [CrossRef]
- Rettler, E.F.; Lambermont-Thijs, H.M.L.; Kranenburg, J.M.; Hoogenboom, R.; Unger, M.V.; Siesler, W.; Schubert, U.S. Water uptake of poly(2-N-alkyl-2-oxazoline)s: Influence of crystallinity and hydrogen-bonding on the mechanical properties. J. Mater. Chem. 2011, 21, 17331–17337. [Google Scholar] [CrossRef]
- Goddeeris, C.; Willems, T.; Houthoofd, K.; Martens, J.A.; Van den Mooter, G. Dissolution enhancement of the anti-HIV drug UC 781 by formulation in a ternary solid dispersion with TPGS 1000 and Eudragit E100. Eur. J. Pharm. Biopharm. 2008, 70, 861–868. [Google Scholar] [CrossRef]
- Yang, Z.; Nollenberger, K.; Albers, J.; Craig, D.; Qi, S. Microstructure of an Immiscible Polymer Blend and Its Stabilization Effect on Amorphous Solid Dispersions. Mol. Pharm. 2013, 10, 2767–2780. [Google Scholar] [CrossRef]
- Marks, J.A.; Wegiel, L.A.; Taylor, L.S.; Edgar, K.J.; Tech, V. Pairwise Polymer Blends for Oral Drug Delivery. J. Pharm. Sci. 2014, 103, 2871–2883. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Hammiche, A.; Pollock, H.M. Modulated differential scanning calorimetry: 4. Miscibility and glass transition behaviour in poly(methyl methacrylate) and poly(epichlorohydrin) blends. Polymer (Guildf) 1996, 37, 5661–5665. [Google Scholar] [CrossRef]
- Moseson, D.E.; Taylor, L.S. The application of temperature-composition phase diagrams for hot melt extrusion processing of amorphous solid dispersions to prevent residual crystallinity. Int. J. Pharm. 2018, 553, 454–466. [Google Scholar] [CrossRef] [PubMed]
- Dudowicz, J.; Douglas, J.F.; Freed, K.F. Two glass transitions in miscible polymer blends? J. Chem. Phys. 2014, 140. [Google Scholar] [CrossRef] [PubMed]
- Lodge, T.P.; Wood, E.R.; Haley, J.C. Two calorimetric glass transitions do not necessarily indicate immiscibility: The case of PEO/PMMA. J. Polym. Sci. Part. B Polym. Phys. 2006, 44, 756–763. [Google Scholar] [CrossRef]
- Dukić, A.; Mens, R.; Adriaensens, P.; Foreman, P.; Gelan, J.; Remon, J.P.; Vervaet, C. Development of starch-based pellets via extrusion/spheronisation. Eur. J. Pharm. Biopharm. 2007, 66, 83–94. [Google Scholar] [CrossRef]
- Lequieu, W.; Van De Velde, P.; Du Prez, F.E.; Adriaensens, P.; Storme, L.; Gelan, J. Solid state NMR study of segmented polymer networks: Fine-tuning of phase morphology via their molecular design. Polymer (Guildf) 2004, 45, 7943–7951. [Google Scholar] [CrossRef]
- Mens, R.; Adriaensens, P.; Lutsen, L.; Swinnen, A.; Bertho, S.; Ruttens, B.; D’Haen, J.; Manca, J.; Cleij, T.; Vanderzande, D.; et al. NMR Study of the Nanomorphology in Thin Films of Polymer Blends Used in Organic PV Devices: MDMO-PPV/PCBM. J. Polym. Sci. Part. A Polym. Chem. 2008, 46, 138–145. [Google Scholar] [CrossRef]
- Bosma, M.; Brinke, G.; Ellist, T.S. Polymer-Polymer Miscibility and Enthalpy Relaxations. Macromolecules 1988, 21, 1465–1470. [Google Scholar] [CrossRef] [Green Version]
- Jorda, R.; Wilkes, G.L. A novel use of physical aging to distinguish immiscibility in polymer blends. Polym. Bull. 1988, 20, 479–485. [Google Scholar] [CrossRef]
- Huggins, M.L. Some properties of solutions of long-chain compounds. J. Phys. Chem. 1942, 46, 151–158. [Google Scholar] [CrossRef]
- Flory, P.J. Themodynamics of high polymer solutions. J. Chem. Phys. 1942, 10, 51–61. [Google Scholar] [CrossRef]
- Chokshi, R.; Shah, N.H.; Sandhu, H.K.; Malick, A.W.; Zia, H. Stabilization of Low Glass Transition Temperature Indomethacin Formulations: Impact of Polymer-Type and Its Concentration. J. Pharm. Sci. 2008, 97, 2286–2298. [Google Scholar] [CrossRef] [PubMed]
- Six, K.; Verreck, G.; Peeters, J.; Augustijns, P.; Kinget, R.; Van den Mooter, G. Characterization of glassy itraconazole: A comparative study of its molecular mobility below Tg with that of structural analogues using MTDSC. Int. J. Pharm. 2001, 213, 163–173. [Google Scholar] [CrossRef]
- Monnery, B.D.; Jerca, V.V.; Sedlacek, O.; Verbraeken, B.; Cavill, R.; Hoogenboom, R. Defined High Molar Mass Poly(2-Oxazoline)s. Angew. Chemie-Int. Ed. 2018, 57, 15400–15404. [Google Scholar] [CrossRef] [PubMed]
- Witte, H.; Seeliger, W. Cyclische Imidsäureester aus Nitrilen und Aminoalkoholen. Justus Liebigs Ann. Chem. 1974, 1974, 996–1009. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample (m/m) | T1H (s) | T1ρH (ms) | |||
|---|---|---|---|---|---|
| T1ρHS | I0S (%) | T1ρHL | I0L (%) | ||
| PEtOx | 2.3 | 0.5 | 18.5 | 6.9 | 81.5 |
| PPrOx | 1.5 | 1.0 | 19.1 | 7.6 | 80.9 |
| PEtOx/PPrOx (1/1) | 1.7 | 0.6 | 17.9 | 6.1 | 82.1 |
| Sample (m/m) | T1H (s) | T1ρH (ms) | |||
|---|---|---|---|---|---|
| T1ρHS | I0S (%) | T1ρHL | I0L (%) | ||
| PEtOx | 1.74 | 0.9 | 14.7 | 7.0 | 85.3 |
| PsecBuOx | 0.78 | 1.3 | 9.4 | 8.9 | 90.6 |
| PEtOx/PsecBuOx (2/1) | 1.28 | 1.5 | 10.0 | 9.3 | 90.0 |
| PEtOx/PsecBuOx (1/1) | 1.10 | 1.9 | 12.0 | 9.7 | 88.0 |
| PEtOx/PsecBuOx (1/2) | 0.97 | 1.6 | 11.0 | 9.6 | 89.0 |
| Drug Loading | 1st Heating Cycle (°C) | 2nd Heating Cycle (°C) | ||||
|---|---|---|---|---|---|---|
| (% m/m) | PEtOx * | PPrOx | PsecBuOx | PEtOx * | PPrOx | PsecBuOx |
| 10% IND | 62.1 (±0.3) | 31.2 (±2.0) | 49.0 (±0.7) | 64.9 (±0.7) | 39.9 (±0.5) | 55.2 (±0.6) |
| 20% IND | 63.5 (±1.4) | 32.8 (±0.2) | 47.8 (±0.9) | 63.7 (±0.3) | 40.6 (±0.5) | 53.7 (±0.6) |
| 30% IND | 60.6 (±0.3) | 33.5 (±0.3) | 49.6 (±0.7) | 62.4 (±0.5) | 41.5 (±0.8) | 52.6 (±0.2) |
| 40% IND | 59.6 (±0.5) | 32.7 (±2.3) | 49.5 (±1.5) | 61.5 (±0.2) | 41.2 (±0.6) | 52.4 (±0.3) |
| Drug Loading | 1st Heating Cycle (°C) | 2nd Heating Cycle (°C) | ||||
|---|---|---|---|---|---|---|
| (% m/m) | PEtOx | PPrOx | PsecBuOx | PEtOx | PPrOx | PsecBuOx |
| 10% ITZ | 56.3 (±1.6) | 32.7 (±0.9) | 55.6 (±1.2) | 64.7 (±0.8) | 42.8 (±0.1) | 56.8 (±0.3) |
| 20% ITZ | 58.9 (±2.0) | 39.2 (±0.9) | 53.8 (±0.7) | 65.3 (±0.4) | 44.0 (±0.2) | 57.1 (±0.9) |
| 30% ITZ | 59.8 (±0.8) | 41.4 (±0.2) | 56.7 (±1.1) | 64.6 (±0.2) | 46.2 (±0.6) | 57.8 (±0.9) |
| 40% ITZ | 60.4 (±0.8) | 43.6 (±0.5) | 56.8 (±0.6) | 65.2 (±0.2) | 47.9 (±0.4) | 59.0 (±0.3) |
| Drug Loading | 1st Heating Cycle (K) | 2nd Heating Cycle (K) | ||||
|---|---|---|---|---|---|---|
| (% m/m) | TgPPrOx | TgPEtOx | TgPPrOx | TgPEtOx | ||
| PEtOx spray dried | / | 336.5 (±0.4) * | 1.10 | / | 336.6 (±0.1) * | 1.07 |
| PPrOx spray dried | 305.0 (±2.2) | / | 314.4 (±1.1) | / | ||
| Spray dried without drug | 306.1 (±0.1) | 330.7 (±1.9) | 1.08 | 317.0 (±0.9) | 336.4 (±0.5) | 1.06 |
| 15% ETR | 333.0 (±4.17) | / | 325.3 (±0.2) | 347.3 (±0.0) | 1.07 | |
| 15% FEN | 291.3 (±0.1) | 313.5 (±1.2) | 1.08 | 297.3 (±0.2) | 323.1 (±0.4) | 1.09 |
| 15% IBU | 287.9 (±0.8) | 305.3 (±0.6) | 1.06 | 295.4 (±0.6) | 314.5 (±0.4) | 1.06 |
| 15% MIC | 295.9 (±2.0) | 317.3 (±2.9) | 1.07 | 302.2 (±1.8) | 326.5 (±0.7) | 1.08 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Everaerts, M.; Tigrine, A.; de la Rosa, V.R.; Hoogenboom, R.; Adriaensens, P.; Clasen, C.; Van den Mooter, G. Unravelling the Miscibility of Poly(2-oxazoline)s: A Novel Polymer Class for the Formulation of Amorphous Solid Dispersions. Molecules 2020, 25, 3587. https://doi.org/10.3390/molecules25163587
Everaerts M, Tigrine A, de la Rosa VR, Hoogenboom R, Adriaensens P, Clasen C, Van den Mooter G. Unravelling the Miscibility of Poly(2-oxazoline)s: A Novel Polymer Class for the Formulation of Amorphous Solid Dispersions. Molecules. 2020; 25(16):3587. https://doi.org/10.3390/molecules25163587
Chicago/Turabian StyleEveraerts, Melissa, Ali Tigrine, Victor R. de la Rosa, Richard Hoogenboom, Peter Adriaensens, Christian Clasen, and Guy Van den Mooter. 2020. "Unravelling the Miscibility of Poly(2-oxazoline)s: A Novel Polymer Class for the Formulation of Amorphous Solid Dispersions" Molecules 25, no. 16: 3587. https://doi.org/10.3390/molecules25163587