
Photooxidation of 2-(tert-Butyl)-3-Methyl-2,3,5,6,7,8-Hexahydroquinazolin-4(1H)-one, an Example of Singlet Oxygen ene Reaction †



Abstract
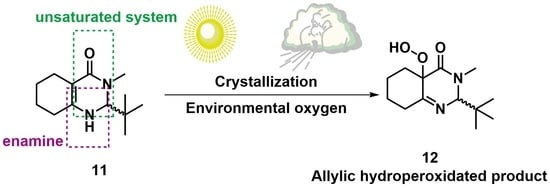
:
1. Introduction
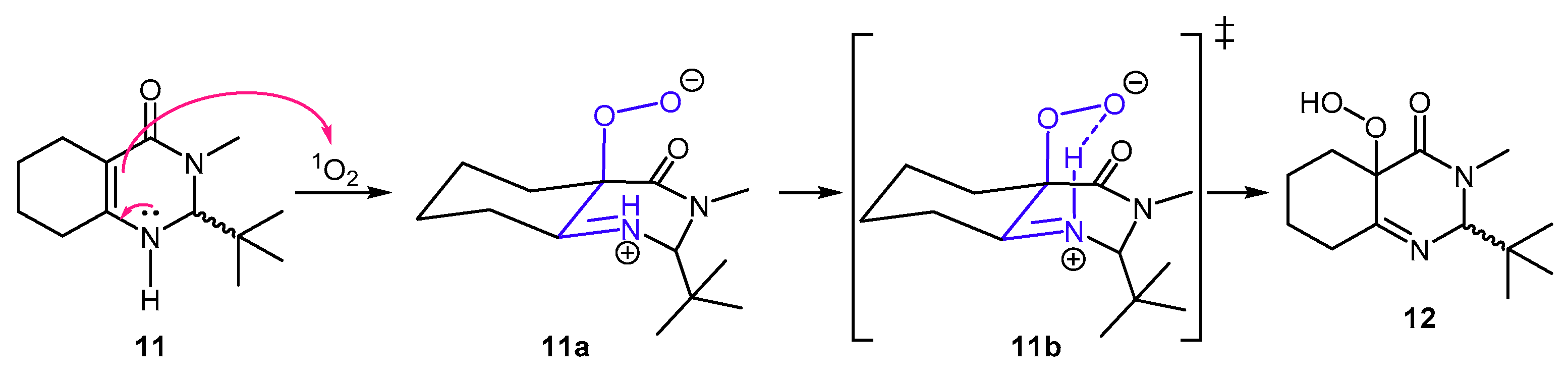
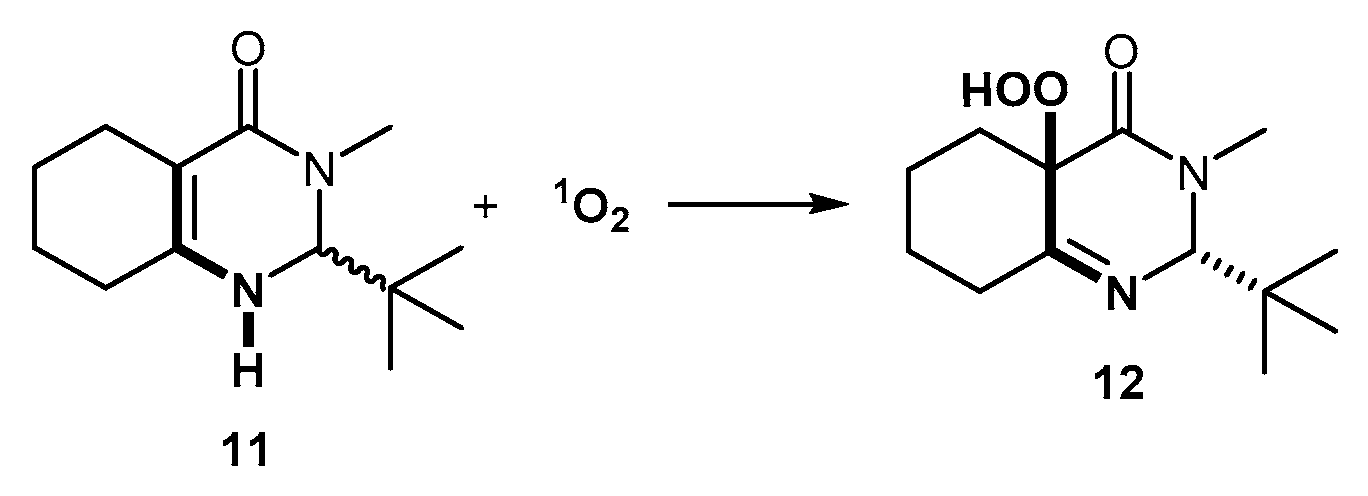
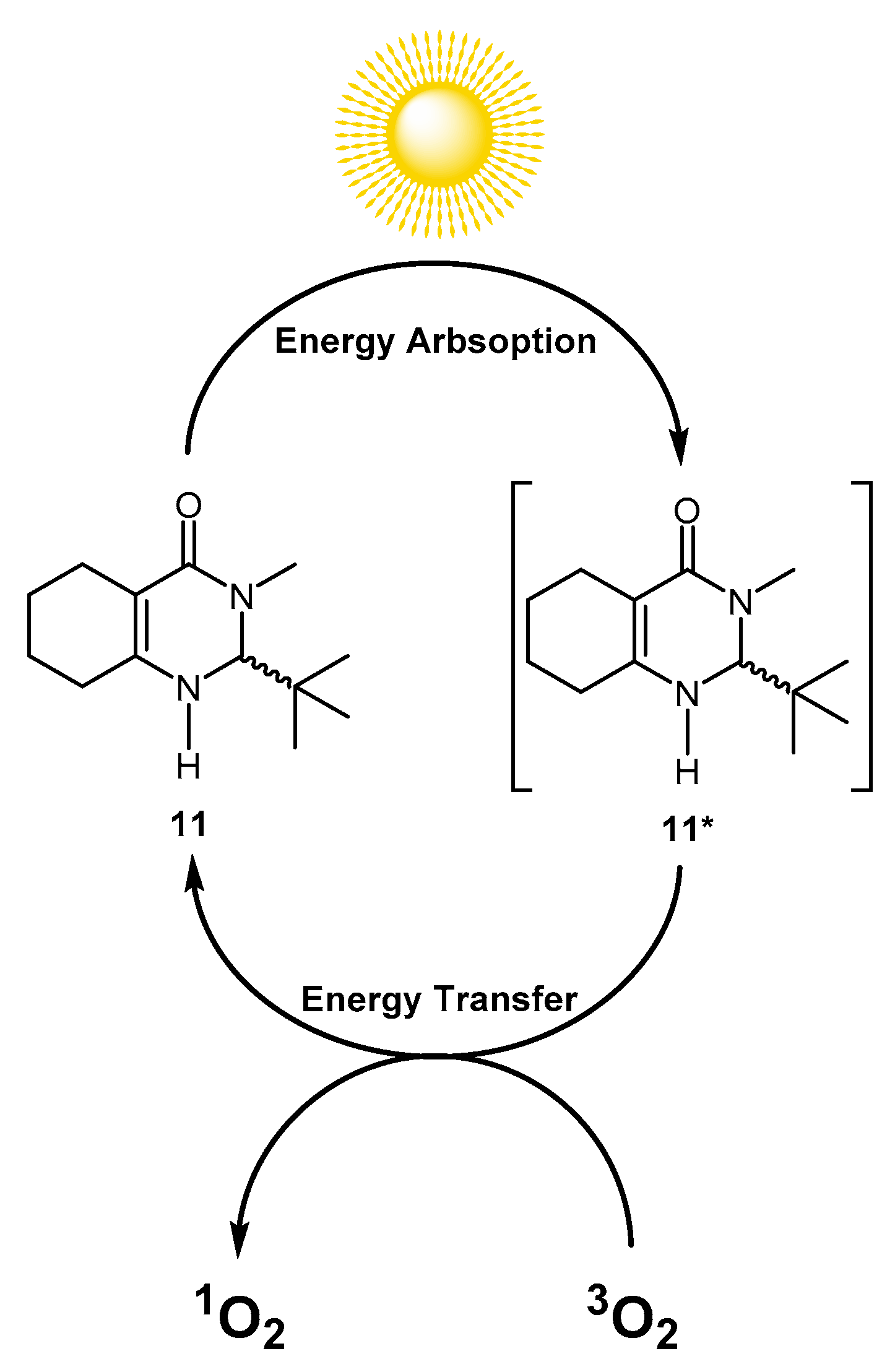
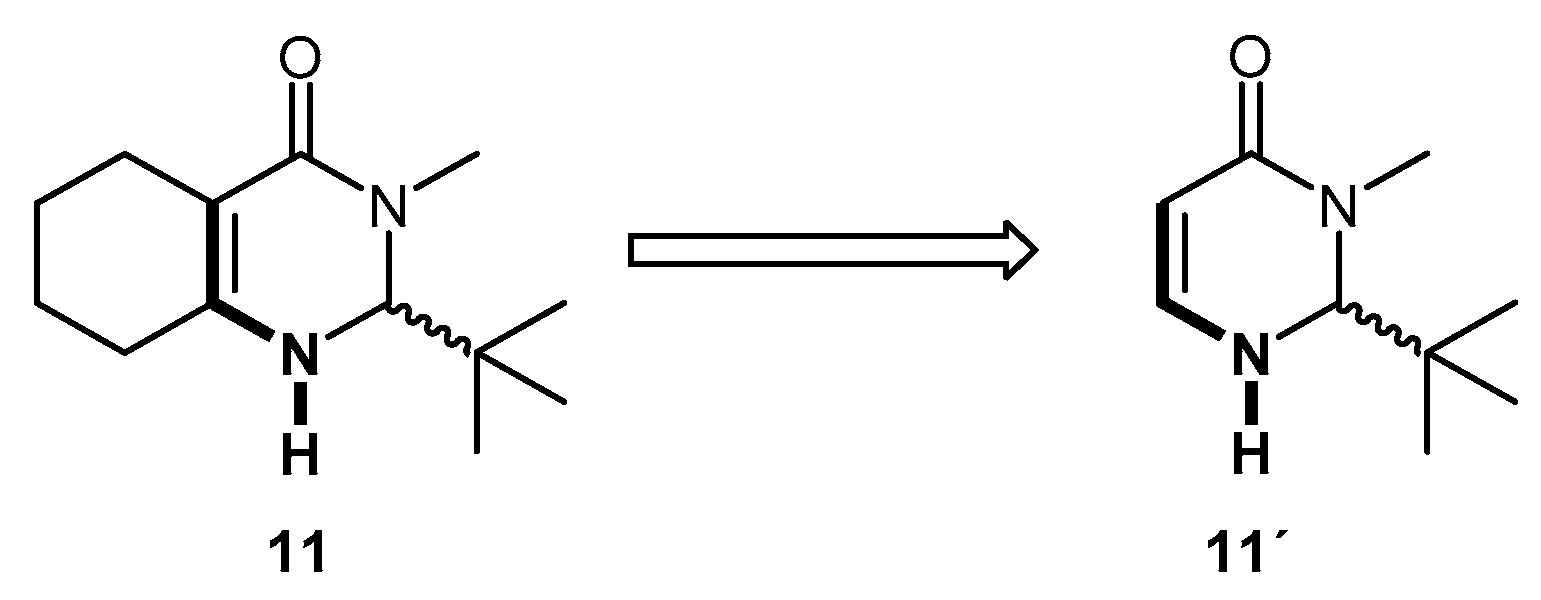
2. Results and Discussion
3. Materials and Methods
3.1. Analytical Methods
3.2. Theorical Study
3.3. Chemistry
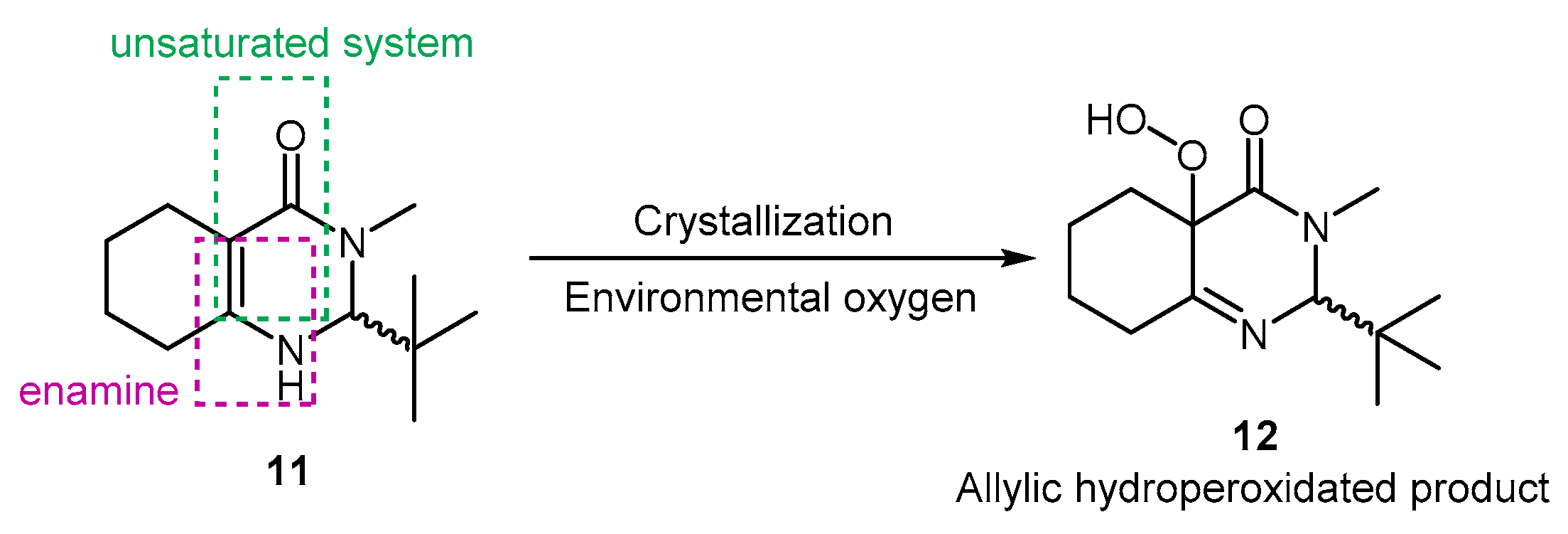
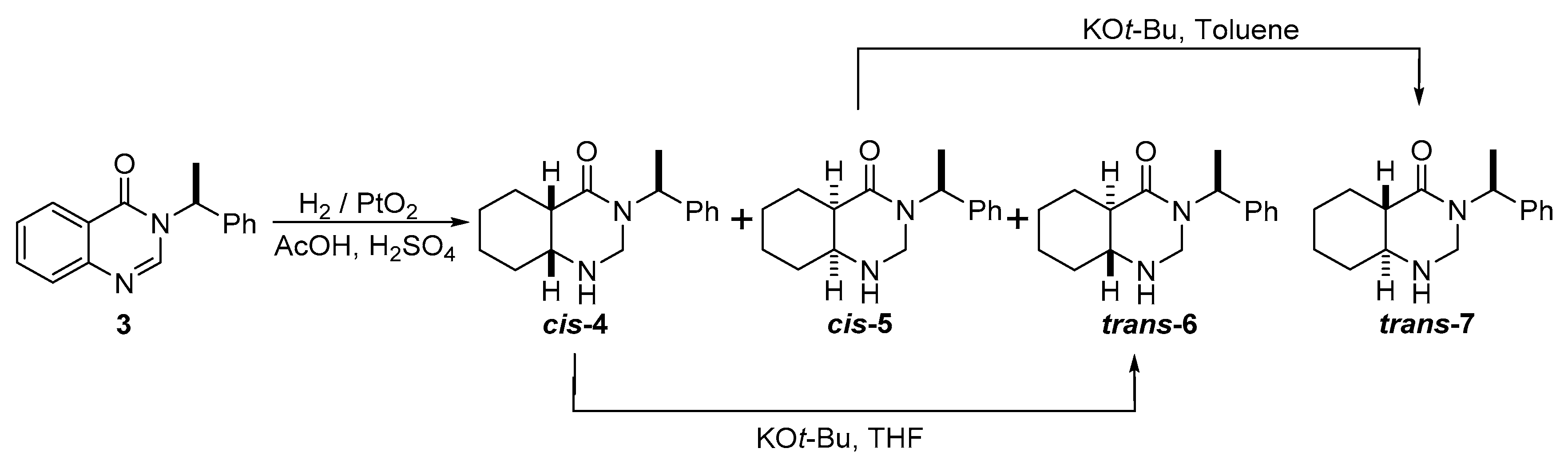
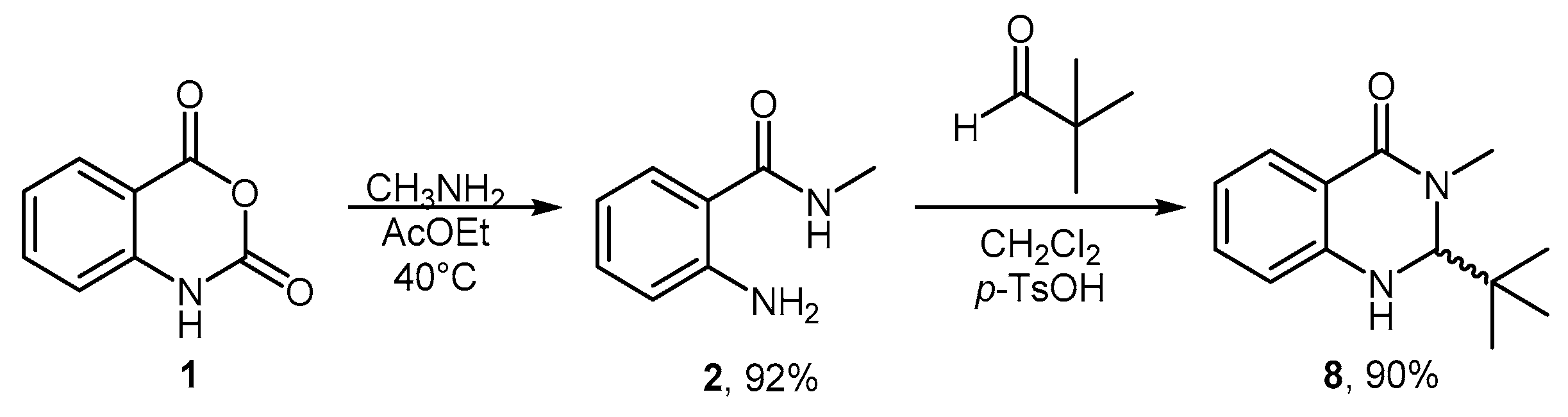
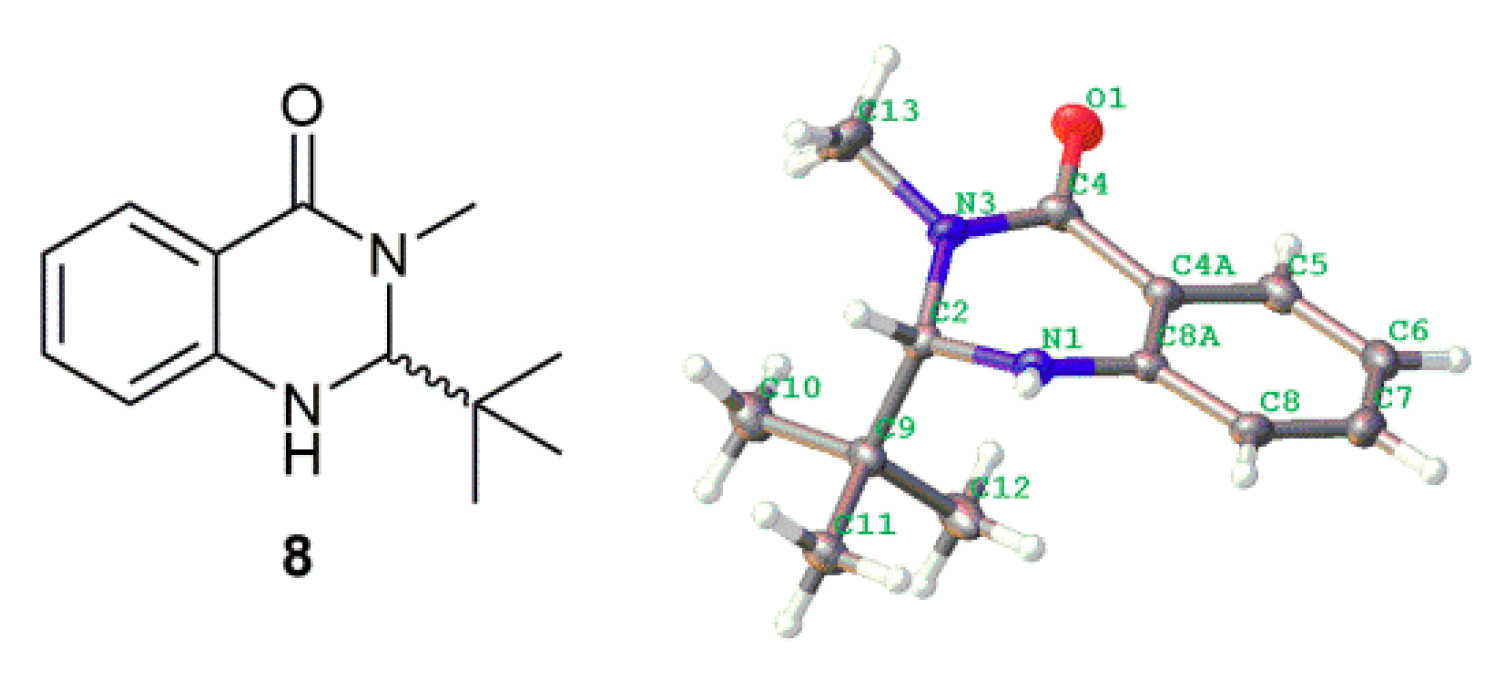
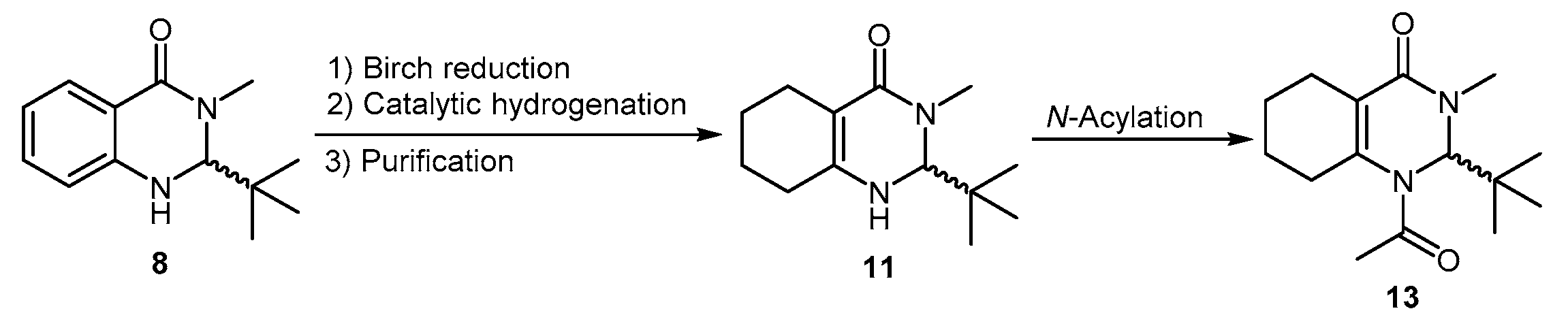
3.3.1. Synthesis of 2-(tert-butyl)-3-methyl-2,3-dihydroquinazolin-4(1H)-one (8)
3.3.2. Synthesis of 2-(tert-butyl)-3-methyl-2,3,5,6,7,8-hexahydroquinazolin-4(1H)-one (11)
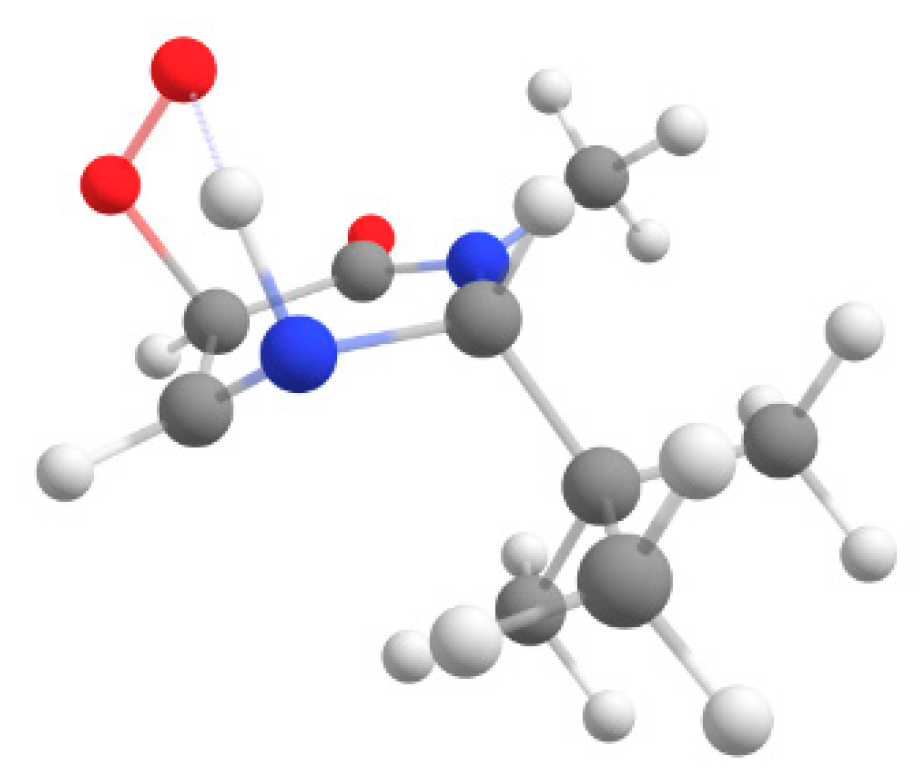
3.3.3. Synthesis of 2-(tert-Butyl)-4a-hydroperoxy-3-methyl-2,4a,5,6,7,8-hexahydroquinazolin- 4(3H)-one (12)
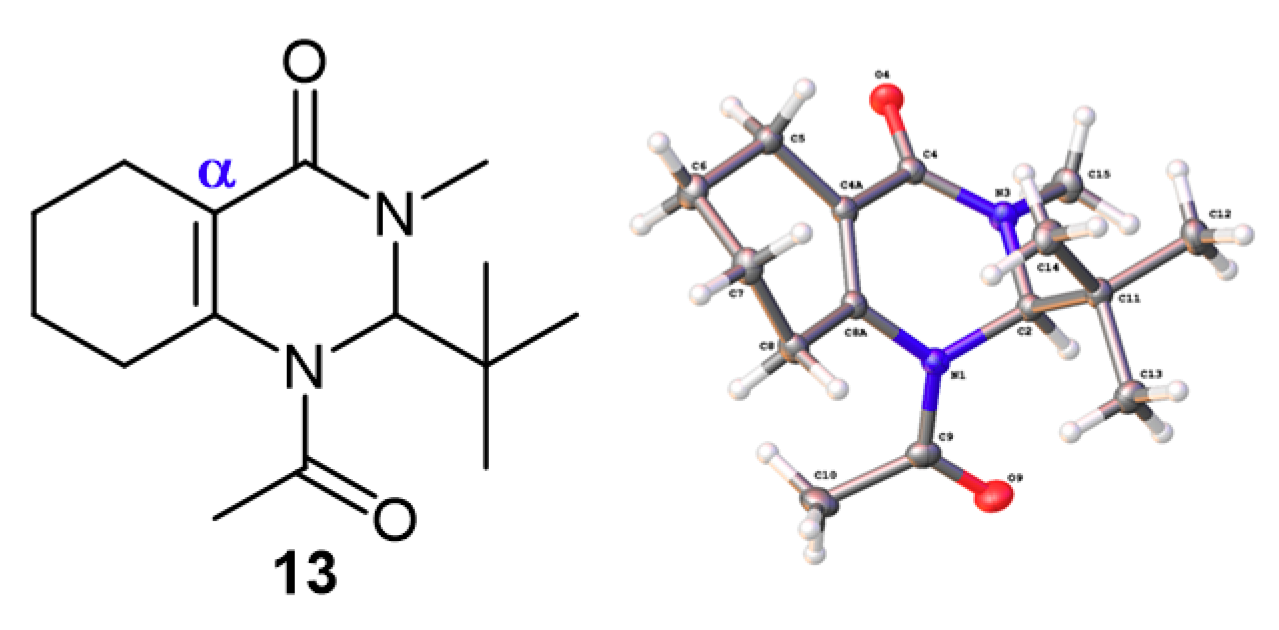
3.3.4. Synthesis of 1-Acetyl-2-(tert-butyl)-3-methyl-2,3,5,6,7,8-hexahydroquinazolin-4(1H)-one (13)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Baklanov, A.V.; Parker, D.H. Weakly Bound Environment of Molecular Oxygen as a Catalyst of Photooxidation. Kinet. Catal. 2020, 61, 174–197. [Google Scholar] [CrossRef]
- Taniguchi, T. Strategy for the Use of Molecular Oxygen in Organic Synthesis. Synlett 2020, 31. [Google Scholar] [CrossRef]
- Bian, C.; Singh, A.K.; Niu, L.; Yi, H.; Lei, A. Visible-Light-Mediated Oxygenation Reactions using Molecular Oxygen. Asian J. Org. Chem. 2017, 6, 386–396. [Google Scholar] [CrossRef]
- Xie, X.; Zhang, Z.; Hu, Y.; Cheng, H. A mechanistic kinetic model for singlet oxygen mediated self-sensitized photo-oxidation of organic pollutants in water. Chem. Eng. J. 2018, 334, 1242–1251. [Google Scholar] [CrossRef]
- Rontani, J.F.; Belt, S.T. Photo-and autoxidation of unsaturated algal lipids in the marine environment: An overview of processes, their potential tracers, and limitations. Org. Geochem. 2020, 139, 103941. [Google Scholar] [CrossRef]
- Davies, M.J. Singlet oxygen-mediated damage to proteins and its consequences. Biochem. Biophs. Res. Commun. 2003, 305, 761–770. [Google Scholar] [CrossRef]
- Srivastava, V.; Singh, P.K.; Singh, P.P. Eosin Y catalysed visible-light mediated aerobic oxidation of tertiary amines. Tetrahedron Lett. 2019, 60, 151041. [Google Scholar] [CrossRef]
- Zhang, M.; Lan, H.; Li, N.; Zhong, Q.; Zhu, H.; Liu, C.; Zhao, H.W. Photocatalyst-Free Singlet Oxygen-Induced Oxygenation: A Strategy for the Preparation of 5-Cyano-2-Pyridones Driven by Blue-Light Irradiation. J. Org. Chem. 2020, 85, 8279–8286. [Google Scholar] [CrossRef]
- Griesbeck, A.G.; Cho, M. Singlet oxygen addition to homoallylic substrates in solution and microemulsion: Novel secondary reactions. Tetrahedron Lett. 2009, 50, 121–123. [Google Scholar] [CrossRef]
- Zhang, Y.; Schilling, W.; Das, S. Metal-Free Photocatalysts for C-H Bond Oxygenation Reactions with Oxygen as the Oxidant. ChemSusChem 2019, 12, 2898–2910. [Google Scholar] [CrossRef]
- Fukuzumi, S.; Lee, Y.M.; Nam, W. Photocatalytic oxygenation reactions using water and dioxygen. ChemSusChem 2019, 12, 3931–3940. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Hu, J.; Mo, X.; Hao, Q.; Fan, Z.; He, G.; He, Q. Novel photocatalyst nitrogen-doped simonkolleite Zn5(OH)8Cl2·H2O with vis-up-conversion photoluminescence and effective visible-light photocatalysis. Appl. Phys. A 2019, 125, 3. [Google Scholar] [CrossRef]
- Alberti, M.N.; Orfanopoulos, M. Unraveling the mechanism of the singlet oxygen ene reaction: Recent computational and experimental approaches. Chem. Eur. J. 2010, 16, 9414–9421. [Google Scholar] [CrossRef]
- Lanteri, D.; Quattrosoldi, S.; Soccio, M.; Basso, A.; Cavallo, D.; Munari, A.; Riva, R.; Lotti, N.; Moni, L. Regioselective photooxidation of citronellol: A way to monomers for functionalized bio-polyesters. Front. Chem. 2020, 8, 85. [Google Scholar] [CrossRef] [PubMed]
- Ghogare, A.A.; Greer, A. Using singlet oxygen to synthesize natural products and drugs. Chem. Rev. 2016, 116, 9994–10034. [Google Scholar] [CrossRef] [PubMed]
- Priego, J.; Flores, P.; Ortiz-Nava, C.; Escalante, J. Synthesis of enantiopure cis-and trans-2-aminocyclohexane-1-carboxylic acids from octahydroquinazolin-4-ones. Tetrahedron Asymmetry 2004, 15, 3545–3549. [Google Scholar] [CrossRef]
- Zimmerman, H.E. A mechanistic analysis of the Birch reduction. Acc. Chem. Res. 2012, 45, 164–170. [Google Scholar] [CrossRef]
- Méndez-Ochoa, A. Síntesis y Resolución de 4-Quinazolinonas y su Posible Utilidad en la Síntesis de Ácidos β-Aminociclohexancarboxílicos. Master’s Thesis, Autonomous Morelos State University, Morelos, Mexico, 2017. [Google Scholar]
- Escalante, J.; Ortíz-Nava, C.; Flores, P.; Priego, J.M.; García-Martínez, C. Synthesis, NMR and crystallographic studies of 2-substituted dihydroquinazolinones derived from (S)-phenylethylamine. Molecules 2007, 12, 173–182. [Google Scholar] [CrossRef] [Green Version]
- Schenck, G.O. Understanding and Manipulating Excited-State Processes. German Patent DE-B 933925, 6 October 1955. [Google Scholar]
- Schenck, G.O.; Eggert, H.; Denk, W. Photochemische Reaktionen III. Über die Bildung von Hydroperoxyden bei photosensibilisierten Reacktionen von O2 mit geeigneten Akzeptoren, insbesondere mit α- und β-Pinen. Liebigs Ann. Chem. 1953, 584, 177. [Google Scholar] [CrossRef]
- Cabrera-Rivera, F.A.; Ortíz-Nava, C.; Escalante, J.; Hernández-Pérez, J.M.; Hô, M. Photoinduced elimination in 2,3-dihydro-2-tert-butyl-3-benzyl-4 (1H)-quinazolinone: Theoretical calculations and radical trapping using TEMPO derivatives. Synlett 2012, 23, 1057–1063. [Google Scholar]
- Aganda, K.C.C.; Hong, B.; Lee, A. Aerobic α-Oxidation of N-Substituted Tetrahydroisoquinolines to Dihydroisoquinolones via Organo-photocatalysis. Adv. Synth. Catal. 2019, 5, 1124–1129. [Google Scholar] [CrossRef]
- Manju, T.; Manoj, N.; Braun, A.M.; Oliveros, E. Self-sensitized photooxidation of N-methyl phenothiazine: Acidity control of the competition between electron and energy transfer mechanisms. Photochem. Photobiol. Sci. 2012, 11, 1744–1755. [Google Scholar] [CrossRef]
- Davies, A.G.; Schiesser, C.H. A PM3 study of the reactions of propene with singlet oxygen and other enophiles. Tetrahedron 1991, 47, 1707–1726. [Google Scholar] [CrossRef]
- Cossy, J.; Belotti, D. Aromatization of enamines promoted by a catalytic amount of Pd/C. Synthesis of aromatic Amines. Org. Lett. 2002, 4, 2557–2559. [Google Scholar] [CrossRef]
- Chen, Z.; Zeng, H.; Girard, S.A.; Wang, F.; Chen, N.; Li, C.J. Formal direct cross-coupling of phenols with amines. Angew. Chem. Int. Ed. 2015, 54, 14487–14491. [Google Scholar] [CrossRef]
- Qiu, Z.; Li, J.-S.; Li, C.-J. Formal aromaticity transfer for palladium-catalyzed coupling between phenols and pyrrolidines/indolines. Chem. Sci. 2017, 8, 6954–6958. [Google Scholar]
- Chen, S.; Liao, Y.; Zhao, F.; Qi, H.; Liu, S.; Deng, G.J. Palladium-catalyzed direct arylation of indoles with cyclohexanones. Org. Lett. 2014, 16, 1618–1621. [Google Scholar] [CrossRef]
- Jumde, V.R.; Petricci, E.; Petrucci, C.; Santillo, N.; Taddei, M.; Vaccaro, L. Domino hydrogenation–reductive amination of phenols, a simple process to access substituted cyclohexylamines. Org. Lett. 2015, 17, 3990–3993. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Revision, A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. IV. A new dynamical correlation functional and implications for exact-exchange mixing. J. Chem. Phys. 1996, 104, 1040–1046. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–652. [Google Scholar] [CrossRef]
- Gill, P.M.W.; Johnson, B.G.; Pople, J.A.; Frisch, M.J. The performance of the Becke—Lee—Yang—Parr (B—LYP) density functional theory with various basis sets. Chem. Phys. Lett. 1992, 197, 499–505. [Google Scholar] [CrossRef] [Green Version]
- Peng, C.; Schlegel, H.B. Combining Synchronous Transit and Quasi-Newton Methods to Find Transition States. Isr. J. Chem. 1993, 33, 449–454. [Google Scholar] [CrossRef]
- Fukui, K. The path of chemical reactions—The IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
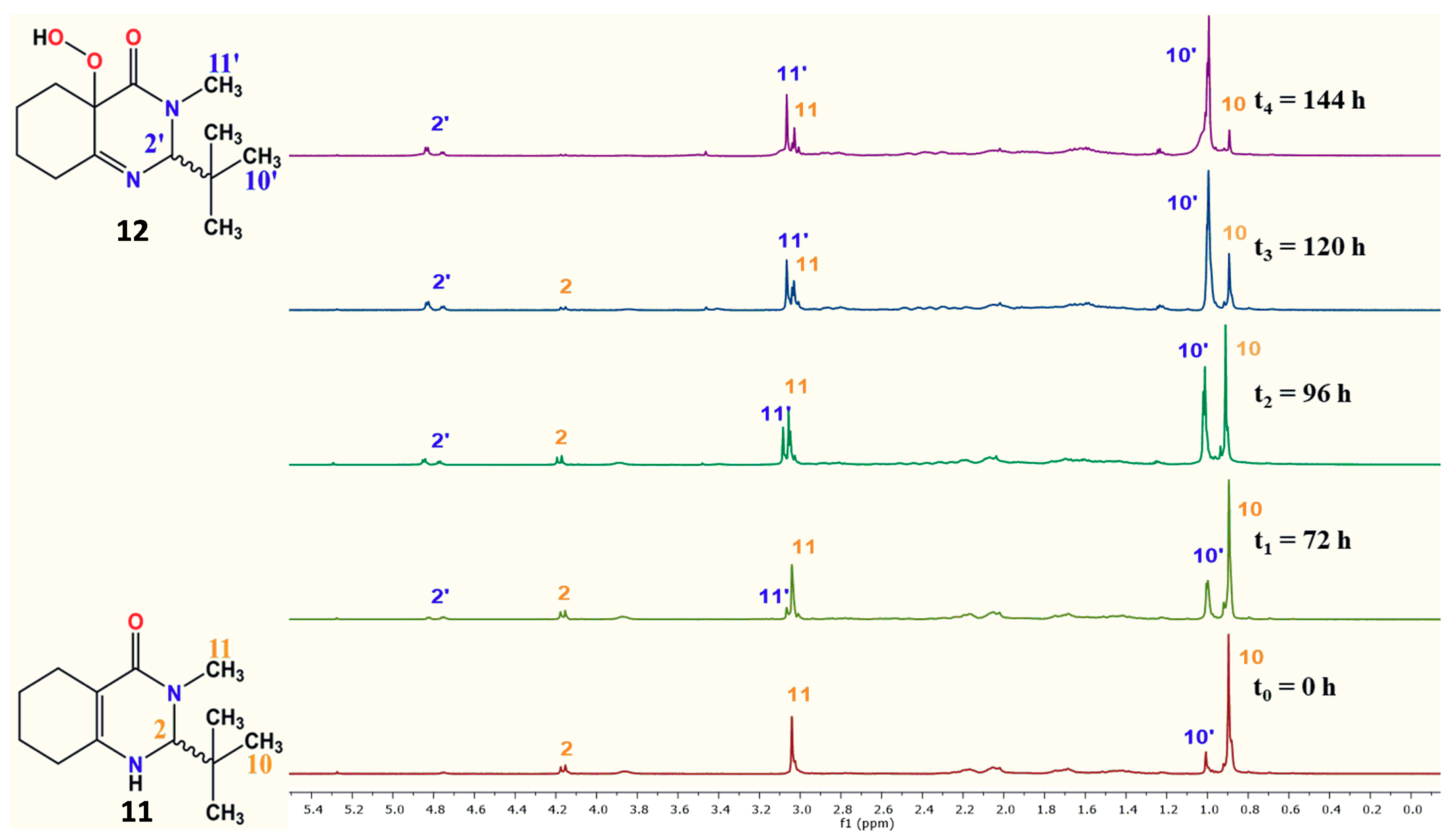
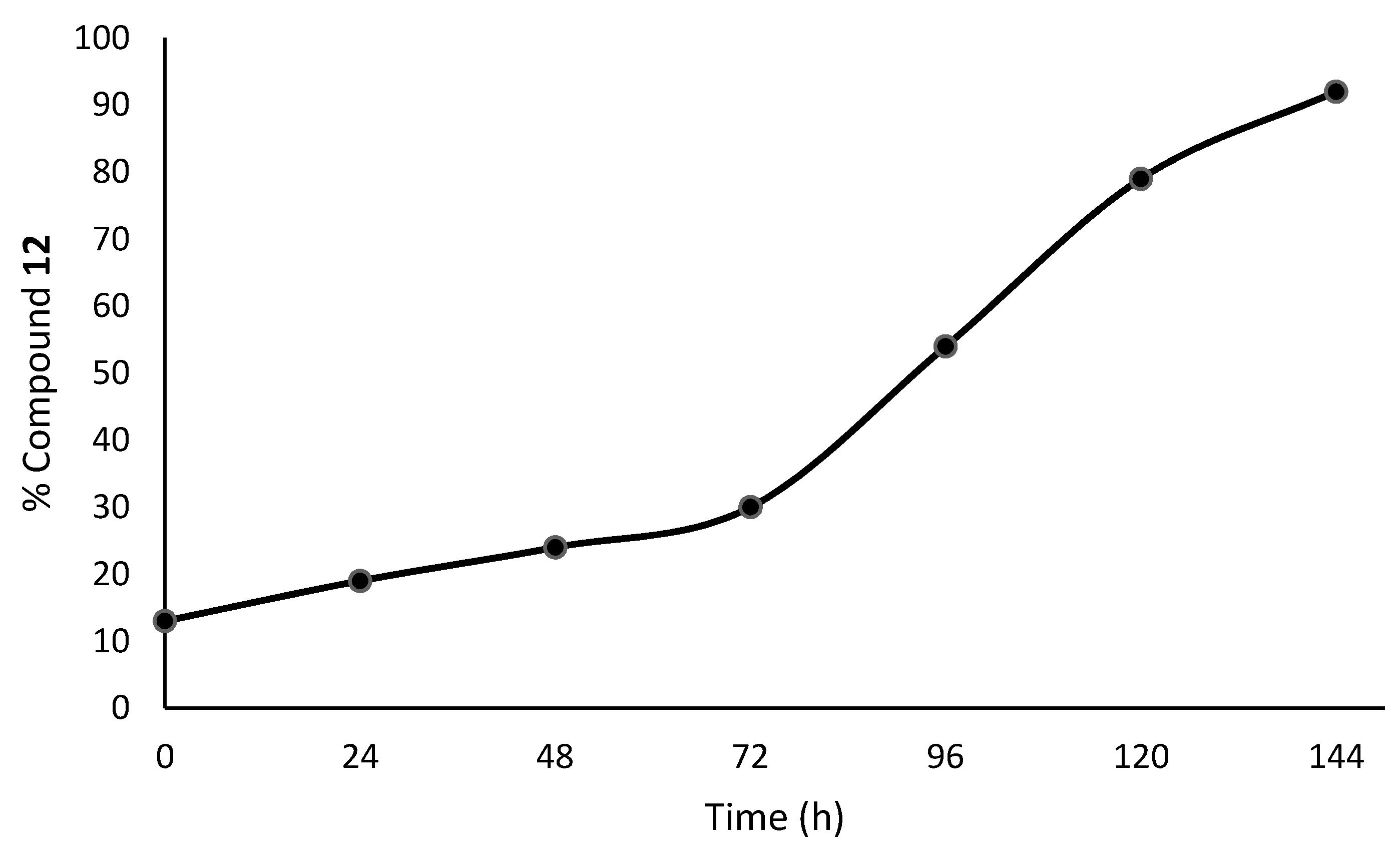
| 1H-NMR Spectra | Exposure Time (h) | 11 (%) | 12 (%) | 12 (%) |
|---|---|---|---|---|
| 1 a | 0 | 87 | 13.0 | 5.6 5.6 5.6 24.2 25 13 |
| 2 b | 24 | 81 | 18.6 | |
| 2 b | 48 | 75 | 24.2 | |
| 2 b | 72 | 70 | 29.8 | |
| 3 | 96 | 46 | 54.0 | |
| 4 | 120 | 21 | 79.0 | |
| 5 | 144 | 8 | 92 |
Sample Availability: Small samples (a few milligrams) of the compounds 8, 12, and 13 are available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Méndez, A.; Valdez-Camacho, J.R.; Escalante, J. Photooxidation of 2-(tert-Butyl)-3-Methyl-2,3,5,6,7,8-Hexahydroquinazolin-4(1H)-one, an Example of Singlet Oxygen ene Reaction. Molecules 2020, 25, 5008. https://doi.org/10.3390/molecules25215008
Méndez A, Valdez-Camacho JR, Escalante J. Photooxidation of 2-(tert-Butyl)-3-Methyl-2,3,5,6,7,8-Hexahydroquinazolin-4(1H)-one, an Example of Singlet Oxygen ene Reaction. Molecules. 2020; 25(21):5008. https://doi.org/10.3390/molecules25215008
Chicago/Turabian StyleMéndez, Adrian, Jonathan Román Valdez-Camacho, and Jaime Escalante. 2020. "Photooxidation of 2-(tert-Butyl)-3-Methyl-2,3,5,6,7,8-Hexahydroquinazolin-4(1H)-one, an Example of Singlet Oxygen ene Reaction" Molecules 25, no. 21: 5008. https://doi.org/10.3390/molecules25215008