5.1. Reagents and Methods
The reagent qualities and sources and general methods were as described before [
3,
25].
5.1.1. 4-Chloro-3,5-Dinitrobenzoic Acid (2)
4-chloro-benzoic acid
1 (20 g, 0.128 mol) was dissolved in H
2SO
4 (d = 1.835 g/mL, 300 mL) at 80 °C, and KNO
3 (66 g, 0.65 mol) was added. The reaction mixture was heated to 125 °C using a high-pressure flask and kept for 2 h, after which the reaction was cooled to rt and poured onto ice. The yield of title compound
2 was 28 g (89%) [
16].
1H NMR (300 MHz, DMSO-
d6) δ = 14.29 (s, 1H), 8.76 (s, 2H).
13C NMR (75 MHz, DMSO-d6) δ 163.61, 149.06, 132.20, 128.70, 122.80. HRMS (ESI):
m/
z calcd. for C
7H
2ClN
2O
6 [M−H]
−: 244.9607; found, 244.9607.
5.1.2. 4-Amino-3,5-Dinitrobenzoic Acid (3)
Compound
2 (20 g, 81 mmol) was dissolved in methanol (100 mL), and aqueous 24% NH
3 (120 mL) was gradually added. The reaction mixture was stirred at rt for 2.5 h, refluxed for 3 h and left at rt for around 14 h. The precipitate that formed was filtered off, and the filtrate was evaporated to dryness. Water (10 mL) and HCl (10 mL) were added to the solid residue, which was combined with the precipitate. After stirring for 10 min, the precipitate was filtered off and washed with water till washings showed neutral. The yield of 4-amino-3,5-dinitrobenzoic acid
3 was 18 g (98%) [
16].
1H NMR (300 MHz, Acetone-
d6) δ = 9.16–9.01 (m, 3H), 9.01–8.84 (s, 2H).
13C NMR (75 MHz, Acetone-
d6) δ 163.49, 143.33, 134.76, 133.75, 128.17, 115.39. HRMS (ESI):
m/
z calcd. for C
7H
4N
3O
6 [M−H]
−: 226.0105; found, 226.0100.
5.1.3. 3,4-Diamino-5-Nitrobenzoic Acid (4)
A suspension of freshly made compound
3 (10 g, 44 mmol) in water (200 mL) was heated to 60 °C with stirring for about 10 min. To this suspension was then added a solution of sodium sulfide (11.2 g, anhydrous) and sodium bicarbonate (11.5 g) in water (250 mL). The reaction mixture was stirred at 75 °C for 1 h and left to cool overnight, after which it was concentrated and subjected to silica gel column chromatography to give a deep red solid compound
4 with a yield of 74% [
17].
1H NMR (300 MHz, DMSO-
d6) δ 12.56 (s, 1H), 7.98 (d,
J = 1.9 Hz, 1H), 7.40 (s, 2H), 7.30 (d,
J = 1.9 Hz, 1H), 5.47 (s, 2H).
13C NMR (75 MHz, DMSO-
d6) δ 166.71, 138.09, 137.74, 130.25, 117.66, 116.01, 115.78. HRMS (ESI):
m/
z calcd. for C
7H
6N
3O
4 [M−H]
−: 196.0364; found, 196.0367.
5.1.4. 4-Nitro-1H-Benzo[d]imidazole-6-Carboxylic Acid (5)
Compound
4 (2 g, 10.15 mmol) was refluxed for 6 h in 50 mL of formic acid. The reaction mixture was concentrated, and then, 10 mL of 30% aqueous NaOH was slowly added to the residue. The resulting suspension was portioned between 30 mL of ethyl acetate and 20 mL of water 3 times; the combined ethyl acetate layer was concentrated and subjected to a silica gel column to obtain 600 mg of
5 with a 29% yield [
30].
1H NMR (300 MHz, DMSO-
d6) δ = 13.54 (s, 2H), 8.64–8.57 (m, 2H), 8.56 (d,
J = 1.5 Hz, 1H).
13C NMR (75 MHz, DMSO-
d6) δ 166.25, 147.16, 133.43, 126.95, 124.45, 119.75. HRMS (ESI):
m/
z calcd. for C
8H
4N
3O
4 [M−H]
−: 206.0207; found, 206.0208.
5.1.5. (4-Nitro-1H-Benzo[d]imidazol-6-yl)-Methanol (6)
Compound 11 (160 mg, 0.72 mmol) was dissolved in methanolic ammonia (7N, 15 mL) and stirred at 25 °C for 1.5 h. After TLC showed the completion of the reaction, the mixture was concentrated, and 10 mL of DCM was added. Undissolved solid compound was filtered, and 123 mg of the title product 6 was collected with a yield of 88%. 1H NMR (300 MHz, DMSO-d6) δ 8.43 (s, 1H), 8.14 (d, J = 1.3 Hz, 1H), 8.06 (d, J = 1.3 Hz, 1H), 4.70 (s, 2H). 13C NMR (75 MHz, DMSO-d6) δ 145.57, 136.64, 133.41, 131.48, 127.20, 124.24, 117.34, 62.28. HRMS (ESI): m/z calcd. for C9H8N3O4 [M + H]+: 194.0560; found, 194.0565.
5.1.6. Methyl 4-Nitro-1H-Benzo[d]imidazole-6-Carboxylate (7)
To a mixture of 100 mg of 5 in 10 mL of methanol was added 3 drops of concentrated sulfuric acid, and the reaction was refluxed for 2 h until TLC indicated the reaction to be completed. A solution of 20 mL of saturated sodium bicarbonate was added to the concentrated reaction mixture, which was extracted with 30 mL of ethyl acetate. The title compound 7 was separated by silica gel chromatography to afford 97 mg (92% yield). 1H NMR (300 MHz, DMSO-d6) δ 8.58 (d, J = 15.1 Hz, 3H), 3.93 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 165.20, 147.55, 145.51, 133.53, 131.11, 126.61, 122.94, 119.45, 52.75. HRMS (ESI): m/z calcd. for C9H8N3O4 [M + H]+: 222.0509; found, 222.0513.
5.1.7. (4-Chloro-3,5-Dinitrophenyl)-Methanol (8)
To a solution of compound 2 (3 g, 12.2 mmol) in dry THF (30 mL) at ambient temperature and under a nitrogen atmosphere was added boron trifluoride diethyl etherate (12 mL), followed by the addition of borane tetrahydrofuran complex (1.0 M solution in THF, 24 mL) over 5 min. The mixture was stirred at ambient temperature overnight, and the reaction was quenched by carefully adding 10 mL of methanol until the gas evolution ceased, followed by 20 mL of water. The mixture was concentrated, the residue was extracted with ethyl acetate (3 × 30 mL), and the combined organic extracts were washed with brine. The title compound 8 was separated with silica gel, affording 2.48 g (87%). 1H NMR (300 MHz, DMSO-d6) δ 8.31 (d, J = 0.9 Hz, 2H), 4.65 (s, 2H). 13C NMR (75 MHz, DMSO-d6) δ 148.75, 145.89, 128.71, 125.89 (two carbons), 116.11, 60.96. HRMS (ESI): m/z calcd. for C7H6ClN2O5 [M + H]+: 232.9960; found, 232.9959.
5.1.8. (4-Amino-3,5-Dinitrophenyl)-Methanol (9)
Compound 8 (2.3 g, 9.95 mmol) was dissolved in methanol (100 mL), and aqueous NH3 (20 mL) was gradually added. The reaction mixture was stirred at rt for 2.5 h, refluxed for 3 h and left at ambient temperature overnight. Following concentration, the reaction mixture was subjected to a silica gel column, affording 1.8 g (92%) of the title compound 9. 1H NMR (300 MHz, DMSO-d6) δ 8.43 (s, 2H), 8.35 (s, 2H), 5.49 (t, J = 5.8 Hz, 1H), 4.48 (d, J = 5.7 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) δ 139.95, 134.68, 131.80 (two carbons), 128.45, 125.87, 60.85. HRMS (ESI): m/z calcd. for C7H6N3O5 [M−H]−: 212.0313; found, 212.0316.
5.1.9. (3,4-Diamino-5-Nitrophenyl)-Methanol (10)
A suspension of 9 (2.1 g, 9.85 mmol) in MeOH (40 mL) was heated to 60 °C with stirring for about 10 min. To this suspension was added a solution of sodium sulfide (5.4 g of the nonahydrate, 22.14 mmol) and sodium bicarbonate (3.5 g, 41.7 mmol) in 60 mL of water. The reaction mixture was stirred at 70–75 °C for 3 h. The reaction was concentrated and subjected to silica gel chromatography, giving 1.34 g (76%) of the title compound 10. 1H NMR (300 MHz, DMSO-d6) δ 7.29 (d, J = 1.7 Hz, 1H), 6.91 (s, 2H), 6.77 (d, J = 1.8 Hz, 1H), 5.24 (s, 2H), 5.07 (t, J = 5.7 Hz, 1H), 4.30 (d, J = 5.6 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) δ 137.74, 134.60, 130.67, 130.09, 116.41, 110.56, 62.52. HRMS (ESI): m/z calcd. for C7H8N3O3 [M−H]−: 182.0571; found, 182.0571.
5.1.10. (4-Nitro-1H-Benzo[d]imidazol-6-yl)-Methyl Formate (11)
A suspension of 10 (100 mg, 0.55 mmol) in 10 mL of formic acid was heated to 115 °C for 3 h, after which the reaction was concentrated, followed by adding 10 mL of ethyl acetate and 1 mL of triethylamine (TEA). The title compound was purified by silica gel column chromatography, and 70 mg of white compound 11 was collected with a yield of 58%. 1H NMR (300 MHz, DMSO-d6) δ 13.36 (s, 1H), 8.49 (s, 1H), 8.36 (s, 1H), 8.20 (d, J = 5.0 Hz, 2H), 5.38 (s, 2H). 13C NMR (75 MHz, DMSO-d6) δ 162.06, 146.13, 129.42, 119.21, 64.46. HRMS (ESI): m/z calcd. for C9H8N3O4 [M + H]+: 222.0509; found, 222.0513.
5.1.11. (2R,3R,4R,5R)-2-(Acetoxymethyl)-5-((4-Nitro-1H-Benzo[d]imidazol-6-yl)methoxy)tetrahydrofuran-3,4-Diyl Diacetate (12)
Compound 6 (110 mg, 0.57 mmol) and 1,2,3,5-tetra-O-acetyl-β–D-ribofuranose (280 mg, 0.63 mmol) were dissolved in 15 mL of dry acetonitrile, followed by adding a solution of stannic chloride (1 M in DCM, 3 mL) into the reaction mixture. After stirring at 25 °C for 16 h, the reaction mixture was diluted with DCM and then poured under stirring into 60 mL of ice-cooled saturated sodium bicarbonate. The resulting suspension was filtered through celite, and the layers were separated. The organic layer was further washed with brine and was dried over Na2SO4, filtered off and evaporated. The residue was purified by silica gel column chromatography to yield 100 mg (40%) of the title compound 12. 1H NMR (300 MHz, DMSO-d6) δ 13.32 (s, 1H), 8.47 (s, 1H), 8.14 (d, J = 4.5 Hz, 2H), 5.34–5.15 (m, 3H), 4.89 (d, J = 11.9 Hz, 1H), 4.74 (d, J = 11.8 Hz, 1H), 4.30 (dt, J = 10.2, 3.7 Hz, 2H), 4.18–3.96 (m, 1H), 2.17–1.92 (m, 9H). 13C NMR (75 MHz, DMSO-d6) δ 170.18, 169.70, 169.53, 145.92, 131.14, 118.74, 104.32, 78.36, 74.20, 71.11, 68.48, 63.92, 20.62, 20.48, 20.44. HRMS (ESI): m/z calcd. for C19H22N3O10 [M + H]+: 452.1300; found, 452.1303.
5.1.12. 6-((Tert-Butyldimethylsilyloxy)methyl)-4-Nitro-1H-Benzo[d]imidazole (13)
The alcohol derivative 6 (100 mg, 0.52 mmol) and TBDMSCl (94 mg, 0.63 mmol) were dissolved in 15 mL of dry acetonitrile, and the reaction mixture was stirred at 25 °C for 20 h. After the completion of the reaction, the mixture was extracted with ethyl acetate (EA) (20 mL) directly, the organic layer was further washed with brine, and the organic layer was subjected to a silica gel column to give 117 mg of title compound 13 with a 74% yield. 1H NMR (300 MHz, DMSO-d6) δ 13.26 (s, 1H), 8.45 (d, J = 2.0 Hz, 1H), 8.10 (d, J = 20.1 Hz, 2H), 4.92 (d, J = 2.1 Hz, 2H), 0.93 (d, J = 2.2 Hz, 9H), 0.12 (d, J = 2.4 Hz, 6H). 13C NMR (75 MHz, DMSO-d6) δ 145.71, 135.26, 116.85, 63.61, 25.94, 18.13, −5.12. HRMS (ESI): m/z calcd. for C14H22N3O3Si [M + H]+: 308.1425; found, 308.1428.
5.1.13. 6-((Tert-Butyldiphenylsilyloxy)methyl)-4-Nitro-1H-Benzo[d]imidazole (14)
This compound was synthesized in analogy to 13 using TBDPSCl from 100 mg (0.52 mmol) of 8. Yield: 89%. 1H NMR (300 MHz, DMSO-d6) δ 8.46 (s, 1H), 8.18 (s, 1H), 8.10 (s, 1H), 7.66 (dt, J = 6.1, 1.7 Hz, 4H), 7.57–7.34 (m, 7H), 4.99 (d, J = 1.8 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) δ 146.38, 145.72, 135.15, 134.52, 132.82, 130.15, 128.13, 124.47, 116.89, 64.48, 26.80, 19.01. HRMS (ESI): m/z calcd. for C24H26N3O3Si [M + H]+: 432.1739; found, 432.1740.
5.1.14. (2R,3R,4R,5R)-2-(Acetoxymethyl)-5-((1-((2R,3R,4R,5R)-3,4-Diacetoxy-5-(Acetoxymethyl)tetrahydrofuran-2-yl)-4-Nitro-1H-Benzo[d]imidazol-6-yl)methoxy)tetrahydrofuran-3,4-Diyl Diacetate (15)
The double sugar-containing compound was obtained using the procedure for 12 by changing the reaction temperature to 60 °C. However, compound 12 remained the major product in this reaction, no matter whether we started from the protected compound 13 or 14. The ratio for compounds 12 and 15 was 3:1, with an overall yield of 40%. 13C NMR (75 MHz, DMSO-d6) δ 170.19, 169.68, 169.62, 169.52, 169.40, 146.09, 138.70, 136.45, 135.39, 132.96, 119.09, 117.32, 104.30, 86.55, 79.92, 78.41, 74.18, 72.40, 71.10, 69.67, 68.33, 63.90, 63.00, 20.62, 20.56, 20.51, 20.46, 20.42, 20.29. HRMS (ESI): m/z calcd. for C30H36N3O17 [M + H]+: 710.2039; found, 710.2040.
5.1.15. Tert-Butyl(4-chloro-3,5-Dinitrobenzyloxy)dimethylsilane (16)
This compound was synthesized in analogy to 13 (which was synthesized from 6) starting from 500 mg (2.59 mmol) of 8. Yield: 84%. 1H NMR (300 MHz, CDCl3) δ 8.57 (d, J = 1.0 Hz, 2H), 5.44 (t, J = 1.0 Hz, 2H), 1.58 (s, 9H), 0.77 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 149.76, 144.05, 124.99, 120.66, 118.50, 63.19, 62.94, 26.08, 18.59, −5.11. HRMS (ESI): m/z calcd. for C13H18ClN2O5Si [M-H]-: 345.0679; found, 345.0674.
5.1.16. 4-((Tert-Butyldimethylsilyloxy)methyl)-2,6-Dinitroaniline (17)
This compound was synthesized from 16 (640 mg 1.85 mmol) in analogy to 9. Yield: 99%. 1H NMR (300 MHz, DMSO-d6) δ 8.45 (s, 1H), 8.37 (s, 1H), 7.33 (s, 2H), 3.35 (s, 2H), 0.92 (s, 5H), 0.10 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 140.08, 134.79, 131.32, 127.05, 62.21, 25.88, 18.06, −5.18. HRMS (ESI): m/z calcd. for C13H20N3O5Si [M−H]−: 326.1178; found, 326.1185.
5.1.17. 5-((Tert-Butyldimethylsilyloxy)methyl)-3-Nitrobenzene-1,2-Diamine (18)
This compound was synthesized in analogy to 10 starting from 600 mg (1.83 mmol) of 17. Yield: 80%. 1H NMR (300 MHz, DMSO-d6) δ 7.28 (d, J = 1.8 Hz, 1H), 6.94 (s, 2H), 6.74 (d, J = 1.9 Hz, 1H), 5.28 (s, 2H), 4.50 (s, 2H), 0.89 (s, 9H), 0.07 (s, 6H). 13C NMR (75 MHz, DMSO-d6) δ 137.91, 134.76, 130.57, 128.64, 115.80, 110.35, 64.04, 25.98, 18.15, −5.06. HRMS (ESI): m/z calcd. for C13H24N3O3Si [M + H]+: 298.1581; found, 298.1571.
5.1.18. 6-((Tert-Butyldimethylsilyloxy)methyl)-4-Nitro-1H-Benzo[d]imidazole (13)
Alternative procedure: A mixture of
18 (410 mg, 1.38 mmol) and triethyl orthoformate (25 mL) was refluxed for about 3 h at 145 °C. After the completion of the reaction as monitored by TLC, 1.5 mL of formic acid was added, and the mixture was refluxed at the same temperature for another 2 h. The solution was evaporated to dryness at reduced pressure, and the residue was dissolved in methanol and stirred at rt, overnight, in the presence of charcoal. Following the removal of the charcoal by vacuum filtration through celite-545, the filtrate was evaporated to give 300 mg of title compound
13 as an off-white solid with a 85% yield without further purification [
31].
1H NMR (300 MHz, DMSO-
d6) δ 13.28 (s, 1H), 8.45 (s, 1H), 8.11 (d,
J = 19.8 Hz, 2H), 4.92 (s, 2H), 0.93 (s, 9H), 0.12 (s, 6H).
13C NMR (75 MHz, DMSO-
d6) δ 146.40, 145.70, 135.20, 133.00, 126.52, 124.53, 116.90, 63.60, 25.93, 18.13, −5.12. HRMS (ESI):
m/
z calcd. for C
14H
22N
3O
3Si [M + H]
+: 308.1425; found, 308.1415.
5.1.19. (2R,3S,4S,5R)-2-(Acetoxymethyl)-5-(5-(((Tert-Butyldimethylsilyl)oxy)methyl)-7-Nitro-1H-Benzo[d]imidazol-1-yl)tetrahydrofuran-3,4-Diyl Diacetate (19)
To a solution of compound
13 (280 mg, 0.9 mmol) in dry 1,2-dichlorethane, an amount of Bis(trimethylsilyl)acetamide (BSA) (366 mg, 1.8 mmol) was added, and the reaction was stirred at 60 °C for 40 min. Hereafter, 1,2,3,5-tetra-
O-acetyl-D-ribofuranose (561 mg, 1.8 mM, dissolved in 8 mL of dry 1,2-dichlorethane), TMSOTf (300 mg, 1.35 mmol) and
N-methylmorpholine (127 mg, 1.08 mmol) were added, and the reaction mixture was stirred at 65 °C for another 2.5 h. After completion, the reaction mixture was concentrated and portioned between EA and saturated NaHCO
3. The residue was subjected to silica gel chromatography, and the title compound
19 was obtained with 330 mg (64% yield) [
30]. 1H NMR (300 MHz, DMSO-
d6) δ 8.78 (s, 1H), 8.05 (dd,
J = 1.6, 0.7 Hz, 1H), 7.99 (d,
J = 1.4 Hz, 1H), 6.46 (d,
J = 3.5 Hz, 1H), 5.78 (dd,
J = 5.6, 3.6 Hz, 1H), 5.41 (t,
J = 6.0 Hz, 1H), 4.97–4.85 (m, 2H), 4.40 (ddd,
J = 6.8, 4.6, 3.1 Hz, 1H), 4.34–4.14 (m, 2H), 2.12 (d,
J = 0.6 Hz, 3H), 2.11–2.05 (m, 3H), 1.91 (d,
J = 0.6 Hz, 3H), 0.92 (d,
J = 0.6 Hz, 9H), 0.11 (d,
J = 0.6 Hz, 6H).
13C NMR (75 MHz, DMSO-
d6) δ 169.89, 169.44, 169.23, 147.29, 144.56, 136.30, 136.14, 123.73, 118.85, 88.50, 79.13, 73.83, 69.31, 63.25, 62.49, 20.40, 20.33, 20.30, 18.09, −5.21. HRMS (ESI):
m/
z calcd. for C
25H
36N
3O
10Si [M + H]+: 566.2164; found, 566.2168.
5.1.20. (2R,3S,4S,5R)-2-(Hydroxymethyl)-5-(5-(((Tert-Butyldimethylsilyl)oxy)methyl)-7-Nitro-1H-Benzo[d]imidazol-1-yl)tetrahydrofuran-3,4-Diol (20)
A solution of compound 19 (327 mg, 0.58 mmol) in methanolic ammonia (7 N, 15 mL) was stirred at 0 °C for 4 h. The reaction was concentrated in the presence of silica gel, and the title compound 20 was purified by silica gel chromatography using EA/methanol (8:2) to afford 254 mg (90% yield). 1H NMR (300 MHz, DMSO-d6) δ 8.69 (s, 1H), 8.02 (d, J = 1.4 Hz, 1H), 7.91 (d, J = 1.4 Hz, 1H), 6.05 (d, J = 3.9 Hz, 1H), 5.70 (d, J = 5.8 Hz, 1H), 5.41 (d, J = 5.6 Hz, 1H), 4.90 (s, 2H), 4.56 (q, J = 4.8 Hz, 1H), 4.18–3.92 (m, 5H), 0.93 (s, 9H), 0.12 (s, 6H). 13C NMR (75 MHz, DMSO-d6) δ 147.17, 144.44, 136.49, 135.95, 123.91, 123.12, 118.36, 89.75, 81.59, 73.83, 70.09, 63.33, 63.12, 25.92, 18.12, −5.14. HRMS (ESI): m/z calcd. for C21H32N3O8Si [M + H]+: 482.1953; found, 482.1962.
5.1.21. (1-(2. R,3S,4S,5R)-(6-(Hydroxymethyl)-2,2-Dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-7-Nitro-1H-Benzo[d]imidazol-5-yl)methanol (21)
To a solution of compound
20 (1.66 g, 3.77 mmol), suspended in dry acetone (500 mL), was added 37 mM dry
p-toluenesulfonic acid (6.5 g, 10 eq.) in one portion. The mixture was stirred under a nitrogen atmosphere and turned into a yellow solution upon dissolution. After 3 h, a saturated NaHCO
3 solution (500 mL) cooled to 0 °C was added with stirring over 5 min. The solvents were removed under reduced pressure, and the residue was portioned between ethyl acetate (200 mL) and water [
32]. The solvent was evaporated, and the title compound
21 was purified by silica gel chromatography (EA/MeOH 9:1) with an 81% yield (1.08 g, 2.95 mM).
1H NMR (300 MHz, DMSO-
d6) δ 8.76 (s, 1H), 8.00 (d,
J = 1.4 Hz, 1H), 7.90 (d,
J = 1.4 Hz, 1H), 6.26 (d,
J = 1.8 Hz, 1H), 5.38 (dd,
J = 5.9, 2.0 Hz, 1H), 4.83 (dd,
J = 5.9, 2.2 Hz, 1H), 4.66 (s, 2H), 4.09 (ddd,
J = 6.2, 4.2, 2.1 Hz, 1H), 3.08 (dd,
J = 11.6, 4.3 Hz, 1H), 2.88 (dd,
J = 11.6, 5.9 Hz, 1H), 1.49 (s, 3H), 1.32 (s, 3H).
13C NMR (75 MHz, DMSO-
d6) δ 147.20, 144.82, 137.44, 136.55, 123.62, 123.41, 118.83, 112.64, 91.20, 86.81, 83.34, 81.11, 62.00, 60.72, 26.79, 25.19. HRMS (ESI):
m/
z calcd. for C
16H
20N
3O
7 [M + H]
+: 366.1296; found, 366.1299.
5.1.22. 5-(Benzyloxy)methyl-2-Chloro-1,3-Dinitrobenzene (22)
To a solution of compound 8 (500 mg, 2.59 mmol) in 20 mL of 1,4-dioxane was added benzyl 2,2,2-trichloroacetimidate (1.3 g, 5.2 mmol) and trifluoromethanesulfonic acid (200 mg, 1.3 mmol), and the reaction mixture was stirred at rt for 1 h. The reaction was portioned between EA and saturated NaHCO3; the organic layer was concentrated and subjected to a silica gel column. The title compound 22 was separated with a 95% yield (680 mg). 1H NMR (300 MHz, CDCl3) δ 8.59 (d, J = 0.9 Hz, 2H), 8.11–7.91 (m, 5H), 5.29 (s, 2H), 5.25 (d, J = 0.9 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 141.13, 136.99, 129.29, 129.00, 128.64, 128.46, 128.24, 128.04, 126.14, 119.09, 73.70, 69.25. HRMS (ESI): m/z calcd. for C14H12ClN2O5 [M + H]+: 323.0429; found, 323.0424.
5.1.23. 4-(Benzyloxy)methyl-2,6-Dinitroaniline (23)
This compound was synthesized in analogy to 9 starting from 680 mg (2.11 mmol) of 22. Yield: 96%. 1H NMR (300 MHz, DMSO-d6) δ 8.48 (s, 2H), 8.40 (s, 2H), 7.40–7.31 (m, 5H), 4.57 (s, 2H), 4.54 (s, 2H). 13C NMR (75 MHz, DMSO-d6) δ 140.33, 134.84, 132.99, 128.54, 128.45, 127.80, 127.71, 127.26, 127.16, 124.22, 71.75, 69.32. HRMS (ESI): m/z calcd. for C14H14N3O5 [M + H]+: 304.0928; found, 304.0930.
5.1.24. 5-(Benzyloxy)methyl-3-Nitrobenzene-1,2-Diamine (24)
This compound was synthesized in analogy to 10 starting from 630 mg (2.08 mmol) of 23. Yield: 80%. 1H NMR (300 MHz, DMSO-d6) δ = 7.50–7.16 (m, 6H), 7.01 (s, 2H), 6.81 (d, J = 1.8, 1H), 5.34 (s, 2H), 4.47 (s, 2H), 4.34 (s, 2H). HRMS (ESI): m/z calcd. for C14H16N3O3 [M + H]+: 274.1186; found, 274.1188.
5.1.25. 6-(Benzyloxy)methyl-4-Nitro-1H-benzo[d]imidazole (25)
This compound was synthesized in analogy to 13 (which was synthesized from 18) starting from 550 mg (2.0 mmol) of 24. Yield: 84%. 1H NMR (300 MHz, DMSO-d6) δ 13.31 (s, 1H), 8.47 (s, 1H), 8.16 (s, 2H), 7.53–7.09 (m, 5H), 4.75 (s, 2H), 4.60 (s, 2H). 13C NMR (75 MHz, DMSO-d6) δ 145.81, 138.35, 132.31, 128.45, 127.72, 127.66, 126.23, 118.39, 71.69, 70.76. HRMS (ESI): m/z calcd. For C15H14N3O3 [M + H]+: 284.1030; found, 284.1021.
5.1.26. (2R,3S,4S,5R)-2-(Acetoxymethyl)-5-(5-((Benzyloxy)methyl)-7-Nitro-1H-Benzo[d]imidazol-1-yl)tetrahydrofuran-3,4-Diyl Diacetate (26)
This compound was synthesized in analogy to 19 starting from 1.44 g (5.09 mmol) of 25. Yield: 64%. 1H NMR (300 MHz, DMSO-d6) δ 8.80 (s, 1H), 8.13 (d, J = 1.4 Hz, 1H), 8.03 (d, J = 1.4 Hz, 1H), 7.50–7.22 (m, 5H), 6.46 (d, J = 3.6 Hz, 1H), 5.79 (dd, J = 5.6, 3.6 Hz, 1H), 5.40 (t, J = 6.0 Hz, 1H), 4.74 (s, 2H), 4.60 (s, 2H), 4.40 (dq, J = 6.4, 3.1 Hz, 1H), 4.34–4.15 (m, 2H), 2.12 (s, 3H), 2.08 (s, 3H), 1.91 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 169.93, 169.46, 169.26, 147.25, 144.66, 138.29, 136.24, 133.42, 128.45, 127.71, 127.67, 125.42, 124.06, 120.28, 88.52, 79.14, 73.81, 71.75, 70.33, 69.32, 62.49, 20.43, 20.37, 20.33. HRMS (ESI): m/z calcd. For C26H27N3O10Na [M + Na]+: 564.1589; found, 564.1607.
5.1.27. (2R,3S,4S,5R)-2-(5-((Benzyloxy)methyl)-7-Nitro-1H-Benzo[d]imidazol-1-yl)-5-(Hydroxymethyl)tetrahydrofuran-3,4-Diol (27)
This compound was synthesized in analogy to 20 starting from 1.55 g (2.86 mmol) of 26. Yield: 66%. 1H NMR (300 MHz, DMSO-d6) δ 8.86 (s, 1H), 8.10 (d, J = 1.4 Hz, 1H), 7.98 (d, J = 1.4 Hz, 1H), 7.47–7.25 (m, 5H), 6.08 (d, J = 5.0 Hz, 1H), 5.51 (d, J = 6.2 Hz, 1H), 5.23 (d, J = 5.0 Hz, 1H), 5.03 (t, J = 5.2 Hz, 1H), 4.73 (s, 2H), 4.60 (s, 2H), 4.29 (q, J = 5.3 Hz, 1H), 4.15–3.98 (m, 1H), 3.95 (d, J = 3.8 Hz, 1H), 3.70–3.42 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 147.33, 144.92, 138.33, 136.24, 132.83, 128.46, 127.71, 127.67, 125.01, 124.36, 119.82, 89.52, 85.34, 75.19, 71.73, 70.45, 69.96, 60.71. HRMS (ESI): m/z calcd. For C20H22N3O7[M + H]+: 416.1452; found, 416.1447.
5.1.28. (2R,3S,4S,5R)-(6-(5-((Benzyloxy)methyl)-7-Nitro-1H-Benzo[d]imidazol-1-yl)-2,2-Dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)Methanol (28)
This compound was synthesized in analogy to 21 starting from 680 mg (1.64 mmol) of 27. Yield: 78%. 1H NMR (300 MHz, CDCl3) δ 9.27 (s, 1H), 8.50 (dd, J = 10.1, 1.5 Hz, 2H), 8.11–7.85 (m, 5H), 7.09 (d, J = 3.2 Hz, 1H), 5.59 (dd, J = 6.1, 2.6 Hz, 1H), 5.50 (dd, J = 6.1, 3.2 Hz, 1H), 5.23 (d, J = 7.5 Hz, 4H), 5.06 (t, J = 2.5 Hz, 1H), 4.55 (dd, J = 12.0, 2.5 Hz, 1H), 4.43 (dd, J = 12.1, 2.7 Hz, 1H), 2.27 (s, 3H), 1.99 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 146.93, 144.57, 137.92, 136.84, 133.52, 128.80, 128.19, 128.13, 125.05, 124.30, 120.77, 114.75, 92.94, 86.61, 85.55, 81.21, 72.91, 71.03, 62.12, 27.45, 25.73. HRMS (ESI): m/z calcd. For C23H26N3O7[M + H]+: 456.1765; found, 456.1759.
5.1.29. (2R,3S,4S,5R)-(6-(5-((Benzyloxy)methyl)-7-Nitro-1H-Benzo[d]imidazol-1-yl)-2,2-Dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl Sulfamate (29)
Chlorosulfonyl isocyanate (630 mg, 4.5 mmol) was taken into a 10 mL flask, and after cooling to 0 °C, formic acid (210 mg, 4.5 mmol) was added and the mixture was stirred for 5 min. The resulting solid was dissolved in dry acetonitrile (6 mL), and the solution was cooled to 0 °C and stirred for another 5 h, gradually reaching rt. Compound 28 (580 mg, 1.27 mmol) was dissolved in 30 mL of dimethylacetamide (DMA) and cooled to 0 °C, followed by adding the obtained sulfamoyl chloride, and the mixture was stirred overnight. The next morning, 3 mL of TEA was added, and the reaction was stirred for 10 min, followed by adding 6 mL of methanol and further stirring for 15 min. The reaction was concentrated, and the residue was partitioned between EA and saturated NaHCO3. The organic layer was further washed with water and brine, and the title compound 29 was purified by silica gel chromatography with a 84% yield (570 mg). 1H NMR (300 MHz, DMSO-d6) δ 8.75 (s, 1H), 8.19–8.07 (m, 1H), 8.01 (d, J = 1.4 Hz, 1H), 7.56 (s, 2H), 7.47–7.25 (m, 5H), 6.39 (d, J = 1.9 Hz, 1H), 5.48 (dd, J = 6.0, 2.0 Hz, 1H), 4.94 (dd, J = 6.0, 3.0 Hz, 1H), 4.74 (s, 2H), 4.60 (s, 2H), 4.36 (m, 1H), 3.75 (m, 2H), 1.55 (s, 3H), 1.37 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 147.16, 144.73, 138.28, 136.50, 133.41, 128.46, 127.75, 127.69, 125.12, 123.80, 120.12, 113.45, 90.92, 83.36, 83.32, 80.50, 71.76, 70.37, 67.44, 26.83, 25.27. HRMS (ESI): m/z calcd. for C23H27N4O9S [M + H]+: 535.1493; found, 535.1503.
5.1.30. (2R,3S,4S,5R)-(6-(5-((Benzyloxy)methyl)-7-Nitro-1H-Benzo[d]imidazol-1-yl)-2,2-Dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl ((Tert-Butoxycarbonyl)glycyl)sulfamate (30a)
Compound 29 (140 mg, 0.26 mM) and Boc-Gly-OSu (140 mg, 0.52 mmol) were dissolved in 16 mL of dry DMF, followed by adding DBU (60 mg, 0.39 mmol), and the reaction was stirred at rt overnight. The reaction was concentrated under vacuum, and the residue was dissolved in EA and washed with saturated NaHCO3. The title compound 30a was purified by silica gel chromatography with a 77% yield (EA:hexane, 2:1). 1H NMR (300 MHz, DMSO-d6) δ 8.81 (s, 1H), 8.10 (d, J = 1.5 Hz, 1H), 7.98 (d, J = 1.4 Hz, 1H), 7.51–7.19 (m, 6H), 6.33 (d, J = 2.1 Hz, 1H), 5.41 (dd, J = 5.9, 2.2 Hz, 1H), 4.95 (dd, J = 5.9, 2.3 Hz, 1H), 4.74 (s, 2H), 4.60 (d, J = 3.1 Hz, 2H), 4.31 (s, 1H), 3.80–3.59 (m, 1H), 3.40 (d, J = 5.8 Hz, 1H), 1.53 (s, 3H), 1.46–1.28 (m, 12H). 13C NMR (75 MHz, DMSO-d6) δ 173.55, 155.61, 147.20, 144.95, 138.31, 136.46, 133.31, 128.44, 127.74, 127.65, 125.00, 123.75, 119.92, 112.94, 91.40, 83.83, 83.41, 81.15, 77.67, 71.75, 70.39, 66.17, 45.63, 28.38, 26.86, 25.29. HRMS (ESI): m/z calcd. for C30H36N5O12S [M−H]−: 690.2086; found, 690.2086.
5.1.31. (2R,3S,4S,5R)-(6-(5-((Benzyloxy)methyl)-7-Nitro-1H-Benzo[d]imidazol-1-yl)-2,2-Dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl (3-(4-(benzyloxy)phenyl)-2-((Tert-Butoxycarbonyl)amino)propanoyl)sulfamate (30b)
This compound was synthesized in analogy to 30a using Boc-Tyr(OBn)-OSu, using 300 mg (0.56 mmol) of starting compound 29. Yield: 99%. 1H NMR (300 MHz, DMSO-d6) δ 8.80 (s, 1H), 8.07 (d, J = 1.4 Hz, 1H), 7.97 (d, J = 1.4 Hz, 1H), 7.50–7.23 (m, 10H), 6.98 (dd, J = 67.8, 8.1 Hz, 5H), 6.35 (d, J = 2.2 Hz, 1H), 6.32–6.06 (m, 1H), 5.47–5.31 (m, 1H), 5.12–4.87 (m, 4H), 4.70 (s, 2H), 4.58 (s, 2H), 4.34 (dt, J = 6.9, 3.5 Hz, 1H), 3.76 (m, 3H), 2.91 (d, J = 13.7 Hz, 1H), 2.69 (dd, J = 13.6, 8.5 Hz, 1H), 1.53 (s, 3H), 1.32 (d, J = 10.5 Hz, 12H). 13C NMR (75 MHz, DMSO-d6) δ 156.89, 155.01, 147.22, 144.93, 138.30, 137.41, 136.41, 133.29, 130.43, 128.50, 128.43, 128.40, 127.83, 127.72, 127.69, 127.64, 125.05, 123.79, 119.95, 114.30, 113.12, 91.32, 83.59, 80.95, 77.78, 71.73, 70.37, 69.27, 57.55, 28.32, 26.89, 25.29. HRMS (ESI): m/z calcd. for C44H48N5O13S [M−H]−: 886.2975; found, 886.3018.
5.1.32. (2R,3S,4S,5R)-(6-(5-((Benzyloxy)methyl)-7-Nitro-1H-Benzo[d]imidazol-1-yl)-2,2-Dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl (O-Benzyl-N-(Tert-Butoxycarbonyl)seryl)sulfamate (30c)
This compound was synthesized in analogy to 30a using the appropriate protected serine, using 347 mg (0.65 mmol) of starting compound 29. Yield: 95%. 1H NMR (300 MHz, DMSO-d6) δ 8.79 (s, 1H), 8.09 (d, J = 1.4 Hz, 1H), 7.98 (d, J = 1.4 Hz, 1H), 7.45–7.18 (m, 10H), 6.33 (d, J = 2.3 Hz, 1H), 6.10 (d, J = 8.1 Hz, 1H), 5.32 (dd, J = 6.1, 2.4 Hz, 1H), 4.92 (dd, J = 5.9, 2.3 Hz, 1H), 4.72 (s, 2H), 4.59 (s, 2H), 4.42 (d, J = 2.1 Hz, 2H), 4.30 (d, J = 6.1 Hz, 1H), 3.94 (s, 2H), 3.83–3.46 (m, 4H), 1.52 (s, 3H), 1.35 (d, J = 11.5 Hz, 12H). 13C NMR (75 MHz, DMSO-d6) δ 173.90, 155.00, 147.22, 145.00, 138.72, 138.30, 136.37, 133.27, 128.44, 128.18, 127.81, 127.73, 127.65, 127.48, 127.31, 125.05, 123.78, 119.94, 113.01, 91.38, 83.74, 83.61, 81.09, 77.78, 71.93, 71.73, 71.24, 70.37, 66.32, 56.62, 28.35, 26.91, 25.29. HRMS (ESI): m/z calcd. for C38H44N5O13S [M−H]−: 810.2662; found, 810.2668.
5.1.33. Tert-Butyl 4-((((6-(5-((Benzyloxy)methyl)-7-Nitro-1H-Benzo[d]imidazol-1-yl)-2,2-Dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methoxy)sulfonyl)amino)-3-((Tert-Butoxycarbonyl)amino)-4-oxobutanoate (30d)
This compound was synthesized in analogy to 30a with the aid of the appropriately protected aspartic acid, using 300 mg (0.56 mmol) of starting compound 29. Yield: 99%. 1H NMR (300 MHz, DMSO-d6) δ 8.77 (s, 1H), 8.10 (d, J = 1.4 Hz, 1H), 7.98 (d, J = 1.4 Hz, 1H), 7.48–7.22 (m, 5H), 6.34 (d, J = 2.3 Hz, 1H), 5.37 (dd, J = 6.2, 2.3 Hz, 1H), 4.93 (dd, J = 5.9, 2.4 Hz, 1H), 4.74 (s, 2H), 4.60 (s, 2H), 4.41–4.26 (m, 1H), 4.19–4.04 (m, 2H), 3.78 (dd, J = 10.9, 4.3 Hz, 1H), 3.63 (s, 0H), 3.18 (s, 2H), 2.70–2.54 (m, 1H), 2.35 (dd, J = 15.3, 8.4 Hz, 1H), 1.53 (s, 3H), 1.46–1.27 (m, 21H). 13C NMR (75 MHz, DMSO-d6) δ 170.02, 154.96, 147.20, 144.95, 138.30, 136.40, 133.28, 128.43, 127.72, 127.65, 125.05, 123.78, 119.97, 113.08, 91.33, 83.62, 83.53, 81.00, 79.73, 77.86, 71.73, 70.37, 48.73, 29.72, 28.31, 27.81, 27.78, 26.89, 25.28. HRMS (ESI): m/z calcd. for C36H46N5O14S [M−H]−: 804.2767; found, 804.2786.
5.1.34. (2R,3S,4S,5R)-(6-(5-((Benzyloxy)methyl)-7-Nitro-1H-Benzo[d]imidazol-1-yl)-2,2-Dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl ((Tert-Butoxycarbonyl)leucyl)sulfamate (30e)
This compound was synthesized in analogy to 30a using Boc-Leu-OSu, using 220 mg (0.41 mmol) of starting compound 29. Yield: 72%. 1H NMR (300 MHz, DMSO-d6) δ 8.80 (s, 1H), 8.10 (d, J = 1.4 Hz, 1H), 7.98 (d, J = 1.4 Hz, 1H), 7.47–7.22 (m, 5H), 6.32 (d, J = 2.3 Hz, 1H), 6.05 (d, J = 8.6 Hz, 1H), 5.36 (d, J = 5.6 Hz, 1H), 4.93 (dd, J = 6.0, 2.2 Hz, 1H), 4.74 (s, 2H), 4.60 (s, 2H), 4.33 (s, 1H), 3.82–3.63 (m, 2H), 3.51 (dd, J = 11.0, 5.7 Hz, 1H), 1.67–1.23 (m, 18H), 0.83 (dd, J = 6.5, 1.5 Hz, 6H). 13C NMR (75 MHz, DMSO-d6) δ 177.14, 155.13, 147.20, 145.00, 138.30, 136.39, 133.26, 128.44, 127.72, 127.65, 125.01, 123.77, 119.92, 112.97, 91.36, 83.79, 83.58, 81.16, 77.47, 71.74, 70.37, 66.21, 54.90, 42.59, 28.36, 26.91, 25.31, 24.54, 23.29, 21.99. HRMS (ESI): m/z calcd. for C34H44N5O12S [M−H]−: 746.2712; found, 746.2708.
5.1.35. (2R,3S,4S,5R)-(6-(5-((Benzyloxy)methyl)-7-Nitro-1H-benzo[d]imidazol-1-yl)-2,2-Dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl ((Tert-Butoxycarbonyl)isoleucyl)sulfamate (30f)
This compound was synthesized in analogy to 30a using Boc-Ile-OSu, using 140 mg (0.26 mmol) of starting compound 29. Yield: 82%. 1H NMR (300 MHz, DMSO-d6) δ 8.80 (s, 1H), 8.10 (d, J = 1.5 Hz, 1H), 7.98 (d, J = 1.4 Hz, 1H), 7.51–7.10 (m, 4H), 6.33 (d, J = 2.2 Hz, 1H), 5.80 (s, 1H), 5.38 (d, J = 5.8 Hz, 1H), 4.93 (dd, J = 5.8, 2.4 Hz, 1H), 4.74 (s, 2H), 4.60 (d, J = 2.8 Hz, 2H), 4.32 (s, 1H), 3.85–3.40 (m, 2H), 2.81 (s, 1H), 1.53 (s, 3H), 1.40 (s, 3H), 1.35 (d, J = 4.9 Hz, 12H), 0.92–0.68 (m, 6H). 13C NMR (75 MHz, DMSO-d6) δ 170.11, 155.15, 147.21, 144.93, 138.31, 136.41, 133.28, 128.44, 127.72, 127.65, 125.03, 123.75, 119.93, 113.01, 91.27, 83.71, 83.52, 81.08, 78.85, 77.71, 71.73, 70.36, 60.37, 28.30, 26.87, 25.62, 25.25, 24.46, 15.74, 11.71. HRMS (ESI): m/z calcd. for C34H44N5O12S [M−H]−: 746.2712; found, 746.2712.
5.1.36. (2R,3S,4S,5R)-(6-(7-Amino-5-Methyl-1H-benzo[d]imidazol-1-yl)-2,2-Dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl ((Tert-Butoxycarbonyl)glycyl)sulfamate (31a)
To a solution of compound 30a (140 mg, 0.187 mmol) in methanol (20 mL) was added 69 mg of Pd/C under argon, after which the argon was exchanged for hydrogen and the mixture was stirred at rt overnight. TLC analysis indicated the reaction to be completed, the Pd/C was filtered, and the filtrate was concentrated. The residue was adsorbed on silica, and title compound 31a was purified by chromatography (MeOH/EA, 1:9) with an 84% yield (95 mg). 1H NMR (300 MHz, CD3OD) δ = 8.31 (s, 1H), 6.89 (dd, J = 1.5, 0.9 Hz, 1H), 6.59 (s, 1H), 6.20 (d, J = 4.2 Hz, 1H), 5.30–5.05 (m, 2H), 4.47 (t, J = 3.1 Hz, 1H), 4.28 (dd, J = 5.5, 3.2 Hz, 2H), 3.69 (s, 2H), 3.37 (s, 1H), 3.33 (p, J = 1.6 Hz, 2H), 2.36 (t, J = 0.6 Hz, 3H), 1.61 (s, 3H), 1.44 (d, J = 1.7 Hz, 10H), 1.39 (s, 3H). 13C NMR (75 MHz, CD3OD) δ 176.21, 156.48, 144.88, 141.33, 133.48, 133.31, 120.29, 114.92, 111.86, 108.59, 91.51, 83.37, 82.20, 79.86, 78.40, 66.53, 45.02, 27.05, 25.59, 23.81, 19.84. HRMS (ESI): m/z calcd. for C23H32N5O9S [M−H]−: 554.1926; found, 554.1924.
5.1.37. (2R,3S,4S,5R)-(6-(7-Amino-5-(Hydroxymethyl)-1H-Benzo[d]imidazol-1-yl)-2,2-Dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl ((Tert-Butoxycarbonyl)tyrosyl)sulfamate (31b)
This compound was synthesized in analogy to 31a starting from 400 mg (0.62 mmol) of 30b. Yield: 98%. 1H NMR (300 MHz, DMSO-d6) δ 9.07 (d, J = 2.0 Hz, 1H), 8.31 (d, J = 2.0 Hz, 1H), 7.07–6.86 (m, 3H), 6.61 (dd, J = 8.5, 2.1 Hz, 3H), 6.24 (s, 1H), 6.04 (d, J = 8.2 Hz, 1H), 5.17 (s, 1H), 5.10–4.80 (m, 4H), 4.58–4.42 (m, 2H), 4.35 (s, 1H), 4.04–3.75 (m, 3H), 2.93 (d, J = 13.7 Hz, 1H), 2.81–2.60 (m, 1H), 1.55 (d, J = 2.0 Hz, 3H), 1.33 (d, J = 2.0 Hz, 12H). 13C NMR (75 MHz, DMSO-d6) δ 175.99, 155.57, 154.98, 145.87, 142.25, 137.98, 134.27, 130.30, 128.99, 121.60, 114.77, 114.32, 109.18, 107.24, 90.37, 83.38, 82.36, 80.25, 77.56, 66.24, 63.55, 57.94, 37.30, 28.37, 27.01, 25.33. HRMS (ESI): m/z calcd. for C30H38N5O11S [M−H]−: 676.2294; found, 676.2291.
5.1.38. (2R,3S,4S,5R)-(6-(7-Amino-5-(Hydroxymethyl)-1H-Benzo[d]imidazol-1-yl)-2,2-Dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl ((Tert-Butoxycarbonyl)seryl)sulfamate (31c)
This compound was synthesized in analogy to 31a starting from 500 mg (0.62 mmol) of 30c. Yield: 80%. 1H NMR (300 MHz, DMSO-d6) δ 8.30 (s, 1H), 6.93 (d, J = 1.3 Hz, 1H), 6.61 (d, J = 1.3 Hz, 1H), 6.23 (d, J = 3.9 Hz, 1H), 6.05 (d, J = 7.9 Hz, 1H), 5.76 (s, 1H), 5.25–5.08 (m, 1H), 5.08–4.96 (m, 2H), 4.80 (d, 3H), 4.46 (d, J = 5.4 Hz, 2H), 4.34 (q, J = 4.0 Hz, 1H), 4.01–3.84 (m, 1H), 3.82–3.67 (m, 1H), 3.58 (d, J = 4.4 Hz, 2H), 2.60 (s, 1H), 1.55 (s, 3H), 1.35 (d, J = 17.8 Hz, 12H). 13C NMR (75 MHz, DMSO-d6) δ 174.73, 155.13, 145.87, 142.28, 137.97, 134.29, 121.60, 114.28, 109.15, 107.22, 90.40, 83.36, 82.33, 80.26, 77.80, 66.04, 63.55, 62.94, 58.57, 28.38, 27.01, 25.33. HRMS (ESI): m/z calcd. for C24H34N5O11S [M−H]−: 600.1981; found, 600.1983.
5.1.39. Tert-Butyl 4-((((6-(7-Amino-5-(Hydroxymethyl)-1H-Benzo[d]imidazol-1-yl)-2,2-Dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methoxy)sulfonyl)amino)-3-((Tert-Butoxycarbonyl)amino)-4-Oxobutanoate (31d)
This compound was synthesized in analogy to 31a starting from 460 mg (0.57 mmol) of 30d. Yield: 68%. 1H NMR (300 MHz, DMSO-d6) δ 8.31 (s, 1H), 6.93 (s, 1H), 6.62 (s, 1H), 6.33 (d, J = 8.5 Hz, 1H), 6.23 (d, J = 4.0 Hz, 1H), 5.17 (dd, J = 6.6, 4.0 Hz, 1H), 5.00 (dd, J = 8.6, 5.6 Hz, 3H), 4.46 (d, J = 5.0 Hz, 2H), 4.34 (d, J = 3.8 Hz, 1H), 4.06 (d, J = 7.1 Hz, 1H), 3.91 (dd, J = 11.1, 4.6 Hz, 1H), 2.64 (dd, J = 15.0, 4.7 Hz, 1H), 2.36 (dd, J = 15.1, 8.5 Hz, 1H), 1.55 (s, 3H), 1.52–1.27 (m, 21H). 13C NMR (75 MHz, DMSO-d6) δ 175.05, 155.03, 145.72, 142.18, 138.03, 134.30, 121.58, 114.27, 109.19, 107.17, 99.65, 90.41, 83.35, 82.41, 80.30, 79.61, 77.72, 66.19, 63.53, 53.56, 28.37, 27.85, 27.01, 25.31. HRMS (ESI): m/z calcd for C29H42N5O12S [M−H]−: 684.2556; found, 684.2569.
5.1.40. (2R,3S,4S,5R)-(6-(7-Amino-5-(Hydroxymethyl)-1H-Benzo[d]imidazol-1-yl)-2,2-Dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl ((Tert-Butoxycarbonyl)leucyl)sulfamate (31e)
This compound was synthesized in analogy to 31a starting from 200 mg (0.27 mmol) of 30e. Yield: 80%. 1H NMR (300 MHz, DMSO-d6) δ 11.97 (s, 1H), 8.30 (s, 1H), 6.92 (d, J = 1.3 Hz, 1H), 6.61 (s, 1H), 6.22 (d, J = 3.9 Hz, 1H), 6.14 (d, J = 8.5 Hz, 1H), 5.16 (dd, J = 6.7, 4.0 Hz, 1H), 5.10–4.75 (m, 2H), 4.46 (d, J = 5.6 Hz, 2H), 4.39–4.24 (m, 1H), 4.02–3.84 (m, 2H), 3.76 (d, J = 5.1 Hz, 1H), 3.18 (d, J = 4.4 Hz, 1H), 1.91 (s, 2H), 1.55 (s, 3H), 1.34 (d, J = 12.9 Hz, 12H), 0.85 (dd, J = 6.5, 1.5 Hz, 8H). 13C NMR (75 MHz, DMSO-d6) δ 177.25, 155.23, 145.89, 142.22, 137.95, 134.26, 121.61, 114.25, 109.14, 107.25, 90.35, 83.35, 82.37, 80.28, 77.50, 66.12, 63.54, 55.03, 42.50, 28.39, 27.01, 25.34, 24.56, 23.33, 21.96. HRMS (ESI): m/z calcd. for C27H40N5O10S [M + H]+: 628.2647; found, 628.2656.
5.1.41. (2R,3S,4S,5R)-(6-(7-Amino-5-(Hydroxymethyl)-1H-Benzo[d]imidazol-1-yl)-2,2-Dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl ((Tert-Butoxycarbonyl)isoleucyl)sulfamate (31f)
This compound was synthesized in analogy to 31a starting from 140 mg (0.176 mmol) of 30f. Yield: 85%. 1H NMR (300 MHz, DMSO-d6) δ 8.31 (s, 1H), 6.93 (s, 1H), 6.62 (d, J = 1.3 Hz, 1H), 6.23 (d, J = 3.9 Hz, 1H), 5.87 (d, J = 8.4 Hz, 1H), 5.09 (dd, J = 6.7, 3.7 Hz, 3H), 4.46 (d, J = 4.8 Hz, 2H), 4.36 (t, J = 3.9 Hz, 1H), 3.96 (dd, J = 12.1, 7.4 Hz, 1H), 3.67 (dd, J = 8.5, 5.0 Hz, 1H), 1.72 (s, 1H), 1.55 (s, 3H), 1.35 (d, J = 15.0 Hz, 12H), 1.05 (dt, J = 15.3, 7.8 Hz, 1H), 0.79 (dt, J = 7.5, 4.0 Hz, 7H). 13C NMR (75 MHz, DMSO-d6) δ 175.79, 155.19, 145.86, 142.17, 137.96, 134.25, 121.61, 114.28, 109.16, 107.25, 99.66, 90.34, 83.37, 82.35, 80.27, 77.71, 66.33, 63.54, 60.58, 59.88, 38.14, 28.33, 26.98, 25.29, 24.45, 15.84, 11.78. HRMS (ESI): m/z calcd. for C27H40N5O10S [M−H]−: 626.2501; found, 626.2496.
5.1.42. (2R,3S,4S,5R)-(5-(7-Amino-5-Methyl-1H-Benzo[d]imidazol-1-yl)-3,4-Dihydroxytetrahydrofuran-2-yl)methyl Glycylsulfamate (32a)
Compound 31a (60 mg, 0.11 mmol) was dissolved in a mixture of TFA and H2O (7 mL, 5:2 v/v), and the reaction mixture was stirred for 40 min. After reaction, the volatiles were evaporated under reduced pressure, followed by co-evaporation with EtOH, and once more with EtOH + 1 mL Et3N to neutralize any remaining acid. The title compound 32a was obtained by reversed phase-HPLC as a white solid with a 60% yield. 1H NMR (300 MHz, D2O) δ 8.20 (s, 1H), 6.87 (s, 1H), 6.50 (s, 1H), 6.00 (d, J = 6.0 Hz, 1H), 4.53 (t, J = 5.8 Hz, 1H), 4.32 (m, 5H), 3.54 (s, 2H), 2.22 (s, 3H). 13C NMR (75 MHz, D2O) δ 172.34, 143.71, 141.14, 134.52, 131.91, 120.76, 113.15, 109.54, 88.63, 81.94, 73.22, 69.30, 67.80, 42.43, 20.03. HRMS (ESI): m/z calcd. for C15H22N5O7S [M + H]+: 416.1234; found, 416.1223.
5.1.43. (2R,3S,4S,5R)-(5-(7-Amino-5-(Hydroxymethyl)-1H-Benzo[d]imidazol-1-yl)-3,4-Dihydroxytetrahydrofuran-2-yl)methyl Tyrosylsulfamate (32b)
This compound was synthesized in analogy to 32a starting from 300 mg (0.44 mmol) of 31b. Yield: 80%. 1H NMR (300 MHz, D2O) δ 8.67 (s, 1H), 7.09–6.82 (m, 3H), 6.81–6.50 (m, 3H), 6.18 (d, J = 2.9 Hz, 1H), 4.61–4.08 (m, 7H), 3.84 (t, J = 6.6 Hz, 1H), 2.86 (qd, J = 14.4, 6.0 Hz, 2H). 13C NMR (75 MHz, D2O) δ 174.41, 154.49, 139.14, 138.81, 137.11, 133.45, 130.41, 125.58, 120.65, 115.21, 112.16, 105.07, 89.76, 82.61, 74.07, 69.49, 67.68, 63.14, 56.24, 35.47. HRMS (ESI): m/z calcd. for C22H26N5O9S [M−H]−: 536.1457; found, 536.1452.
5.1.44. (2R,3S,4S,5R)-(5-(7-Amino-5-(Hydroxymethyl)-1H-Benzo[d]imidazol-1-yl)-3,4-Dihydroxytetrahydrofuran-2-yl)methyl Serylsulfamate (32c)
This compound was synthesized in analogy to 32a starting from 115 mg (0.19 mmol) of 31c. Yield: 62%. 1H NMR (300 MHz, D2O) δ 8.47 (s, 1H), 6.98 (d, J = 1.4 Hz, 1H), 6.65 (d, J = 1.4 Hz, 1H), 6.05 (dd, J = 5.8, 1.3 Hz, 1H), 4.52 (d, J = 8.4 Hz, 3H), 4.42–4.17 (m, 4H), 3.99–3.69 (m, 3H). 13C NMR (75 MHz, D2O) δ 172.83, 140.46, 140.26, 137.76, 132.93, 121.36, 111.56, 106.78, 89.12, 82.33, 73.59, 69.39, 67.80, 63.35, 59.94, 56.67. HRMS (ESI): m/z calcd. for C16H22N5O9S [M−H]−: 460.1144; found, 460.1146.
5.1.45. 3-Amino-4-((((5-(7-Amino-5-(Hydroxymethyl)-1H-Benzo[d]imidazol-1-yl)-3,4-Dihydroxytetrahydrofuran-2-yl)methoxy)sulfonyl)amino)-4-Oxobutanoic Acid (32d)
This compound was synthesized in analogy to 32a starting from 120 mg (0.18 mmol) of 31d. Yield: 48%. 1H NMR (300 MHz, D2O) δ 9.19 (s, 1H), 7.13 (s, 1H), 6.89 (s, 1H), 6.33 (d, J = 4.9 Hz, 1H), 4.70–4.20 (m, 7H), 3.98 (t, J = 5.3 Hz, 1H), 2.90 (d, J = 5.4 Hz, 2H). 13C NMR (75 MHz, D2O) δ 173.71, 173.43, 140.87, 137.35, 134.06, 133.04, 119.85, 117.95, 114.08, 112.91, 103.02, 90.54, 83.20, 74.53, 69.74, 67.62, 62.90, 51.41, 34.36. HRMS (ESI): m/z calcd. for C17H22N5O10S [M−H]−: 488.1093; found, 488.1064.
5.1.46. (2R,3S,4S,5R)-(5-(7-Amino-5-(Hydroxymethyl)-1H-Benzo[d]imidazol-1-yl)-3,4-Dihydroxytetrahydrofuran-2-yl)methyl Leucylsulfamate (32e)
This compound was synthesized in analogy to 32a starting from 80 mg (0.13 mmol) of 31e. Yield: 82%. 1H NMR (300 MHz, D2O) δ 8.26 (d, J = 2.3 Hz, 1H), 7.10 (d, J = 1.9 Hz, 1H), 6.70 (q, J = 1.4 Hz, 1H), 6.06 (dd, J = 6.6, 2.3 Hz, 1H), 4.64–4.19 (m, 7H), 3.59 (td, J = 5.4, 2.5 Hz, 1H), 1.66–1.21 (m, 3H), 0.71 (dd, J = 6.0, 2.1 Hz, 6H). 13C NMR (75 MHz, D2O) δ 176.02, 144.13, 141.89, 136.45, 132.51, 122.55, 111.30, 108.93, 88.19, 82.12, 73.10, 69.52, 68.03, 63.60, 53.80, 39.81, 23.62, 21.39, 20.41. HRMS (ESI): m/z calcd. for C19H28N5O8S [M−H]−: 486.1664; found, 486.1666.
5.1.47. (2R,3S,4S,5R)-(5-(7-Amino-5-(Hydroxymethyl)-1H-Benzo[d]imidazol-1-yl)-3,4-Dihydroxytetrahydrofuran-2-yl)methyl Isoleucylsulfamate (32f)
This compound was synthesized in analogy to 32a starting from 158 mg (0.25 mmol) of 31f. Yield: 84%. 1H NMR (300 MHz, D2O) δ 8.35 (s, 1H), 7.12 (s, 1H), 6.74 (d, J = 1.3 Hz, 1H), 6.10 (d, J = 6.5 Hz, 1H), 4.67–4.19 (m, 7H), 3.54 (dd, J = 4.3, 1.2 Hz, 1H), 3.11 (d, J = 7.3 Hz, 1H), 1.76 (s, 1H), 1.37–1.09 (m, 2H), 1.09–0.54 (m, 6H). 13C NMR (75 MHz, D2O) δ 174.77, 144.16, 141.79, 136.43, 132.51, 122.59, 111.24, 108.92, 88.14, 82.12, 73.06, 69.59, 68.05, 63.61, 59.73, 35.97, 23.63, 14.02, 10.44. HRMS (ESI): m/z calcd. for C19H28N5O8S [M−H]−: 486.1664; found, 486.1664.



 ,
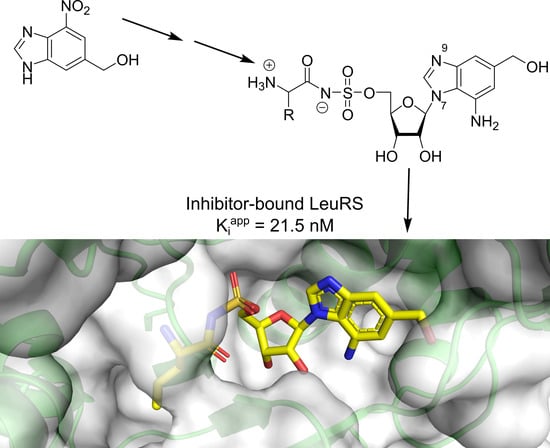
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








