
Molecular Modeling of µ Opioid Receptor Ligands with Various Functional Properties: PZM21, SR-17018, Morphine, and Fentanyl—Simulated Interaction Patterns Confronted with Experimental Data

 , , and
, , and
Abstract
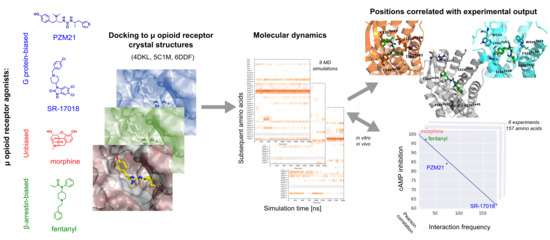
:
1. Introduction
2. Results and Discussion
2.1. Comparison of Modeled Ligands and µ Opioid Receptor Crystal Structures
2.2. Docking
2.3. Molecular Dynamics and Correlation Studies
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Grim, T.W.; Acevedo-Canabal, A.; Bohn, L.M. Toward Directing Opioid Receptor Signaling to Refine Opioid Therapeutics. Biol. Psy. 2020, 87, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Manglik, A.; Lin, H.; Aryal, D.K.; McCorvy, J.D.; Dengler, D.; Corder, G.; Levit, A.; Kling, R.C.; Bernat, V.; Hübner, H.; et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016, 537, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Kudla, L.; Bugno, R.; Skupio, U.; Wiktorowska, L.; Solecki, W.; Wojtas, A.; Golembiowska, K.; Zádor, F.; Benyhe, S.; Buda, S.; et al. Functional characterization of a novel opioid, PZM21, and its effects on the behavioural responses to morphine. Br. J. Pharmacol. 2019, 176, 4434–4445. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.; Disney, A.; Conibear, A.; Sutcliffe, K.; Dewey, W.; Husbands, S.; Bailey, C.; Kelly, E.; Henderson, G. The novel μ-opioid receptor agonist PZM21 depresses respiration and induces tolerance to antinociception. Br. J. Pharmacol. 2018, 175, 2653–2661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, C.L.; Kennedy, N.M.; Ross, N.C.; Lovell, K.M.; Yue, Z.; Morgenweck, J.; Cameron, M.D.; Bannister, T.D.; Bohn, L.M. Bias factor and therapeutic window correlate to predict safer opioid analgesics. Cell 2017, 171, 1165–1175.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grim, T.W.; Schmid, C.L.; Stahl, E.L.; Pantouli, F.; Ho, J.H.; Acevedo-Canabal, A.; Kennedy, N.M.; Cameron, M.D.; Bannister, T.D.; Bohn, L.M. A G protein signaling-biased agonist at the μ-opioid receptor reverses morphine tolerance while preventing morphine withdrawal. Neuropsychopharm 2020, 45, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, V.S.; Srinivas, K. Modern drug discovery process: An in silico approach. J. Bioinform. Seq. Anal. 2011, 2, 89–94. [Google Scholar]
- Reddy, A.S.; Pati, S.P.; Kumar, P.P.; Pradeep, H.N.; Sastry, G.N. Virtual screening in drug discovery—A computational perspective. Curr. Protein Pept. Sci. 2007, 8, 329–351. [Google Scholar] [CrossRef]
- Bittemcourt, J.A.H.M.; Neto, M.F.A.; Lacerda, P.S.; Bittencourt, R.C.V.S.; Silva, R.C.; Lobato, C.C.; Silva, L.B.; Leite, F.H.A.; Zuliani, J.P.; Rosa, J.M.C.; et al. In silico Evaluation of ibuprofen and two benzoylpropionic acid derivatives with potential anti-inflammatory activity. Molecules 2019, 24, 1476. [Google Scholar] [CrossRef] [Green Version]
- Dellafiora, L.; Galaverna, G.; Cruciani, G.; Dall’Asta, C.; Bruni, R. On the mechanism of action of anti-inflammatory activity of hypericin: An in silico study pointing to the relevance of janus kinase inhibition. Molecules 2018, 23, 3058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Aguilar, J.M.; Kang, S.-G.; Zhang, L.; Zhou, R. Modeling and structural characterization of the sweet taste receptor heterodimer. ACS Chem. Neurosci. 2019, 10, 4579–4592. [Google Scholar] [CrossRef] [PubMed]
- Geppert, H.; Vogt, M.; Bajorath, J. Current trends in ligand-based virtual screening: Molecular representations, data mining methods, new application areas, and performance evaluation. J. Chem. Inf. Model. 2010, 2, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 2014, 12, 4977–5010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.Y. Pharmacophore modeling and applications in drug discovery: Challenges and recent advances. Drug Discov. Today 2010, 15, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Stumpfe, D.; Bajorath, J. Similarity searching. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 2, 260–282. [Google Scholar] [CrossRef]
- Anderson, A.C. The process of structure-based drug design. Chem Biol. 2003, 9, 787–797. [Google Scholar] [CrossRef] [Green Version]
- Sousa, F.; Fernandes, P.A.; Ramos, M.J. Protein—Ligand docking: Current status and future challenges. Proteins 2006, 1, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Daddam, J.R.; Sreenivasulu, B.; Peddanna, K.; Umamahesh, K. Designing, docking and molecular dynamics simulation studies of novel cloperastine analogues as anti-allergic agents: Homology modeling and active site prediction for the human histamine H1 receptor. RCS Adv. 2020, 10, 4745. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Lu, D.; Zhang, H.; Zheng, M.; Yang, H.; Xu, Y.; Luo, C.; Zhu, W.; Yu, K.; Jiang, H. Applying high-performance computing in drug discovery and molecular simulation. Nat. Sci. Rev. 2016, 3, 49–63. [Google Scholar] [CrossRef]
- Pándy-Szekeres, G.; Munk, C.; Tsonkov, T.M.; Mordalski, S.; Harpsøe, K.; Hauser, A.S.; Bojarski, A.J.; Gloriam, D.E. GPCRdb in 2018: Adding GPCR structure models and ligands. Nucleic Acids Res. 2017, 46, D440–D446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Instant JChem 6.3.0. ChemAxon, 2014. Available online: http://www.chemaxon.com (accessed on 28 September 2020).
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the {mu}-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Manglik, A.; Venkatakrishnan, A.J.; Laeremans, T.; Feinberg, E.N.; Sanborn, A.L.; Kato, H.E.; Livingston, K.E.; Thorsen, T.S.; Kling, R.C.; et al. Structural insights into mu-opioid receptor activation. Nature 2015, 524, 315–321. [Google Scholar] [CrossRef] [Green Version]
- Koehl, A.; Hu, H.; Maeda, S.; Zhang, Y.; Qu, Q.; Paggi, J.M.; Latorraca, N.R.; Hilger, D.; Dawson, R.; Matile, H.; et al. Structure of the mu-opioid receptor-Giprotein complex. Nature 2018, 558, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Bajusz, D.; Racz, A.; Heberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 2015, 7, 20. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Huang, T.; Li, J. Molecular Dynamics Simulations to Investigate How PZM21 Affects the Conformational State of the µ-Opioid Receptor Upon Activation. Int. I. Mol. Sci. 2020, 21, 4699. [Google Scholar] [CrossRef]
- Mordalski, S.; Witek, J.; Smusz, S.; Rataj, K.; Bojarski, A.J. Multiple conformational states in retrospective virtual screening—Homology models vs. crystal structures: Beta-2 adrenergic receptor case study. J. Cheminform. 2015, 7, 13. [Google Scholar] [CrossRef] [Green Version]
- Pasternak, G.W.; Pan, Y.-X. Mu opioids and their receptors: Evolution of a concept. Pharmacol. Rev. 2013, 65, 1257–1317. [Google Scholar] [CrossRef] [Green Version]
- Kaserer, T.; Lantero, A.; Schmidhammer, H.; Spetea, M.; Schuster, D. µ opioid receptor: Novel antagonists and structural modeling. Sci. Rep. 2016, 6, 21548. [Google Scholar] [CrossRef] [Green Version]
- Gillis, A.; Gondin, A.B.; Kliewer, A.; Sanchez, J.; Lim, H.D.; Alamein, C.; Manandhar, P.; Santiago, M.; Fritzwanker, S.; Schmiedel, F.; et al. Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists. Sci. Signal. 2020, 13, eaaz3140. [Google Scholar] [CrossRef]
- Marcou, G.; Rognan, D. Optimizing fragment and scaffold docking by use of molecular interaction fingerprints. J. Chem. Inf. Model. 2007, 47, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.A. Frequency distribution of the values of the correlation coefficient in samples from an indefinitely large population. Biometrika 1915, 10, 507–521. [Google Scholar] [CrossRef]
- LigPrep. Schrödinger Release 2020-1; LLC: New York, NY, USA, 2020. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glide. Schrödinger Release 2020-1; LLC: New York, NY, USA, 2020. [Google Scholar]
- Schrödinger Release 2020-1: Desmond Molecular Dynamics System; D.E. Shaw Research: New York, NY, USA, 2020.
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 26. [Google Scholar] [CrossRef]
Sample Availability: Not available. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Symbol | Compound Structure | µ Opioid Receptor Activity | Molecular Weight * | LogP ** | # H Bond Acceptors | # H Bond Donors | # Rotatable Bonds |
|---|---|---|---|---|---|---|---|
| PZM21 |  | G protein-biased agonist | 361.50 | 2.85 | 3 | 3 | 8 |
| SR-17018 |  | G protein-biased agonist | 410.73 | 4.75 | 2 | 1 | 3 |
| morphine |  | Unbiased agonist | 285.34 | 0.90 | 4 | 2 | 0 |
| fentanyl |  | β-arrestin-2-biased agonist | 336.47 | 3.82 | 2 | 0 | 6 |
| PDB ID | Receptor State | Resolution (Å) | Co-Crystallized Ligand |
|---|---|---|---|
| 4DKL | inactive | 2.8 | BF0 (Antagonist) |
| 5C1M | active | 2.1 | BU72 (Agonist) |
| 6DDF | active | 3.5 | DAMGO (Peptide agonist) |
| Modeled Ligand | BF0 | BU72 |
|---|---|---|
| PZM21 | 0.306 | 0.318 |
| SR-17018 | 0.297 | 0.331 |
| morphine | 0.764 | 0.595 |
| fentanyl | 0.289 | 0.322 |
| Crystal Structure/Parameter. | Gai2 Activation | cAMP Inhibition | bArr2 Recruitment | Rab5 Trafficking | GIRK Activation | GRK2 Recruitment |
|---|---|---|---|---|---|---|
| 4DKL | T1202×56, I3227×38 | T1202×56, V2365×43 | F1523×37, R211 | |||
| 5C1M | I2966×51 | L1212×57, I3227×38 | Y1483×33 | I3227×38 | ||
| 6DDF | W3187×34 | W3187×34 | L1212×57, W133, I1443×29, C3217×37, G3257×41 | W133 | W133, I1443×29 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Podlewska, S.; Bugno, R.; Kudla, L.; Bojarski, A.J.; Przewlocki, R. Molecular Modeling of µ Opioid Receptor Ligands with Various Functional Properties: PZM21, SR-17018, Morphine, and Fentanyl—Simulated Interaction Patterns Confronted with Experimental Data. Molecules 2020, 25, 4636. https://doi.org/10.3390/molecules25204636
Podlewska S, Bugno R, Kudla L, Bojarski AJ, Przewlocki R. Molecular Modeling of µ Opioid Receptor Ligands with Various Functional Properties: PZM21, SR-17018, Morphine, and Fentanyl—Simulated Interaction Patterns Confronted with Experimental Data. Molecules. 2020; 25(20):4636. https://doi.org/10.3390/molecules25204636
Chicago/Turabian StylePodlewska, Sabina, Ryszard Bugno, Lucja Kudla, Andrzej J. Bojarski, and Ryszard Przewlocki. 2020. "Molecular Modeling of µ Opioid Receptor Ligands with Various Functional Properties: PZM21, SR-17018, Morphine, and Fentanyl—Simulated Interaction Patterns Confronted with Experimental Data" Molecules 25, no. 20: 4636. https://doi.org/10.3390/molecules25204636