
Scaffold Modifications in Erythromycin Macrolide Antibiotics. A Chemical Minireview

Department of Chemistry, University of Oslo, 0315 Oslo, Norway
Molecules 2020, 25(17), 3941; https://doi.org/10.3390/molecules25173941
Submission received: 27 May 2020
/
Revised: 25 August 2020
/
Accepted: 25 August 2020
/
Published: 28 August 2020
(This article belongs to the Special Issue Medicinal Chemistry in Europe II)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Clarithromycin and congeners are important antibacterial members of the erythromycin A 14-membered macrocyclic lactone family. The macrolide scaffold consists of a multifunctional core that carries both chemically reactive and non-reactive substituents and sites. Two main approaches are used in the preparation of the macrolides. In semisynthesis, the naturally occurring macrocycle serves as a substrate for structural modifications of peripheral substituents. This review is focused on substituents in non-activated positions. In the total synthesis approach, the macrolide antibiotics are constructed by a convergent assembly of building blocks from presynthesized substrates or substrates prepared by biogenetic engineering. The assembled block structures are linear chains that are cyclized by macrolactonization or by metal-promoted cross-coupling reactions to afford the 14-membered macrolactone. Pendant glycoside residues are introduced by stereoselective glycosylation with a donor complex. When available, a short summary of antibacterial MIC data is included in the presentations of the structural modifications discussed.
1. Introduction
Clarithromycin (B, Figure 1) and congeners are antibacterials from the erythromycin A 14-membered lactone family. The macrolides exert their drug action by inhibition of bacterial protein synthesis in sensitive pathogens [1,2,3]. Members of each antibiotic class share a common core structure or scaffold. Several reports describe efforts to improve antimicrobial efficacies, widen the microbial spectra, and improve activity against pathogens that have become partly resistant to the macrolides. In most structural investigations, the core of the antibiotic is left intact to preserve the natural activity of the scaffold. The chemical groups at the periphery of the scaffold are modified. The multifunctional erythromycin scaffold carries chemically reactive as well as chemically inert substituents. This review describes work on modifications of chemically inert carbon substituents and non-activated sites in the scaffold congeners of clarithromycin ketolides [4,5,6].
The semisynthetic drug, clarithromycin, is a 6-methyl ether of the parent 14-membered erythromycin A. Removal of the 3-(L)-desosamine sugar residue in erythromycin and oxidation of the resultant free 3-hydroxy group afford highly active 3-oxo antibacterials. The potencies of some of these compounds are further improved after annulation reactions that afford cyclic 11,12-carbamates, where the 11,12-cyclic carbamate linkage occupies the positions of the two 11,12-hydroxy groups in erythromycin (Scheme 1). The methodology for the preparation of cyclic 11,12-carbamate macrolides was developed by Baker and coworkers [7].
Erythromycin derivatives are preferably prepared by semisynthetic methodology. Total synthesis has remained a challenge. Recent work, however, has opened up for total syntheses of modified structures (vide infra). Intermediates from biogenetic engineering are incorporated into the products.
The presentation in this report is arranged according to the increase in numbering of the sites in the macrolide core, starting with the ester carbonyl carbon as the 1-position (Structure A in Figure 1). Clarithromycin is formally formed by replacement of the 6-hydroxy group in erythromycin by a methoxy group. In telithromycin (C) and solithromycin (D), the 3-glycocyl function has been cleaved by hydrolysis and the resultant 3-hydroxy group oxidized to afford the 3-ketolide. In a similar way, cethromycin (E) is a 3-ketolide, but the 6-hydroxy group has been converted into an ether function, where the O-sidechain is bridged by an allyl group onto the 3-position in quinoline. Clarithromycin 3-ketolide (F) is available as above from clarithromycin.
2. Synthesis
A series of reactions, starting at the C12-hydroxy group in a substrate such as compound 1 (Scheme 1) to afford a cyclic C11,C12-carbamate 3, is frequently referred to as the Baker protocol. The carbamoyl reaction of the 10,11-anhydroerythromycin substrate 1 (Scheme 1) is carbamoylated at the 12-hydroxy group by carbonyl diimidazole (CDI) using NaH in DMF or THF to afford the carbamate 2. A subsequent treatment with aqueous ammonia in acetonitrile leads to a 2-step cyclization reaction and formation of 11-deoxy-11-carboxyamino-6-O-methylerythromycin A 11,12-cyclic ester 3. The Michael reaction requires base catalysis and the rate of addition is fastest in polar solvents such as 10% aqueous acetonitrile or DMF. With primary amine reactants, a variety of N-substituted products (3) is formed. The major product from the intramolecular Michael reaction has the natural (R)-configuration of erythromycin at the C10-position. The stereochemistry at C10 can be established by NMR spectroscopy (vide infra) [7].
Scheme 1.
Reaction and conditions: (i) CDI, NaH, DMF, THF, 0 °C to rt, 4–5 h; (ii) R1NH2, DMF, or MeCN (10% aq), rt, 1–3 d. Abbreviation: CDCI, 1,1′-carbonyldiimidazole.
Scheme 1.
Reaction and conditions: (i) CDI, NaH, DMF, THF, 0 °C to rt, 4–5 h; (ii) R1NH2, DMF, or MeCN (10% aq), rt, 1–3 d. Abbreviation: CDCI, 1,1′-carbonyldiimidazole.

2.1. C2-Derivatives
The acylimidazolide 5 is prepared in a similar manner from the corresponding C12-hydroxy 2,3- and 10,11-diene 4 by reactions with CDI (Scheme 2) [8]. Further reactions with ammonia or primary amines in aqueous acetonitrile or DMF afford cyclocondensation and formation of the fused cyclocarbamate 6. Methanolysis at room temperature cleaves off the 2′-protecting group. The product 7 is an epimeric mixture at C10-position, but frequently, the product formation shows high preference for the natural (10R)-epimer. Epimeric mixtures can be isomerized under basic conditions towards the more stable (10R)-isomer. N-Alkylated carbamates are obtained when a primary amine is used instead of ammonia. Reductive alkylation of the carbamate nitrogen after an ammonia cyclization will also yield N-substituted derivatives.
Antibacterials: The cyclic carbamates show good antibacterial activity against inducibly MLS-resistant S. aureus A 5177, against macrolide-resistant S. Pyogenes PIU 2548 and S. pneumonia 5649 m, but are inactive against Gram-negative organisms. The C10-epi analogues are significantly less active than their natural C10-counterparts. In vivo activity is inferior to the activity of clarithromycin.
Carbazates are formed when hydrazine is the amine reactant. The carbamate 5 reacts with hydrazine in DMF to afford 2,3-anhydro-5-O-desosaminyl-11-hydrazo-6-O-methylerythronalide A 11,12-carbamate 8 (Scheme 3). Formation of an epimeric mixture of the (10R)-carbazate (39%) and the (10S)-carbazate (49%) 8 is shown [9]. Reductive alkylations with aldehydes using NaBH3CN and acetic acid afford N-alkylated products. With benzaldehyde as the reactant, the N-benzyl-2,3-anhydro derivative 9 is formed from the carbazate 8 (Scheme 3) [10].
Antibacterials: The antibacterial potency of 11,12-cyclic carbazates is comparable to the potency of erythromycin A. N-Alkylation improves their potency as antibacterials. C10-epimers are generally less active than their natural C10-epimers. The compounds are moderately active in in vivo studies in mice [9,10].
2.2. Reactivity of the C2-Methyl Group
The C2-position in the 1,3-dioxo substrate 10 is chemically activated by the adjacent oxo groups and is readily enolized. The 6-O-protected ketolide 10, as an enolate, reacts readily with electrophilic reagents to afford 2-halo, 2-benzenesulfenyl, or 2-benzeneselenyl intermediates (11) (Scheme 4). The latter are substrates for nucleophilic displacements [11]. Sulfides, and especially selenides, are highly activated by oxidation, and the oxides readily suffer base promoted elimination with formation of a 2-methylene product 12 (Scheme 4). Further oxidations of the C2-methylene compound 12 using hydrogen peroxide reagents afford diols as well as epoxide 13.
Antibacterials: The compounds are, in general, active antibacterials. MIC values are available.
2.3. Fluoro Derivatives
In medicinal chemistry, fluorine as a hydrogen bioisostere is used to alter the polarity, lipophilicity, or metabolic stability of a drug. The inclusion of fluorine may be beneficial for activity, protein binding, and cell penetration. Several drugs contain fluoro substituents. Fluorides are included in this review. In a macrolactone 3-ketolide, the C2-position is activated for electrophilic substitutions after initial enol ether or ester formation. The enolized substrate is halogenated and fluorinated. The 2-fluoro derivative is a stable compound, whereas corresponding chloro or bromo halides readily undergo substitution or elimination reactions. The C2-F substituent prevents the 3-keto function from enolization. A synthesis of a 2-fluoro-6-O-propargyl-11,12-cabamate 16 is shown in Scheme 5 [12]. The 6-O-propargylic ketones and electron-deficient 6-propargylic aromatic derivatives resist enolization of the β-keto ester. The propargyl 2-fluoroketolide 15 in Scheme 5 is prepared directly from enol species arising from the ketone 14 by treatment with NaH under ionic conditions using N-fluorobenzene sulfonamide for the fluorination. The product 15 is a single diastereomer. Trans-coupling reactions with acyl bromides or chlorides are promoted by Pd-catalysis under Sonogashira conditions to deliver carbocyclic or heterocyclic alkynyl ketones. Subsequent deacylation of the 2′-protected species is effected by dissolution of the product in cold methanol to afford the target compound 16.
Antibacterials: The MIC data for the compounds are consistent with high antibacterial activity.
Solithromycin (D, Figure 1) is a powerful 2-fluoroantibiotic that can be prepared by click chemistry from the azide 17 and 3-ethynylaniline. The (3 + 2) dipolar cycloaddition is effected by Cu(I)-catalysis (Scheme 6) [13,14]. The catalyst is generated in situ from CuSO4, sodium ascorbate, and the terminal alkyne in tBuOH:water (1:1) at rt, 24 h, yield 70–90% of the anti-(1,4)-triazole 19. 2-Fluoro-11-pendent-ω-azidobutyl-ketolides such as the azide 17 is converted to 4-substituted [1,2,3]-triazole as a single regional isomer by reaction with the corresponding acetylene to afford 2-fluoro-ketolide 19 [13]. Hetaryl derivatives such as 3-thienyl derivatives are prepared in the same manner.
Antibacterials: Some of the compounds possess equipotent MIC values with solithromycin and are active against wild-type and resistant isolates of Gram-positive S. aureus strain, E. coli strains, and drug multiresistant pathogens.
Ribosome-templated azide-alkyne cycloadditions are achieved in situ by click chemistry in a similar manner. With ruthenium catalysis, the regiochemistry is changed from structure 19 to the syn-(1,5)-triazole 20 [14].
Antibacterials: Solithromycin is a very potent antibiotic. Four analogues from the ribosome-templated derivatives in situ display similar therapeutics indices as solithromycin.
2.4. 9-Aza Derivatives
2.5. Synthesis of 4-Desmethyl
Extensive synthetic organic chemistry is involved in the total syntheses of 14-membered macrolide scaffolds and falls outside the scope of this review. The presentation herein is restricted to syntheses that start at the step for the cyclization reactions and afford 14-membered macrolide scaffolds. The antibacterial importance of methyl groups hinged to the macrocycle in non-activated positions is determined indirectly after total syntheses of desmethyl macrolide scaffolds. Scheme 8 shows the preparation of the 4-desmethyl macrolide 28. The telithromycin intermediate 24 is prepared from the linear hydroxy acid 23 by a macrolactonization reaction to afford 24 in 65% yield [16]. The hydroxy acid 23 is prepared by a multistep process. Stereoselective glycosylation of 24 with the donor of the desosamine sugar 25 affords the glycoside 26. The Baker protocol subsequently delivers the macrolactone framework that affords the ketolide 28 [16].
Antibacterials: 4-Desmethyl telithromycin is equipotent with telithromycin against wild-type bacteria but is 4-fold less potent against the A2058G mutant.
2.6. 4,10-Didesmethyl-Telithromycin Derivatives
Total synthesis involves several reaction steps to afford a linear chemo and stereoselectively substituted substrate 29 for the synthesis of macrolide 33 without methyl groups in the 4- and 10-positions (Scheme 9). The intermediate 29 is cyclized by a Grubbs(II)-catalyzed RCM (ring closing metathesis) reaction to afford the cyclic structure 30 in 60% yield [17]. Reduction of the 9-oxo group and protection of the resultant hydroxy group afford intermediate 31 that is glycosylated using the desosamine donor 25 to generate intermediate 32. The cyclic C11-C12 carbamate moiety is installed by reactions with CDI and NaH, and subsequently, by reactions with a 1,4 diaminobutane. Methanolysis removes the protecting groups in the product and delivers the desmethyl macrocycle 33.
Antibacterials: 4,10-Didesmethyl telithromycin is less potent than telithromycin but four times more active than the 4,8,10-tridesmethyl congener.
2.7. 4,8,10-Tridesmethyl-Telithromycin Derivatives
Total synthesis of (-)-4,8,10-tridesmethyl-telithromycin analogue of the telithromycin ketolide involves a ring closing reaction for construction of the 14-membered ring. Glycosylation by the donor complex 25, the Baker sequential one-pot carbamoylation, and intramolecular amine reactions afford the analogue 40 (Scheme 10) [18].
Antibacterials: The products demonstrate antibacterial activity against several wild-types and resistant bacterial strains but are, in general, less potent than the parent telithromycin.
2.8. Cethromycin. Ketolides
The 14-membered macrolactone 4,8,10-tridesmethyl of cethromycin (E, Figure 1) is prepared from the linear chain substrate 41 that is synthesized by a multistep process (Scheme 11) [19]. Cyclization is achieved by the addition of CrCl2 and catalytic amount of NiCl2 to a solution of 41 in degassed DMSO with stirring at rt for 20 h. The product 42 (51%) is a 1:1 mixture of diastereomeric allylic alcohols at C9. Subsequent oxidation of 42 with DMP affords the 9-oxo product 43 that is glycosylated by a reaction with the thiopyrimidine desosamine donor 25 to afford 44. In the installation of the cethromycin 3-quinolinyl sidechain, 44 is subjected to a modification of the Heck reaction with 3-bromoquinoline to afford 45 in 60% yield. Fluoride mediated cleavage of silyl ether protecting groups using TBAF and a subsequent DMP oxidation of the resulting hydroxy intermediate afford the corresponding 3,9-dioxo compound. The Baker protocol with NaH and CDI followed by ammonium hydroxide treatment afford the oxazolidinone 46.
Antibacterials: The inhibitory activity against E. coli and S. aureus in the telithromycin series increases with the number of methyl groups. The desmethyl cethromycin analogue exhibits similar potency to that of telithromycin and is more active than the desmethyl telithromycin analogues against wild-type E. coli and A2058G mutant strains.
A summary of the synthetic achievements in the total synthesis program of desmethyl macrolide antibiotics is available [20].
Antibacterials: Activity data for the influence of the C4-, C8-, and C10-methyl groups in the macrolactones are available from screening programs against E. coli and S. aureus bacteria. All compounds tested are inactive against E. coli. A significant increase in antibacterial potency results from methyl group additions to the macrolactone scaffold (vide supra). This finding is attributed to changes in the macrolactone conformation caused by introduction of the C8- and C10-methyl groups that serve to rigidify the molecule [20].
2.9. Synthesis of C8-Fluoro Derivatives
Fluorination in the 8-position in erythromycin is achieved via the cyclic carbamate 48. The latter is prepared in a reaction between 47 and phosgene with pyridine as the base. In the acylation process, the sugar dimethylamino substituent loses one of the methyl groups (Scheme 12). 1-(Chloromethyl)-4-fluoro-1,4-diazabicyclo-[2,2,2]octane bistetrafluoroborate 49 (SelectfluorTM) is an effective agent for fluorination. In reactions with 8,9-anhydro-N-desmethylerythromycin A 2′,3′-carbamate-11,12-carbonate-6,9-hemiacetal, a mixture of the tautomers of hemiketal 50a and the hydroxy-ketone 50b is obtained. Cleavage of the product with a weak acid provides compound 51 in 88% yield. DMP will oxidize the 3-hydroxy derivative 51 to the corresponding 3-ketolide, which is a potential intermediate for the preparation of 8-fluorinated erythromycin cyclic 2´, 3´-carbamates (vide supra) [21].
Antibacterials: The MIC values against ATTCC 25923 and E. coli 2592 show low activity in comparison with clarithromycin as the reference macrolide. The low potency may in part be due to loss of the basic tertiary amino center in the desosamine sugar moiety that has been converted to the N-acylated species 51.
2.10. C9–C10 Unsaturation
A Claisen rearrangement of the 9-vinyl ether 52 affords C9-C10 unsaturation when compound 52 is thermolyzed in an aprotic solvent to afford the 9,10-alkene 53 (Scheme 13) [22]. Further treatment with acid at elevated temperature removes the C3-cladinose sugar. Selective oxidation of the 3-hydroxy group by DMP affords the aldehyde 54. Methanolysis removes the protecting group at the 2′-position. Reductive amination using sodium cyanoborohydride in ethanol affords the amine 55 that is deprotected by methanolysis to furnish the 9,10-unsaturated macrolide 56.
Antibacterials: Biodata are not presented in the report.
2.11. 9-Azamacrolides
Replacement of the 9-keto carbon by an amino nitrogen atom provides 9-azamacrolides. A total synthesis of the 9-azamacrolide 60 is shown in Scheme 14. The building blocks 57 and 58 are joined in a reductive amination reaction of the keto function to provide the linear chain product 59. The latter is a substrate for macrocyclization by thermolysis (1 mM in chlorobenzene) to yield the macrolactone 60 in 78% yield [15]. The cyclization occurs without significant negative influence from the secondary amino function or the secondary alcohol. The reaction is promoted by the rigid cyclic carbamate function on the left-hand side of the molecule that brings the acyl ketene function and the reactive secondary alcohol into favorable proximity. The thermolysis proceeds through a transient acyl intermediate. Incorporation of the building block for azide-alkyne dipolar cycloaddition delivers structure 61 (which corresponds to 2-desfluorosolithromycin).
2.12. Transformations in the C10-Position
2.12.1. Activation of the C10-Methyl Group
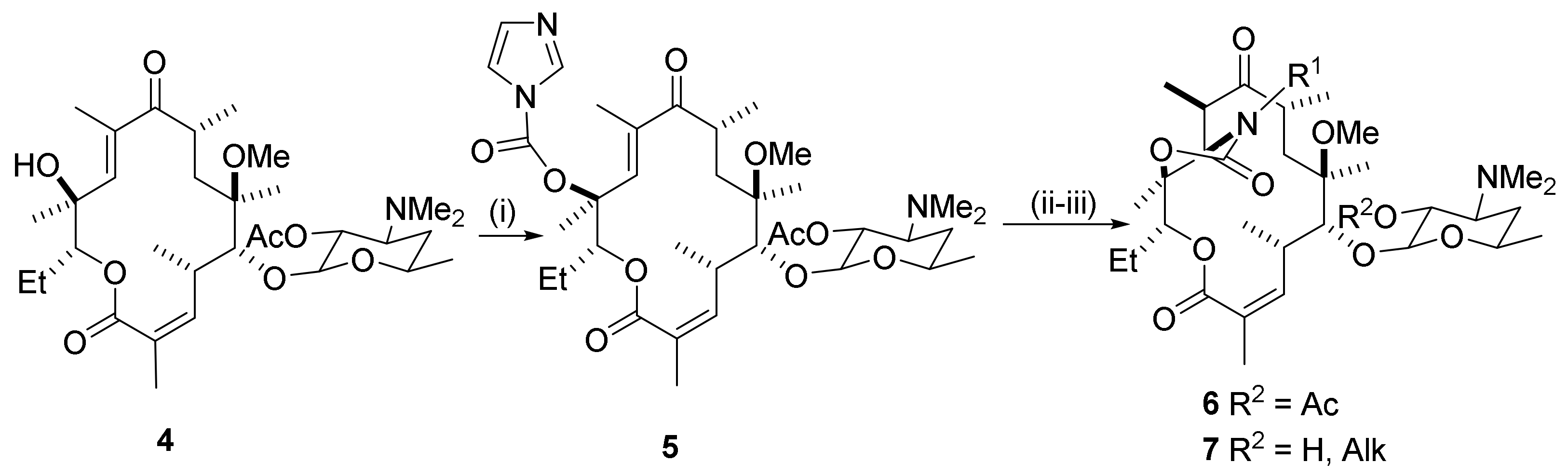
Reactions leading to transformations of the C10-methyl group in the erythromycin scaffold are illustrated in Scheme 15. The 6-methoxy function in clarithromycin may be regarded as a protected 6-hydroxy group in erythromycin. Before the reaction, clarithromycin is transformed into the 10,11-anhydro-O6-methylerythromycin substrate 62 (Scheme 15) and the tertiary 3′-amino group is oxidized chemoselectively to the N-oxide 63 using hydrogen peroxide in methanol [23]. The reaction in methanol proceeds almost to completion, affording the oxide in 84% yield. The N-oxide function serves to protect the amino-nitrogen in the ensuing reaction. Use of NBS or NCS for halogenation of the oxide 63 affords the allylic acetate 64. NCS seems to be the better reagent (88% yield). This is a key reaction for the chemical activation of the C10-methyl group. With NBS, some brominated byproducts may be isolated. Deoxygenation of the N-oxide by 1,2-bis (diphenylphosphanyl) ethane affords the amine 65. Triphenylphosphane is an alternative reagent for deoxygenation. The 2′-hydroxy sugar group is acetyl-protected (66) and the acetate is oxidized chemoselectively by DMP to afford the ketolide 67. CDI in the presence of NaH in THF affords the 12-acylimidazole 68. Further treatment with aqueous ammonia in acetonitrile leads to a two-step cyclization by the Baker procedure. The internal Michael addition results in acetate elimination to deliver the 10-methylene product 69 [23,24]. The stereochemistry in the cyclic carbamate formation is established by NMR spectroscopy and has been verified by single-crystal X-ray analysis. N-Substituted cyclic carbamates are formed (vide infra) with primary amines. Other allylic leaving groups at the C10-carbon will afford 10-methylene products. An example is provided by the 10-azidomethyl derivative 70 in a reaction with aqueous ammonia (Scheme 16) [23].
2.12.2. C10-Methyl Aminations
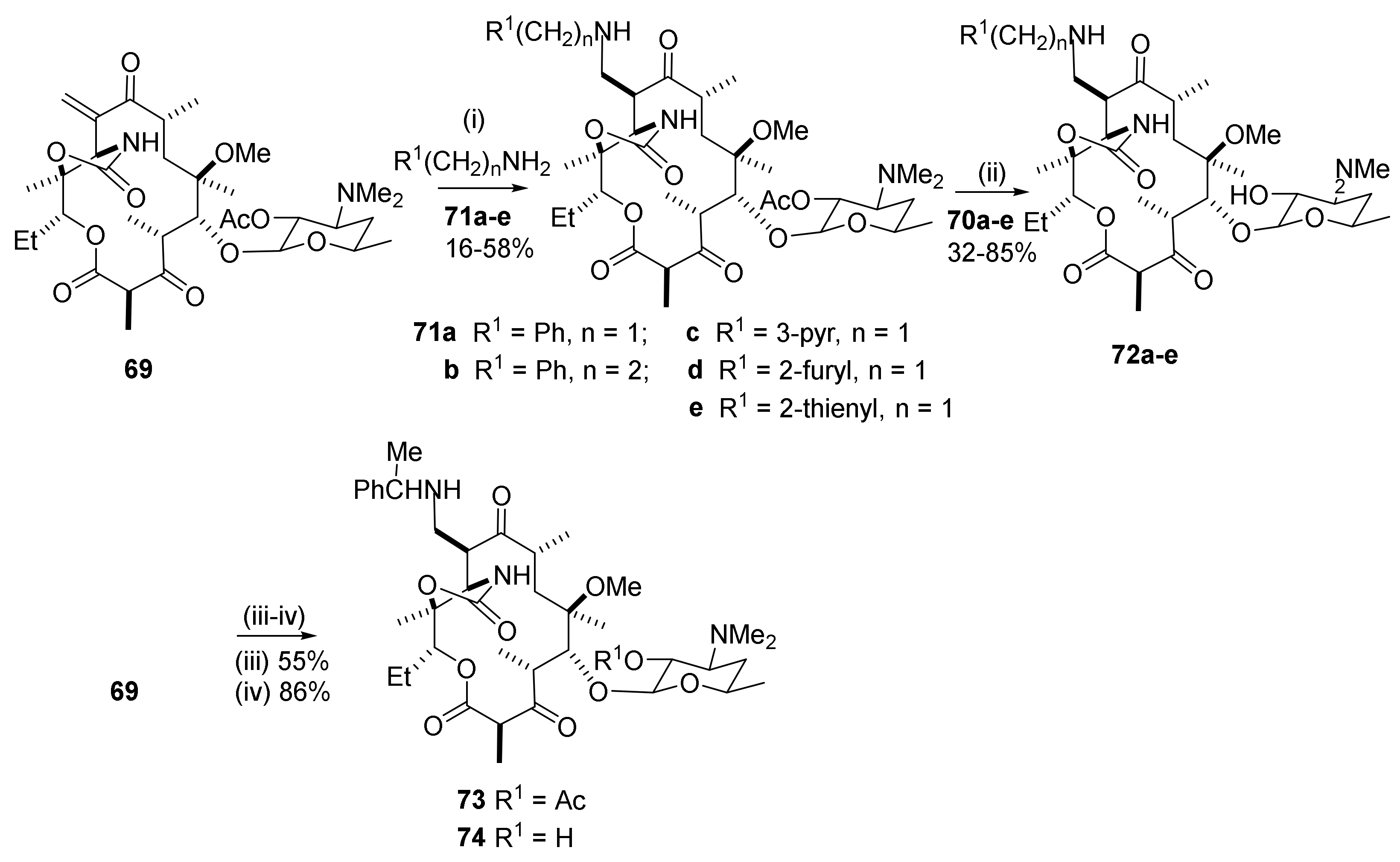
The C10-methylene group in structure 69 is part of a π-electron-deficient α,β-unsaturated carbonyl system that reacts with heteroatom nucleophiles (Scheme 17) [25]. Benzylamine is a good nucleophile in the conjugated amine addition. Phenethylamine reacts in the same manner. Pyridine-3-amine has a π-deficient system with deactivation of the amino group. The yield of the adduct 71c is low. The furyl-2- and thienyl-2-amines are π-excessive systems and afford the furyl derivative 71d and the thienyl derivative 71e in high yields. Cleavage of the ester group in compound 71 by methanolysis affords the 2′-hydroxy compound 72. Only one C10-stereoismer is obtained. The product is identified by its diagnostic signal in 1H NMR from H-11, which resonates as a singlet at ca. 3.9 ppm (3.82, 3.91, and 3 × 3.93 ppm) and is, therefore, assigned the (R)-configuration. In closely related clarithromycin derivatives, it has been observed that the vicinal H-11 proton in the (10S)-isomer resonates as a doublet, but as a singlet in the (10R)-isomer. The C10-epimeric preference will depend on structural features as well as experimental conditions. The natural (10R)-isomer is thermodynamically more stable than its (10S)-isomer (vide supra). The reactivity is affected by the nucleophilicity of the amino nitrogen. Aniline fails to form adducts under the conditions used in the reactions with alkylamines. Non-bonded interactions are also important. The secondary N-methylbenzylamine fails to form adducts. Shifting the methyl group to the α-benzyl carbon, however, gives an active amine affording the adduct 73.
Antibacterials: The in vitro MIC values against strains of the respiratory pathogens S. pneumonia and S. aureus show antibacterial potencies and profiles close to the values of clarithromycin together with improved activities against efflux-resistant S. pneumonia BAA1402M (mef), as well as against inducibly resistant strains of S. aureus BAA976M (mef), and some improvement towards BAA977 iMLS (erm). The MIC values show that the C10-methylene compound is less active than the reference compound, clarithromycin. Activity is greatly improved for the more lipophilic benzylated N-carbamoyl homologue.
2.12.3. C10-α-Heteraspirane Formation
The C10-carbon is transformed into a quaternary spirocarbon by (3 + 2) dipolar cycloaddition reactions [25]. Scheme 18 shows a reaction between trimethylsilyl diazomethane and substrate 69 in dichloromethane. With electron-deficient alkenes, the nucleophilic diazoalkene adds to the more electrophilic β-carbon of the alkene. The initially formed 3H-4,5-dihydropyrazole is unstable and is isomerized to 1H-4,5-dihydropyrazole. The cycloadduct 75 is obtained in 33% yield after 48 h at 0 °C. A significant non-bonded interaction from the β-substituent of the macrolide core may, in part, explain the slow reaction. An elimination reaction in the initially formed spirane affords the final product 75. H-11 in 75 has no vicinal protons for coupling in NMR and resonates as a singlet at 3.56 ppm. Only one singlet is seen for the H-11 proton in support of a pure epimer. Two NH signals are present as singlets at 5.50 and 6.33 ppm, in accordance with structure 75.
Antibacterials: The MIC values for the C10-spiroketolide do not differ significantly from the figures for the reference compound, clarithromycin.
2.12.4. C10-Carbylation Reactions
Stabilized carbanions from the sodium salts of diethyl malonate, malononitrile, or ethyl 3-oxo-3-phenylpropanoate react at the C10-hinged methylene carbon to afford adducts 76 (Scheme 19). N-Benzyl-2-amino-2-cyanoacetamide reacts similarly with formation of the adduct 78 that is hydrolyzed to the alcohol 79 [25].
Antibacterials: The MIC values for the malonyl esters are close to the values for the reference compound, clarithromycin.
2.12.5. C10-Trans-Coupling by Transition Metal Catalysis
Palladium-mediated trans-coupling reactions under Stille conditions with the acetate 62 afford aryl, alkenyl, and alkynyl products. Formation of 10-phenyl, 10-phenylethynyl, or 10-phenylethenyl products 80 is shown in Scheme 20. Trans-coupling under Suzuki conditions using organoboranes may be preferable to the Stille conditions when it is important to exclude any toxic tin-containing impurities in products that are to be used for clinical studies. The coupling product 80 is protected by acetylation of the 2′-hydroxy group before the 3-OH group is oxidized under DMP conditions to afford the 3-oxo derivative 81. A subsequent reaction with CDI gives the corresponding 12-imidazolylcarbonyl ester that affords target compound 82 in aqueous ammonia. Methanolysis provides the deprotected product 83 as a mixture of the two C10-epimers. Separation or partial separation is achieved by chromatography. The major isomer has the (10R)-configuration. The isomer distribution in 83a and 83c is 2:1, and in 83b, it is close to 1:1. [25].
Antibacterials: The MIC values for the phenyl derivative are similar to the values for the reference compound, clarithromycin.
Abbreviation: NMP, N-methylpyrrolidin-2,5-dione. Unsaturation in the bridge can be removed by catalytic hydrogenation, as in the saturation of substrate 84 [24]. The C10-epimeric mixture 84 affords the saturated C10-epimers 85a and 85b (Scheme 21).
Structure 90 in Scheme 22 has the same ether-sidechain as cethromycin (E, Figure 1) compound E) [26]. The synthesis of substrate 86 follows largely the methodology developed for the preparation of the clarithromycin ketolide 67 outlined in Scheme 13. Acylation with CDI in the presence of sodium hydride and cyclization of the product with ammonia affords the cyclic carbamate 87. The triple bond in the propargyl ether moiety is reduced to a double bond under Lindlar conditions. The resultant allyl intermediate 88 is subjected to trans-coupling with 3-bromoquinoline in acetonitrile in a reaction promoted by Pd(OAc) and tri-o-tolylphosphine to produce the 6-(quinolin-3-yl) Heck product 89. Adduct formation between diamine reactants and 89 affords the C10-diamine 90 by analogy to the procedure in Scheme 17.
Antibacterials: The in vitro MIC values show little or no alteration in potency compared to the values for cethromycin against the strains S. pneumonia ATCC49619, S. pneumonia ATCC1402, and S. pneumonia ATCC1407.
2.12.6. Reactions in the C12-Position
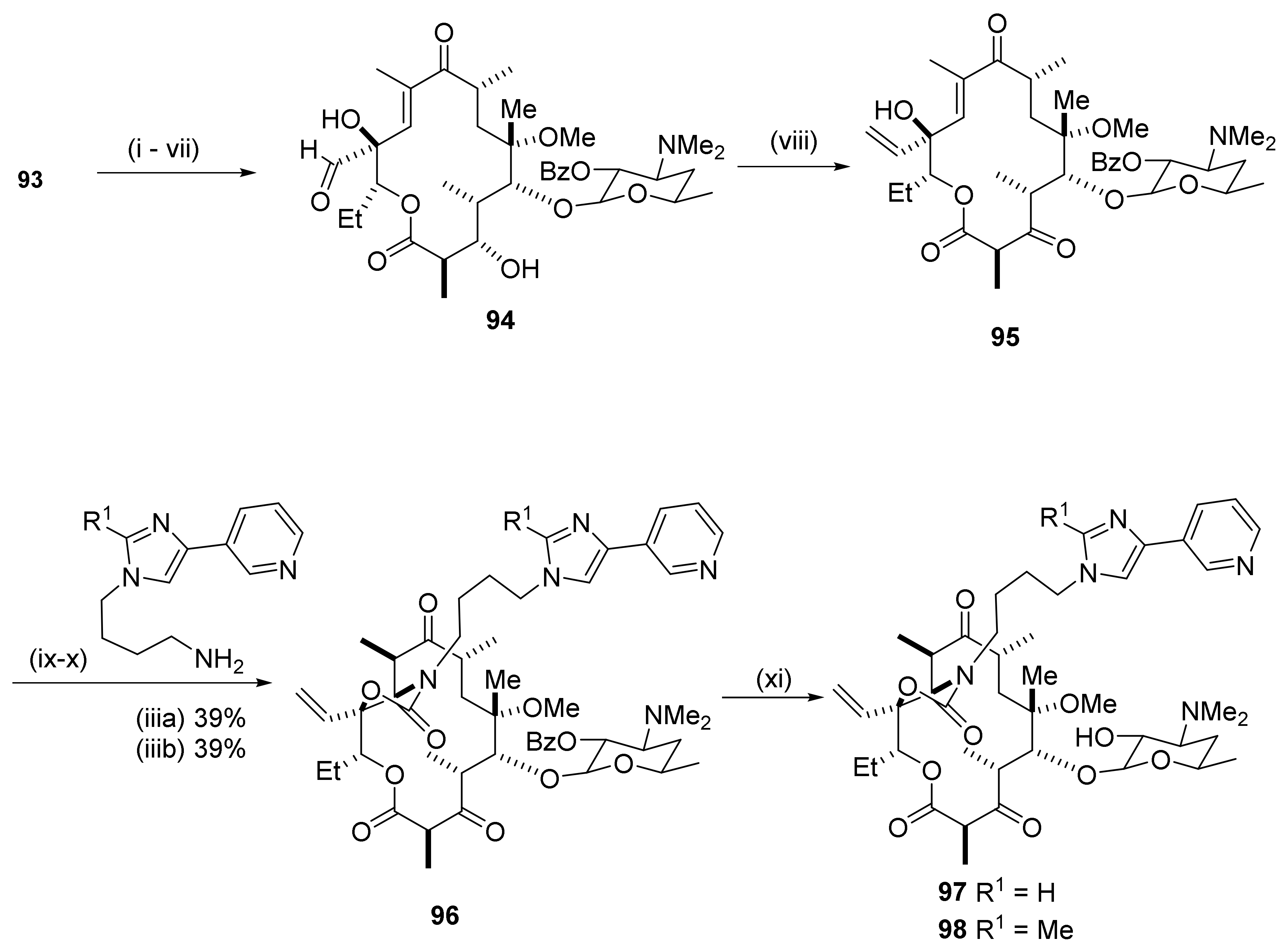
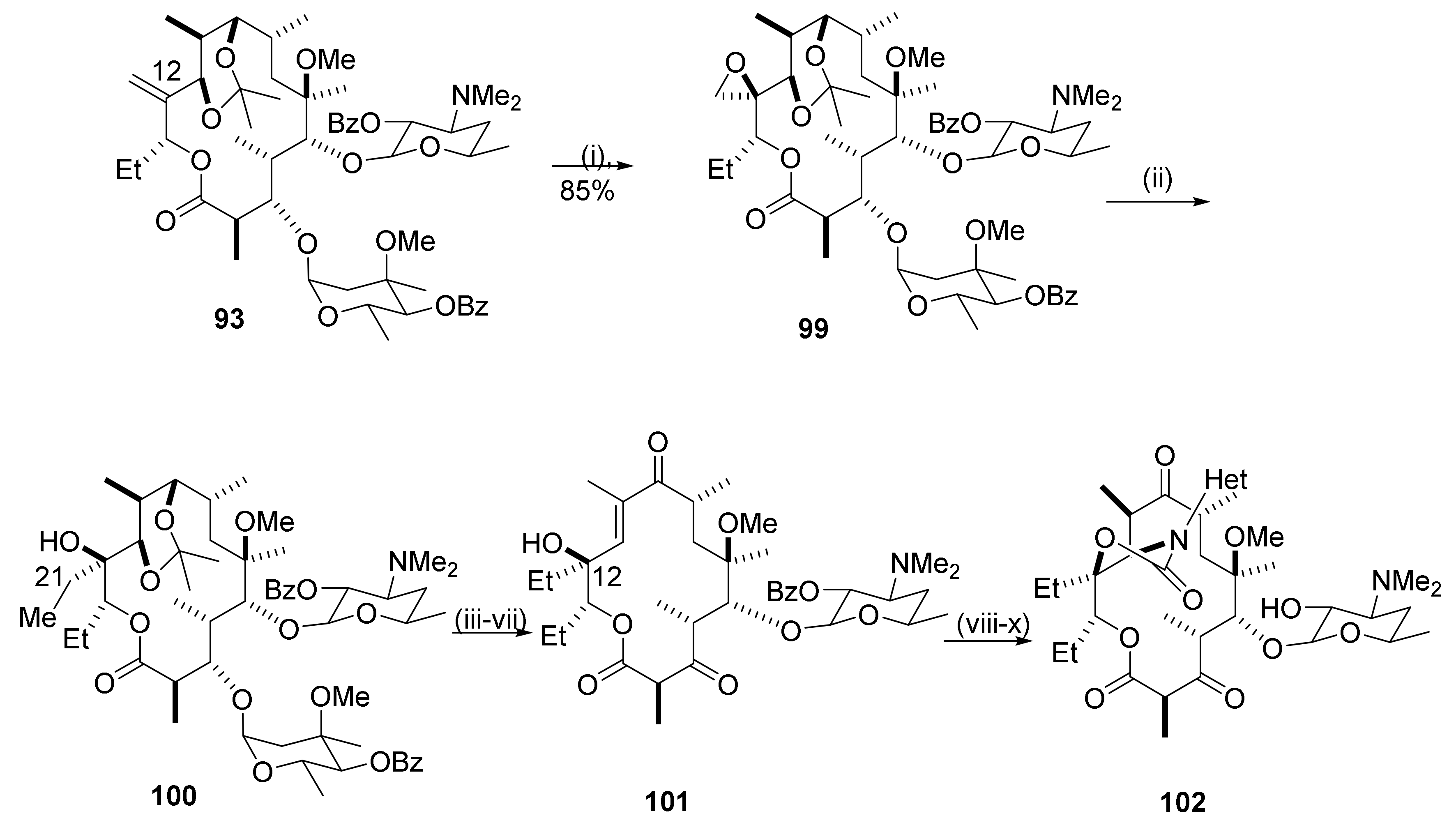
Scheme 23 shows synthesis of a C12-methylene ketolide 93 [27]. For the synthesis, the 2′,4″-dihydroxy groups in clarithromycin are protected by benzoylation and the 9-keto group is reduced by sodium borohydride to afford the 9-hydroxy intermediate 91. The latter is protected as a cyclic ketal 92 using TsOH as the catalyst under reflux in acetone. The ketal in ethyl acetate is reacted with sulfonyl chloride and triethylamine and subsequently, with TsOH in MeCN at 60 °C to afford the C12-methylene ketal 93. The C12–C21 double bond provides a handle for introduction of novel groups into the C12-position in the macrolide core.
Introduction of a vinyl group into the 12-position is shown in Scheme 24. A number of reaction steps from the 12-methylene compound 93 (Scheme 23) affords the 12-formyl-12-hydroxy intermediate 94 [27]. A Wittig reaction at the 12-formyl group using (methyl) triphenyl phosphonium bromide together with potassium bis (trimethylsilyl) amide as base affords the vinyl derivative 95. Further reactions using the Baker protocol afford the C12-vinyl, C11-C12 carbamate ketolide 96. Methanolysis provides the deprotected 12-vinyl compound 97 and its 2-methyl homologue 98. The former is a 12-vinyl homologue of telithromycin. Several analogues are available where the sidechain is substituted in either the 5-membered or the 6-membered rings.
Antibacterials: The C12-methylene ketolides possess potent antibacterial activity against a range of pathogens. The spectrum includes activity against S. pneumonia strains with the mef and erm genes. The heterocyclic moieties are tethered to the cyclocarbamate nitrogen atom via a four-carbon spacer affording compounds with potencies similar to that of telithromycin against S. aureus and susceptible S. pneumonia.
A formal replacement of the natural C12-methyl group in the erythromycin core with an ethyl group is illustrated in Scheme 25 [28]. The structural modifications start with epoxidation of the C12-methylene macrolide 93 using MCPBA [29]. Oxidation of the tertiary amino function occurs concurrently. The N-oxide is reduced by sodium thiosulfate in aqueous THF to afford epoxide 99. The epoxidation is stereoselective and occurs from the beta face of the macrocycle. Opening of the epoxide ring with dimethylcuprate adds a methyl group at C21 to afford the 12-ethyl derivative 100. Further transformations, as indicated in Scheme 25, afford the unsaturated intermediate 101 that is subjected to the Baker protocol for conversion to the 12-ethyl target compound 102.
Antibacterials: C12-ethyl ketolides with tethered heteroaromatic fused bicycles have potencies similar to telithromycin against S. aureus, S. pyogenes, the susceptible S. pneumonia mef gene containing S. pneumonia strains, and H. influenzae. They have a weaker potency than the reference compound, telithromycin, against S. epidermidis, E. faecalis, and the erm gene containing S. pneumonia strain A in an in vivo mouse infection model.
2.12.7. Reactions in the C13-Position
The C13-ethyl group is chemically inactive in the naturally occurring members of the erythromycins. A C13-vinyl analogue, however, provides a versatile intermediate for incorporation of several novel groups at the C13-position in the macrolide (Scheme 26). Fortunately, 13-vinyl compound 103 can be prepared indirectly by fermentation in a precursor-directed biosynthesis [29]. This methodology allows for preparation of 13-substituted derivatives that include the parent 13-ethyl compound. The 2′-O-benzoyl macrolide 103 is a substrate in the preparation of the 14,15-dehydroerythromycin product 107 (Scheme 26). Mesylation provides the 11-O-mesylate 104 that is also mesylated at the 4″-hydroxy group. The 11-O-mesyl group is eliminated by treatment with a base such as DBU to afford the 10,11-alkene 105. Hydrolysis under acidic conditions removes the 3-glycosyl function to afford the 3-hydroxy derivative 106. A subsequent DMP oxidation of the 3-OH group provides the 14,15-dehydroerythromycin A ketolide 107.
Antibacterials: MIC data not accessible.
2.12.8. Reactions in the C15-Position
Chemobiosynthesis has been used to prepare analogues of erythromycins with functional groups in the 15-position. The reactions are illustrated by the preparations of 15-fluoro, 15-chloro, and 15-azido derivatives (Scheme 27) [30]. The preparation starts with synthesis of the racemic diketide thioesters 108–110 that are converted into the corresponding 6-deoxyerythronolide B analogues 111–113 using streptomyces coelicolor CH999/pJRJ2. Conversion of the chloride 112 into azide 113 is effected by a reaction with NaN3 in DMF. An engineered mutant strain Saccharopolyspora erythraea K39-14V is used for the subsequent bioconversion of 111 and 113 into the erythromycin A glycosides 114 and 115. Inaccessible analogues of erythromycin by traditional chemical methods are available by chemobiosynthetic techniques.
Antibacterials: The in vitro MIC values are comparable to corresponding data for erythromycin A. The data show that introduction of a chemical handle at C15 is not deleterious to antibacterial activity
2.12.9. 15-Amino Macrolides
Preparation of 15-pharmacophoric amides is illustrated in Scheme 28. Selective reduction of the azido group in C15-azides affords corresponding primary amines. The 15-azido clarithromycin-derived macrolide 115 is 2′-O-protected by benzoylation and mesylated at the 11-hydroxy group. Treatment of the mesylate with DBU results in the elimination and formation of the 10,11-alkene 118, which is an appropriate unsaturated intermediate for annulation by the Baker protocol to afford the 15-azido ketolide 119 [31]. The azide function is reduced selectively to amine by trimethylphosphine, and the amine is acylated using a carbodiimide coupling procedure for amide bond formation to afford pharmacophoric amides such as illustrated by structure 120.
Antibacterials: The compounds possess antibacterial activity against Gram-positive, Gram-negative, and anaerobic bacteria, with special reference to S. aureus, S. epidermidis, S. pneumonia, S. pyogenes, and H. influence.
Scheme 29 illustrates synthesis of a 2,15-difluoro derivative 124. The substrate 121 has a chemically stable C15-fluoro substituent and a chemically reactive 6-allyloxy function. The 6-allylic ether function reacts in a Pd-promoted Heck coupling with the triflate of 7-hydroxyquinoline. The C15-fluoro product 122 is enolized and converted into a silyl ether at C2 using triethylsilane. The silyl ether is chlorinated at the C2-position using NCS and is subsequently fluorinated. The product after solvolysis affords the 2,15-difluoride 124 [32].
Antibacterials: The antibacterial tests of the compounds include a rat lung pneumonia lower respiratory tract infection model. In vitro hepatotoxicity of the compounds is reported.
Funding
The project received no external funding.
Conflicts of Interest
The author declares no conflict of interest
References
- Katz, L.; Ashley, G.W. Translation and protein synthesis: Macrolides. Chem. Rev. 2005, 105, 499–527. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.N. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat. Rev. 2014, 12, 35–48. [Google Scholar] [CrossRef]
- Dinos, G.P. The macrolide antibiotic renaissance. Br. J. Pharmacol. 2017, 174, 2967–2983. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, M.A.; Walsh, C.T. Antibiotics for emerging pathogens. Science 2009, 325, 1089–1093. [Google Scholar] [CrossRef]
- Fernandes, P.; Martens, E.; Bertrand, D.; Pereira, D. The solithromycin journey: Its all in the chemistry. Bioorg. Med. Chem. 2016, 24, 6420–6428. [Google Scholar] [CrossRef] [Green Version]
- Wright, P.M.; Seiple, I.B.; Myers, A.G. The evolving role of chemical synthesis in antibacterial drug discovery. Angew. Chem. Int. Ed. 2014, 53, 8840–8869. [Google Scholar] [CrossRef] [Green Version]
- Baker, W.R.; Clark, J.D.; Stephens, R.L.; Kim, K.H. Modification of macrolide antibiotics: Synthesis of 11-dioxy-11-(carboxyamino)-6-O-methylerythromycin A 11,12-(cyclic esters) via an Intramolecular Michael reaction of O-carbamates with an α,β-unsaturated ketone. J. Org. Chem. 1988, 53, 2340–2345. [Google Scholar] [CrossRef]
- Elliott, R.L.; Pireh, D.; Griesgraber, G.; Nilius, A.M.; Ewing, P.J.; Bui, M.H.; Raney, P.M.; Flamm, R.K.; Kim, K.; Henry, R.F.; et al. Anhydrolide macrolides: 1. Synthesis and antibacterial activity of 2,3-anhydro-6-O-methyl 11,12-carbamate erythromycin A analogues. J. Med. Chem. 1998, 41, 1651–1659. [Google Scholar] [CrossRef]
- Griesgraber, G.; Kramer, M.J.; Elliott, R.L.; Nilius, A.M.; Ewing, P.J.; Raney, P.M.; Bui, M.-H.; Flamm, R.K.; Chu, D.T.W.; Plattner, J.J.; et al. Anhydrolide macrolides. 2. Synthesis and antibacterial actity of 2,3-anhydro-6-O-methyl 11,12-carbazate erythromycin A analogues. J. Med. Chem. 1998, 41, 1660–1670. [Google Scholar] [CrossRef]
- Griesgraber, G.; Or, Y.-S.; Chu, D.T.W.; Nilius, A.M.; Johnson, R.K.; Flamm, R.K.; Henry, R.F.; Plattner, J.J. 3-Keto-11,12-carbazate derivatives of 6-O-methylerythromycin A: Synthesis and in vitro activity. J. Antibiot. 1996, 49, 465–477. [Google Scholar] [CrossRef] [Green Version]
- Phan, L.T.; Or, Y.S.; Ma, Z. Preparation of macrolide erythromycin 6-O-alkyl-2-nor-2-substituted agents. Patent WO2001040241, 7 June 2001. [Google Scholar]
- Phan, L.T.; Clark, R.F.; Rupp, M.; Or, Y.S.; Chu, D.T.W.; Ma, Z. Synthesis of 2-fluoro-6-O-propargyl-11,12-carbamate ketolides: A novel class of antibiotics. Org. Lett. 2000, 2, 2951–2954. [Google Scholar] [CrossRef] [PubMed]
- Daher, S.; Jin, X.; Patel, J.; Freundlich, J.S.; Buttaro, B.; Andrade, R.B. Synthesis and biological evaluation of solithromycin analogues against multidrug resistant pathogens. Bioorg. Med. Chem. Lett. 2019, 29, 1386–1389. [Google Scholar] [CrossRef] [PubMed]
- Glassford, I.; Teijaro, C.N.; Daher, S.S.; Weil, A.Q.; Small, M.C.; Redhu, S.K.; Colussi, D.J.; Jacobson, M.A.; Childers, W.E.; Buttaro, B.; et al. Ribozome-templated azide alkyne cycloadditions: Synthesis of potent macrolide antibiotics by in situ click chemistry. J. Am. Chem. Soc. 2016, 138, 3136–3144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seiple, I.B.; Zhang, Z.; Jakubec, P.; Langlois-Mercier, A.; Wright, P.M.; Hog, D.T.; Yabu, K.; Allu, S.R.; Fukuzaki, T.; Carlsen, P.N.; et al. A platform for the discovery of new macrolide antibiotics. Nature 2016, 533, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Glassford, I.; Lee, M.; Wagh, B.; Velvadupu, V.; Paul, T.; Sandelin, G.; Debrosse, C.; Klepacki, D.; Small, M.C.; Nackerell, A.D.; et al. Desmethyl macrolides: Synthesis and evaluation of 4-desmethyl telithromycin. Med. Chem. Lett. 2014, 5, 1021–1026. [Google Scholar] [CrossRef]
- Velvadapu, V.; Glassford, I.; Lee, M.; Paul, T.; Debrosse, C.; Klepacki, D.; Small, M.C.; MacKerell, A.D.; Andrade, R.B. Desmethyl macrolides: Synthesis and evaluation of 4,10-didesmethyl telithromycin. Med. Chem. Lett. 2012, 3, 211–215. [Google Scholar] [CrossRef]
- Velvadapu, V.; Paul, T.; Wagh, B.; Glassford, I.; DeBrosse, C.; Andrade, R.B. Total Synthesis of (−)−2,8,10-tridesmethyl telithromycin. J. Org. Chem. 2011, 76, 7516–7527. [Google Scholar] [CrossRef] [Green Version]
- Wagh, B.; Paul, T.; DeBrosse, C.; Klepacki, D.; Small, M.C.; MacKerell, A.D.; Andrade, R.B. Desmethyl macrolides: Synthesis and evaluation of 4,8,10-tridesmetyl cetromycin. Med. Chem. Lett. 2013, 4, 1114–1118. [Google Scholar] [CrossRef]
- Andrade, R.B. Total synthesis of desmethyl macrolide antibiotics. Synlett 2015, 26, 2199–2215. [Google Scholar] [CrossRef]
- Heggelund, A.; Undheim, K. Synthesis of 8-fluorinated erythromycin cyclic 2′,3′-carbamates. Synth. Commun. 2009, 39, 1903–1913. [Google Scholar] [CrossRef]
- Phan, L.T.; Farmer, J.J.; Or, Y.S. Preparation of antibiotic macrolide erythromycin 11-C-Substituted ketolides. Patent WO2004000864, 31 December 2004. [Google Scholar]
- Gunnes, S.; Undheim, K. Chemoselective synthesis of erythromycin A ketolides substituted in the C10-methyl group. Bioorg. Med. Chem. 2007, 15, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Gunnes, S.; Rømming, C.; Undheim, K. Selective substitutions in the C10-methyl group in erythromycin derivatives. Tetrahedron 2006, 62, 6090–6099. [Google Scholar] [CrossRef]
- Anwar, H.F.; Andrei, M.; Undheim, K. Synthesis of clarithromuycin ketolides chemically modified at the unreactive C10-methyl group. Bioorg. Med. Chem. 2017, 25, 2313–2326. [Google Scholar] [CrossRef] [PubMed]
- Lociuro, S.; Ege, T.; Andreotti, D.; Arena, A.; Gagliardi, S.; Palombi, G.; Presenti, C. Erythromycin ketolide derivatives bearing C-10 modifications. Patent WO2014049356, 3 April 2014. [Google Scholar]
- Burger, M.T.; Lin, X.; Chu, D.T.; Hiebert, C.; Rico, A.C.; Seid, M.; Caroll, G.L.; Barker, L.; Huh, K.; Langhorne, M.; et al. Synthesis and antibacterial activity of novel C12 vinyl ketolides. J. Med. Chem. 2006, 49, 1730–1743. [Google Scholar] [CrossRef] [PubMed]
- Burger, M.T.; Hiebert, C.; Seid, M.; Chu, D.T.; Barker, L.; Langhorne, M.; Shawar, R.; Kidney, J.; Desai, M.C.; Plattner, J.J. Synthesis and antibacterial activity of novel C12 ethyl ketolides. Bioorg. Med. Chem. 2006, 14, 5592–5604. [Google Scholar] [CrossRef]
- Fardis, M.; Ashley, G.W.; Carney, J.R.; Chu, D.T. Synthesis of 14,15-dehydroerythromycin A ketolides: Effect of the 13-substituent on erythromycin tautomerism. J. Antibiot. 2001, 54, 278–284. [Google Scholar] [CrossRef]
- Ashley, G.W.; Burlingame, M.; Desai, R.; Fu, H.; Leaf, T.; Licari, P.J.; Tran, C.; Abbanat, D.; Bush, K.; Macielag, M. Preparation of erythromycin analogs having functional groups at C-15. J. Antibiot. 2006, 59, 392–401. [Google Scholar] [CrossRef]
- Ashley, G.; Shaw, S.J.; Li, Y. Preparation of erythromycin A derived amido-macrolides for use in treatment of bacterial infections. Patent WO2003061671, 31 July 2003. [Google Scholar]
- Shaw, S.J.; Ashley, G.W.; Burlingame, M.A. Preparation of 7-quinolyl 15-fluoro-ketolide erythromycin derivatives as antibacterial agents. Patent WO2007070621, 21 June 2007. [Google Scholar]
Figure 1.
Erythromycin A (A), Clarithromycin (B), Telithromycin (C), Solithromycin (D), Cethromycin (E), and Clarithromycin 3-ketolide (F).
Figure 1.
Erythromycin A (A), Clarithromycin (B), Telithromycin (C), Solithromycin (D), Cethromycin (E), and Clarithromycin 3-ketolide (F).

Scheme 2.
Reagents and conditions: (i) CDI, NaH, DMF, THF, 0 °C to rt, 4–5 h; (ii) R1NH2, DMF, or MeCN (10% aq.), rt, 1–3 d; (iii) MeOH, rt, 14 h.
Scheme 2.
Reagents and conditions: (i) CDI, NaH, DMF, THF, 0 °C to rt, 4–5 h; (ii) R1NH2, DMF, or MeCN (10% aq.), rt, 1–3 d; (iii) MeOH, rt, 14 h.

Scheme 3.
Reagents and conditions: (i) NH2NH2, DMF, 60 °C; (ii) MeOH, rt; (iii) PhCHO, NaBH3CN, AcOH, MeOH, rt, 3 h.
Scheme 3.
Reagents and conditions: (i) NH2NH2, DMF, 60 °C; (ii) MeOH, rt; (iii) PhCHO, NaBH3CN, AcOH, MeOH, rt, 3 h.

Scheme 4.
Reagents and conditions; (i) NaH, DMF, PhSeCl, 0 °C, 3 h; (ii) Oxone, THF, H2O, rt, 26 h; (iii) H2O2. Abbreviation: Oxone, potassium peroxymonosulfate.
Scheme 4.
Reagents and conditions; (i) NaH, DMF, PhSeCl, 0 °C, 3 h; (ii) Oxone, THF, H2O, rt, 26 h; (iii) H2O2. Abbreviation: Oxone, potassium peroxymonosulfate.

Scheme 5.
Reagents and conditions; (i) (a) NaH, DMF, 0 °C, 1 h, (b) (PhSO2)2NF, −5 °C, 3 h; (ii) R1COBr(Cl), PPh3, CuI, NEt3, MeCN, 80 °C; (iii) MeOH, rt.
Scheme 5.
Reagents and conditions; (i) (a) NaH, DMF, 0 °C, 1 h, (b) (PhSO2)2NF, −5 °C, 3 h; (ii) R1COBr(Cl), PPh3, CuI, NEt3, MeCN, 80 °C; (iii) MeOH, rt.

Scheme 6.
Reagents and conditions. (i) NaH, (PhSO2)2NF, THF; (ii) MeOH, rt; (iii) CuI (cat), toluene, 86 °C.
Scheme 6.
Reagents and conditions. (i) NaH, (PhSO2)2NF, THF; (ii) MeOH, rt; (iii) CuI (cat), toluene, 86 °C.

Scheme 7.
Reagents and conditions: (i) (PhSO2)2NF, tBuOK, −78 °C.

Scheme 8.
Reagents and conditions: (i) CCl3COCl, iPrNEt2, DMAP, benzene, 12 h; (ii) CeCl3.7H2O, NaBH4, MeOH, −15 °C to rt, 45 min; (iii) 2,6-lutidine, TMSOTf, CH2Cl2, −7 °C, 30 min: (iv) DDQ, H2O, CH2Cl2, 0 °C, 30 min; (v) AgOTf, DTBMT, 4 Å mol. sieve, toluene, CH2Cl2, 12 h; (vi) THF, pyridine, 0 °C to 15 °C, 3 h; (vii) DMP, CH2Cl2, 3 h; (viii) NaH, CDI, DMF, THF, −20 °C to 0 °C, 45 min; (ix) MeCN, H2O, 72 h¸ (x) TASF, DMF, H2O, 14 h; (xi) Me2S, NCS, NEt3, CH2Cl2, −20 °C; (xii) MeOH, rt, 14 h. Abbreviation: DTBMP, 2,6-di-tbutyl-4-methylpyridine; TASF, tris(dimethylamino)sulfonium difluorotrimethylsilicate; DMP, Dess-Martin periodinane.
Scheme 8.
Reagents and conditions: (i) CCl3COCl, iPrNEt2, DMAP, benzene, 12 h; (ii) CeCl3.7H2O, NaBH4, MeOH, −15 °C to rt, 45 min; (iii) 2,6-lutidine, TMSOTf, CH2Cl2, −7 °C, 30 min: (iv) DDQ, H2O, CH2Cl2, 0 °C, 30 min; (v) AgOTf, DTBMT, 4 Å mol. sieve, toluene, CH2Cl2, 12 h; (vi) THF, pyridine, 0 °C to 15 °C, 3 h; (vii) DMP, CH2Cl2, 3 h; (viii) NaH, CDI, DMF, THF, −20 °C to 0 °C, 45 min; (ix) MeCN, H2O, 72 h¸ (x) TASF, DMF, H2O, 14 h; (xi) Me2S, NCS, NEt3, CH2Cl2, −20 °C; (xii) MeOH, rt, 14 h. Abbreviation: DTBMP, 2,6-di-tbutyl-4-methylpyridine; TASF, tris(dimethylamino)sulfonium difluorotrimethylsilicate; DMP, Dess-Martin periodinane.

Scheme 9.
Reagents and conditions: (i) 20 Mol% Grubbs(II) catalyst; (ii) NaBH4, CeCl3.7H2O; (iii) TESCl, imidazole, (iv) AgOTf, DTBMP; (v) DMP, CH2Cl2: (vi) NaH, CDI, THF, DMF; (vii) amine; (vii) TAS, DMF, H2O; (viii) NCS, Me2S, NEt3; (ix) MeOH.
Scheme 9.
Reagents and conditions: (i) 20 Mol% Grubbs(II) catalyst; (ii) NaBH4, CeCl3.7H2O; (iii) TESCl, imidazole, (iv) AgOTf, DTBMP; (v) DMP, CH2Cl2: (vi) NaH, CDI, THF, DMF; (vii) amine; (vii) TAS, DMF, H2O; (viii) NCS, Me2S, NEt3; (ix) MeOH.

Scheme 10.
Reagents and conditions: (i) CrCl2, NiCl2(cat), DMSO; (ii) DMP; (iii) NaBH4, CeCl3.7H2O; (iv) TESOTf, 2,6-lutidine; (v) TsOH; (vi) TESCl, imidazole; (vii) 25, AgOTf, DTBMP; (viii) 1 M TBAF, THF; (ix) DMP, CH2Cl2; (x) NaH, CDI, THF, DMF, then diamine (A); (xi) TASF, DMF, H2O; (xii) NCS, DMS, NEt3; (xiii) MeOH. Abbreviation: TES, triethylsilyl.
Scheme 10.
Reagents and conditions: (i) CrCl2, NiCl2(cat), DMSO; (ii) DMP; (iii) NaBH4, CeCl3.7H2O; (iv) TESOTf, 2,6-lutidine; (v) TsOH; (vi) TESCl, imidazole; (vii) 25, AgOTf, DTBMP; (viii) 1 M TBAF, THF; (ix) DMP, CH2Cl2; (x) NaH, CDI, THF, DMF, then diamine (A); (xi) TASF, DMF, H2O; (xii) NCS, DMS, NEt3; (xiii) MeOH. Abbreviation: TES, triethylsilyl.

Scheme 11.
Reagents and conditions: (i) CrCl2, NiCl2 (100:1), DMSO; (ii) DMP, NaHCO3, CH2Cl2; (iii) AgOTf, DTBMP, 4 Å mol sieve, toluene, CH2Cl2, rt, 24 h; (iv) Pd(OAc)2, NaHCO3, 3-Br-quinoline, Bu4NCl, DMF, 80 °C; (v) TBAF, THF, 0 oC, 15 min; (vi) DMP, CH2Cl2; (vii) NaH, CDI, DMF, THF, −20 °C–0 °C, (viii) NH4OH, MeCN, H2O.
Scheme 11.
Reagents and conditions: (i) CrCl2, NiCl2 (100:1), DMSO; (ii) DMP, NaHCO3, CH2Cl2; (iii) AgOTf, DTBMP, 4 Å mol sieve, toluene, CH2Cl2, rt, 24 h; (iv) Pd(OAc)2, NaHCO3, 3-Br-quinoline, Bu4NCl, DMF, 80 °C; (v) TBAF, THF, 0 oC, 15 min; (vi) DMP, CH2Cl2; (vii) NaH, CDI, DMF, THF, −20 °C–0 °C, (viii) NH4OH, MeCN, H2O.

Scheme 12.
Reagents and conditions: (i) COCl2, pyridine, CH2Cl2, rt, 6 h; (ii) MeCN, H2O, rt, 40 min; (iii) AcOH, H2O, reflux, 30 min.
Scheme 12.
Reagents and conditions: (i) COCl2, pyridine, CH2Cl2, rt, 6 h; (ii) MeCN, H2O, rt, 40 min; (iii) AcOH, H2O, reflux, 30 min.

Scheme 13.
Reagents and conditions: (i) Toluene, heat; (ii) acid; (iii) oxidation; (iv) R1R2NH, NaBH3CN, EtOH; (v) acid; (vi) oxidation; (vii) MeOH.
Scheme 13.
Reagents and conditions: (i) Toluene, heat; (ii) acid; (iii) oxidation; (iv) R1R2NH, NaBH3CN, EtOH; (v) acid; (vi) oxidation; (vii) MeOH.

Scheme 14.
Reagents and conditions: (i) NaBH3CN; (ii) PhCl, 1 mM, 132 °C; (iii) Cu2SO4, Na-L-ascorbate.
Scheme 14.
Reagents and conditions: (i) NaBH3CN; (ii) PhCl, 1 mM, 132 °C; (iii) Cu2SO4, Na-L-ascorbate.

Scheme 15.
Reagents and conditions: (i) H2O2, MeOH, rt, 9 h; (ii) NCS, AcOH, rt, 20 h; (iii) PPh2PCH2CH2PPh2, THF, reflux, 20 h; (iv) Ac2O, NEt3, rt, 17 h; (v) DMP, CH2Cl2, rt, 3 h; (vi) CDI, NaH, 0 °C to rt, 17 h; (vii) NH3(aq), MeCN, THF, rt, 48 h.
Scheme 15.
Reagents and conditions: (i) H2O2, MeOH, rt, 9 h; (ii) NCS, AcOH, rt, 20 h; (iii) PPh2PCH2CH2PPh2, THF, reflux, 20 h; (iv) Ac2O, NEt3, rt, 17 h; (v) DMP, CH2Cl2, rt, 3 h; (vi) CDI, NaH, 0 °C to rt, 17 h; (vii) NH3(aq), MeCN, THF, rt, 48 h.

Scheme 16.
Reagent and conditions: (i) NH3(aq), MeCN, THF, rt, 17 h.

Scheme 17.
Reagents and conditions: (i) R1CH2NH2, THF, reflux, 48 h; (ii) MeOH, rt. 14 h; (iii) PhCH(Me)NH2, THF, reflux 60 h; (iv) MeOH, rt, 14 h.
Scheme 17.
Reagents and conditions: (i) R1CH2NH2, THF, reflux, 48 h; (ii) MeOH, rt. 14 h; (iii) PhCH(Me)NH2, THF, reflux 60 h; (iv) MeOH, rt, 14 h.

Scheme 18.
Reagents and conditions: (i) TMSCHN2, CH2Cl2, 0 °C- rt, 72 h.

Scheme 19.
Reagents and conditions: (i) R1CH2R2, NaH, THF, 0 °C to rt; MeOH, rt, 14 h; BnNHCOCH2CN, NaH, THF, 0 °C to rt.; (iii) PhCH(Me)NH2, THF, reflux, 60 h; (iv); MeOH, rt.
Scheme 19.
Reagents and conditions: (i) R1CH2R2, NaH, THF, 0 °C to rt; MeOH, rt, 14 h; BnNHCOCH2CN, NaH, THF, 0 °C to rt.; (iii) PhCH(Me)NH2, THF, reflux, 60 h; (iv); MeOH, rt.

Scheme 20.
Reagents and conditions: (i) Bu3SnPh, Pd2dba3, CHCl3, NMP, 90 °C, 14 h; (ii) Ac2O, NEt3, CH2Cl2, rt, 17 h; (iii) DMP, CH2Cl2, rt, 3 h; (iv) (a) CDI, NaH, THF, 0 °C, 20 h, (b) NH3(aq), MeCN, THF, rt, 48 h; (v) MeOH, rt, 14 h.
Scheme 20.
Reagents and conditions: (i) Bu3SnPh, Pd2dba3, CHCl3, NMP, 90 °C, 14 h; (ii) Ac2O, NEt3, CH2Cl2, rt, 17 h; (iii) DMP, CH2Cl2, rt, 3 h; (iv) (a) CDI, NaH, THF, 0 °C, 20 h, (b) NH3(aq), MeCN, THF, rt, 48 h; (v) MeOH, rt, 14 h.

Scheme 21.
Reagents and conditions: (i) Pd/C, H2, MeOH, rt, 24 h.

Scheme 22.
Reagents and conditions. (i) NaH, CDI, THF, rt, 3 h; (ii) NH3(aq), MeCN, THF; (iii) Lindlar cat., quinoline, EtOAc, H2, rt, 6 h; (iv) 3-Br-quinoline, MeCN, Pd(OAc)2, P(toluene)3, NEt3, 90 °C, 22 h; (v) MeOH, rt, 22 h; (vi) NH2C2NR1R2, MeCN, rt, −100 °C.
Scheme 22.
Reagents and conditions. (i) NaH, CDI, THF, rt, 3 h; (ii) NH3(aq), MeCN, THF; (iii) Lindlar cat., quinoline, EtOAc, H2, rt, 6 h; (iv) 3-Br-quinoline, MeCN, Pd(OAc)2, P(toluene)3, NEt3, 90 °C, 22 h; (v) MeOH, rt, 22 h; (vi) NH2C2NR1R2, MeCN, rt, −100 °C.

Scheme 23.
Reagents and conditions: (i) Me2C(OMe)2,TsOH-pyridine, acetone, reflux; (ii) SOCl2, NEt3, EtOAc; (iii) TsOH, MeCN, H2O, 68 °C.
Scheme 23.
Reagents and conditions: (i) Me2C(OMe)2,TsOH-pyridine, acetone, reflux; (ii) SOCl2, NEt3, EtOAc; (iii) TsOH, MeCN, H2O, 68 °C.

Scheme 24.
Reagents and conditions: (i) TsOH, MeCN, H2O, 65 °C; (ii) OsO4, NMO, acetone, H2O; (iii) AcCl, DMAP, pyridine, (iv) DMP, CH2Cl2, −10 °C; (v) MsCl, pyridine; (vi) DBU, acetone, rt, −65 °C, 16 h; (vii) Me2S, NCS, THF, −13 °C (viii) MeP(Ph)3Br, KN(TMS)2, THF, toluene, −60 °C to rt; (ix) CDI, THF, NaH, 0 °C; (x) Het-amine, MeCN, H2O. 14 h; (xi) MeOH, 70 °C. Abbreviations: NMO, N-methylmorpholine oxide; DBU, 8-diazabicyclo[4,5.0]undec-7-ene.
Scheme 24.
Reagents and conditions: (i) TsOH, MeCN, H2O, 65 °C; (ii) OsO4, NMO, acetone, H2O; (iii) AcCl, DMAP, pyridine, (iv) DMP, CH2Cl2, −10 °C; (v) MsCl, pyridine; (vi) DBU, acetone, rt, −65 °C, 16 h; (vii) Me2S, NCS, THF, −13 °C (viii) MeP(Ph)3Br, KN(TMS)2, THF, toluene, −60 °C to rt; (ix) CDI, THF, NaH, 0 °C; (x) Het-amine, MeCN, H2O. 14 h; (xi) MeOH, 70 °C. Abbreviations: NMO, N-methylmorpholine oxide; DBU, 8-diazabicyclo[4,5.0]undec-7-ene.

Scheme 25.
Reagents and conditions: (i) (a) MCPBA, CH2Cl2, rt, 21 h, (b) 2 M NaH/SO3(aq), rt, 30 min; (ii) (a) Me2S.CuBr, −78 °C, (b) MeLi, −78 °C, (c) epoxide, 0–5 °C, 12 h; (iii) TsOH/pyridine, MeCN:H2O (2:1), 78 °C 48 h; (iv) DMP, CH2Cl2, −5 to −10 °C, 30 h; (v) (a) MsCl, pyridine, 0 °C—rt, 18 h, (b) DMU, acetone, rt, −40 °C, 40 h; (vi) 3 M HCl, MeCN. rt, 22 h: (vii) DMP, CH2Cl2, rt, 2 h; (viii) NaH, CDI, THF, −15 to 0 °C, 25 min; (ix) Het-amine, 78 °C, 40 h; (x) MeOH, 70 °C, 22 h. Abbreviation: MCPBA, m-chloroperbenzoic acid.
Scheme 25.
Reagents and conditions: (i) (a) MCPBA, CH2Cl2, rt, 21 h, (b) 2 M NaH/SO3(aq), rt, 30 min; (ii) (a) Me2S.CuBr, −78 °C, (b) MeLi, −78 °C, (c) epoxide, 0–5 °C, 12 h; (iii) TsOH/pyridine, MeCN:H2O (2:1), 78 °C 48 h; (iv) DMP, CH2Cl2, −5 to −10 °C, 30 h; (v) (a) MsCl, pyridine, 0 °C—rt, 18 h, (b) DMU, acetone, rt, −40 °C, 40 h; (vi) 3 M HCl, MeCN. rt, 22 h: (vii) DMP, CH2Cl2, rt, 2 h; (viii) NaH, CDI, THF, −15 to 0 °C, 25 min; (ix) Het-amine, 78 °C, 40 h; (x) MeOH, 70 °C, 22 h. Abbreviation: MCPBA, m-chloroperbenzoic acid.

Scheme 26.
Reagents and conditions: (i) MeSO2Cl, pyridine, rt, 24 h; (ii) DBU, acetone, rt, 24 h; (iii) 3 M HCl, MeCN, rt, 22 h; (iv) DMP, CH2Cl2, rt, 1 h.
Scheme 26.
Reagents and conditions: (i) MeSO2Cl, pyridine, rt, 24 h; (ii) DBU, acetone, rt, 24 h; (iii) 3 M HCl, MeCN, rt, 22 h; (iv) DMP, CH2Cl2, rt, 1 h.

Scheme 27.
Reagents and conditions: (i) Strept. coelicolor CH999/pJRJ2; (ii) Sac. Erythraea K39-14V.
Scheme 27.
Reagents and conditions: (i) Strept. coelicolor CH999/pJRJ2; (ii) Sac. Erythraea K39-14V.

Scheme 28.
Reagents and conditions: (i) MsCl, pyridine, rt, 16 h; (ii) DBU, acetone, rt, 8 h; (iii) NaH, CDI, THF, −15 °C, 15 min; (iv) MeCN, THF, NH3(aq), rt, 16 h; (v) PMe3, THF, rt, 45 min; (vi) RCO2H, Me2N(CH2)3N = C = NEt; (vii) NEt3, MeOH, 50 °C, 14 h.
Scheme 28.
Reagents and conditions: (i) MsCl, pyridine, rt, 16 h; (ii) DBU, acetone, rt, 8 h; (iii) NaH, CDI, THF, −15 °C, 15 min; (iv) MeCN, THF, NH3(aq), rt, 16 h; (v) PMe3, THF, rt, 45 min; (vi) RCO2H, Me2N(CH2)3N = C = NEt; (vii) NEt3, MeOH, 50 °C, 14 h.

Scheme 29.
Reagents and conditions: (i) PdBr2, dppf, DiPEA, DMF; (ii) Et3SiH, TFA, CH2Cl2; (iii) NCS, Me2S, NEt3; (iv) NaHMDS, (PhSO2)2NF; (v) MeOH, rt. Abbreviations: DiPEA, N,N-diisopropylethylamine; dppf, bis(diphenylphosphino)ferrocene; NCS, N-chlorosuccinimide.
Scheme 29.
Reagents and conditions: (i) PdBr2, dppf, DiPEA, DMF; (ii) Et3SiH, TFA, CH2Cl2; (iii) NCS, Me2S, NEt3; (iv) NaHMDS, (PhSO2)2NF; (v) MeOH, rt. Abbreviations: DiPEA, N,N-diisopropylethylamine; dppf, bis(diphenylphosphino)ferrocene; NCS, N-chlorosuccinimide.

© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Undheim, K. Scaffold Modifications in Erythromycin Macrolide Antibiotics. A Chemical Minireview. Molecules 2020, 25, 3941. https://doi.org/10.3390/molecules25173941
AMA Style
Undheim K. Scaffold Modifications in Erythromycin Macrolide Antibiotics. A Chemical Minireview. Molecules. 2020; 25(17):3941. https://doi.org/10.3390/molecules25173941
Chicago/Turabian StyleUndheim, Kjell. 2020. "Scaffold Modifications in Erythromycin Macrolide Antibiotics. A Chemical Minireview" Molecules 25, no. 17: 3941. https://doi.org/10.3390/molecules25173941