
Comprehensive Review on Alzheimer’s Disease: Causes and Treatment



Abstract
:1. Introduction
2. Alzheimer’s Disease Diagnostic Criteria
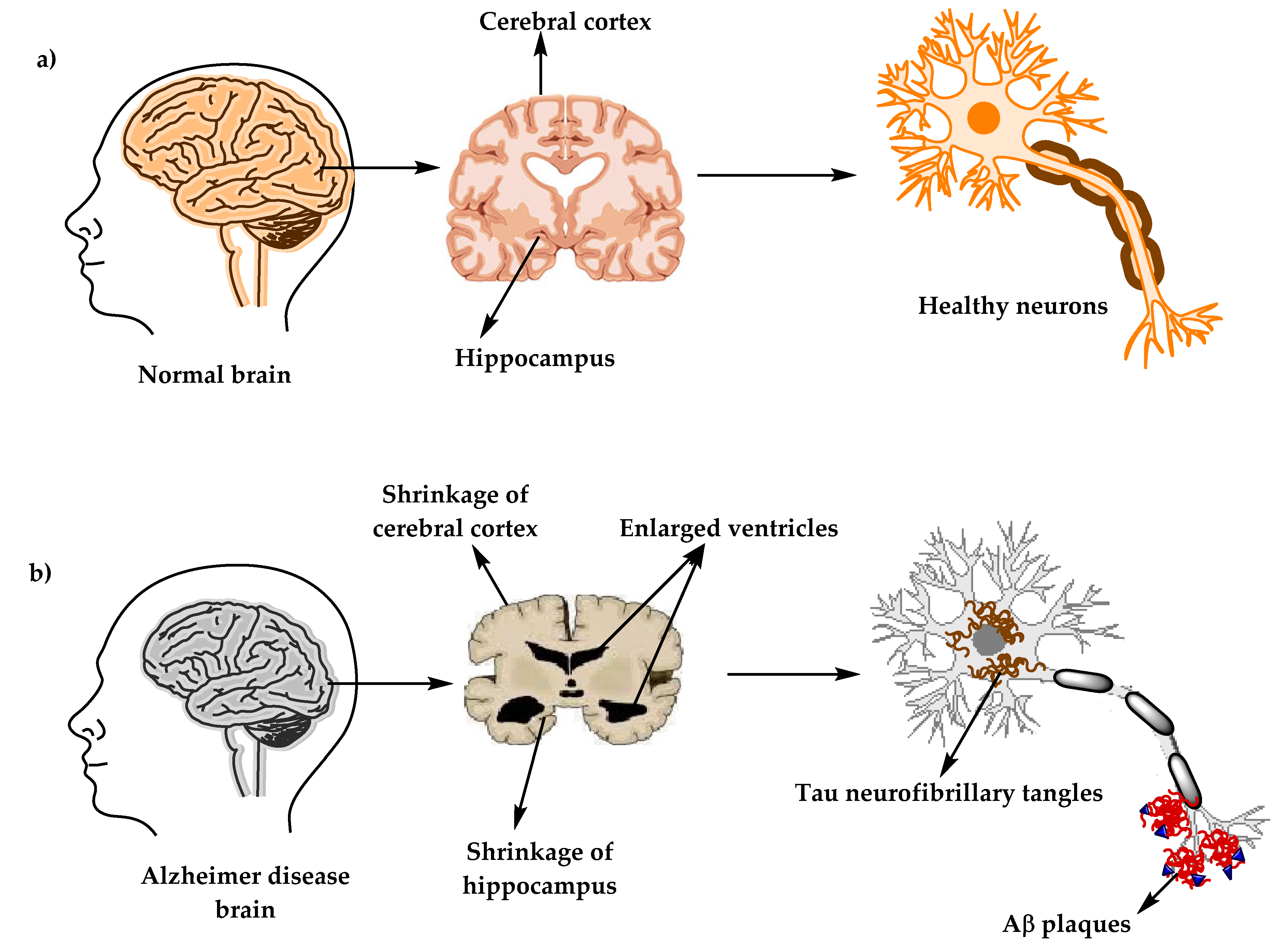
3. Alzheimer’s Disease’s Neuropathology
3.1. Senile Plaques (SP)
3.2. Neurofibrillary Tangles (NFTs)
3.3. Synaptic Loss
4. The Stages of Alzheimer’s Disease
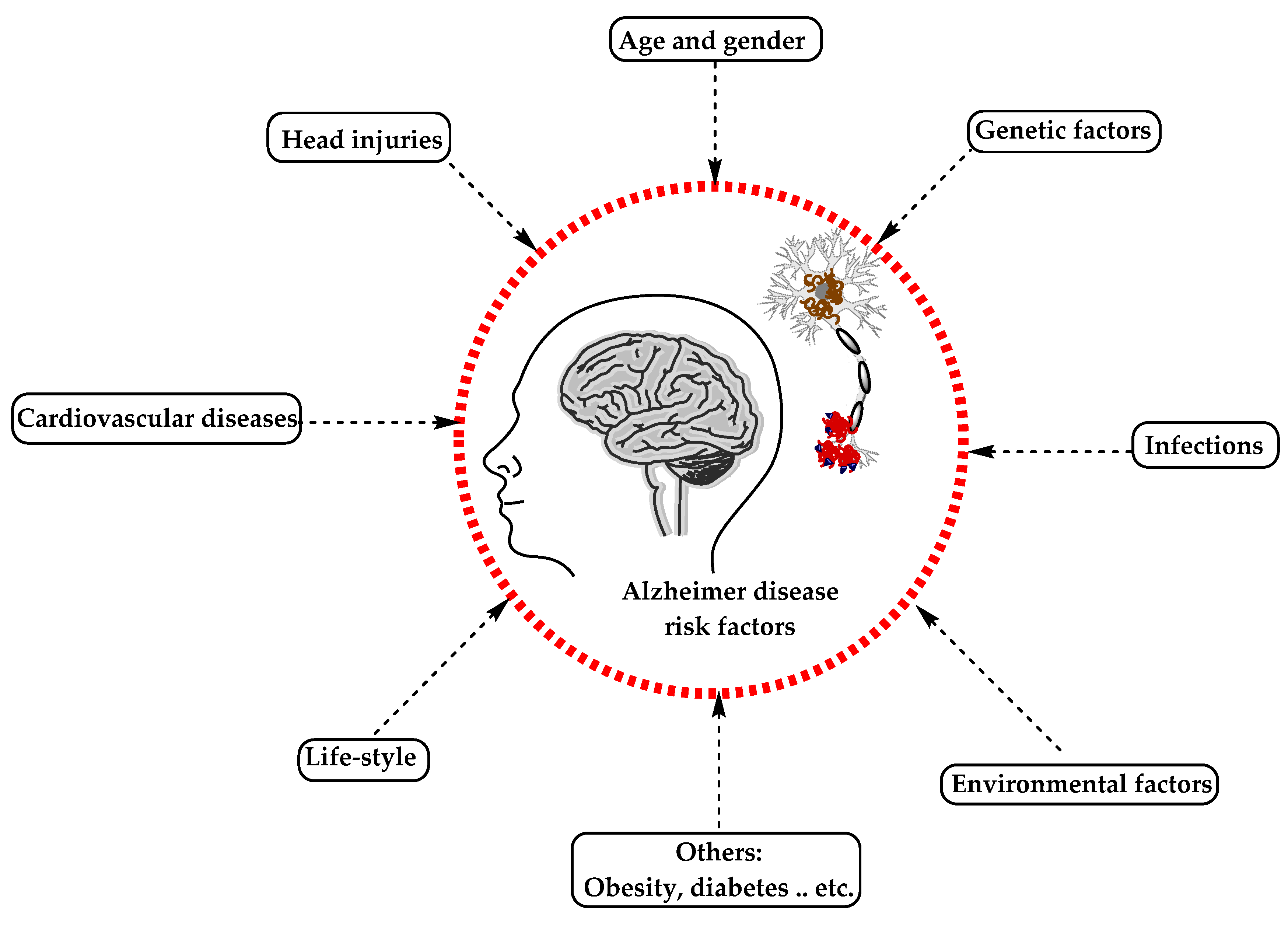
5. Causes and Risk Factors of Alzheimer’s Disease
5.1. Alzheimer’s Disease Hypotheses
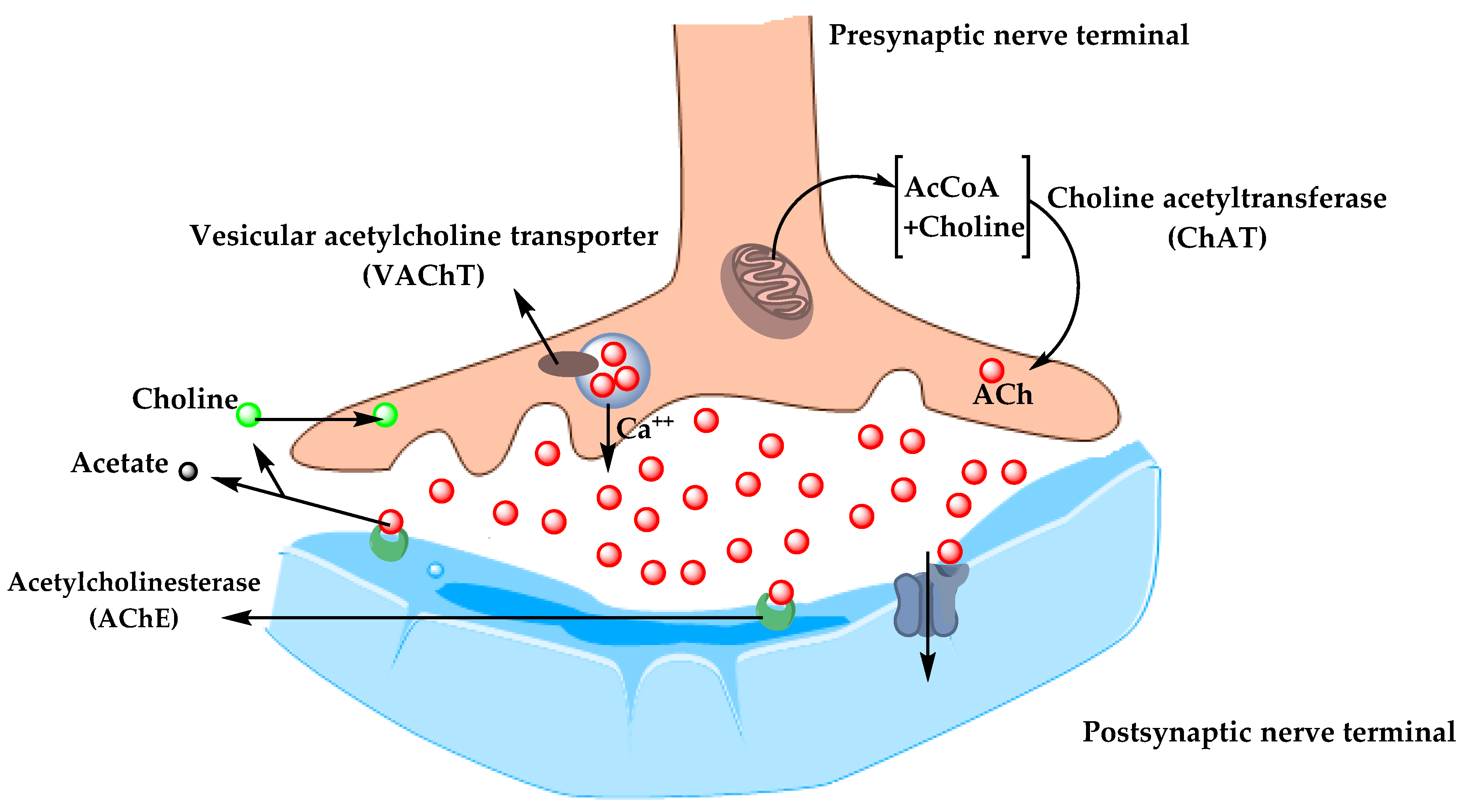
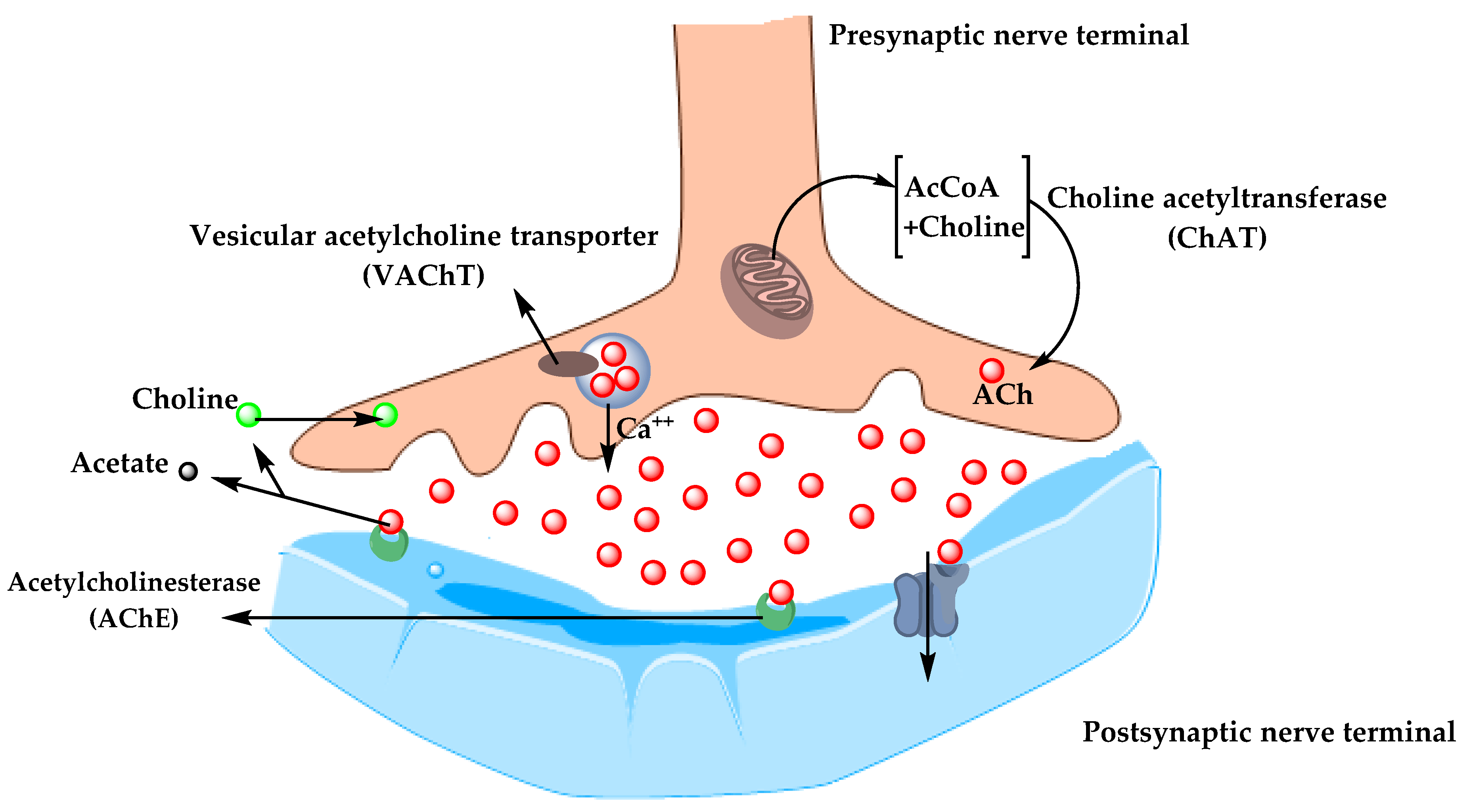
5.1.1. Cholinergic Hypothesis
5.1.2. Amyloid Hypothesis
5.2. Alzheimer’s Disease Risk Factors
5.2.1. Aging
5.2.2. Genetics
- Amyloid Precursor Protein (APP)
- Presenilin-1 (PSEN-1) and Presenilin-2 (PSEN-2)
- Apolipoprotein E (ApoE)
- ATP Binding Cassette Transporter A1 (ABCA1)
- Clusterin Gene (CLU) and Bridging Integrator 1 (BIN1)
- Evolutionarily Conserved Signaling Intermediate in Toll pathway (ECSIT)
- Estrogen Receptor Gene (ESR)
- Other Genes
5.2.3. Environmental Factors
- Air Pollution
- Diet
- Metals
- Infections
5.2.4. Medical Factors
- Cardiovascular Disease (CVDs)
- Obesity and Diabetes
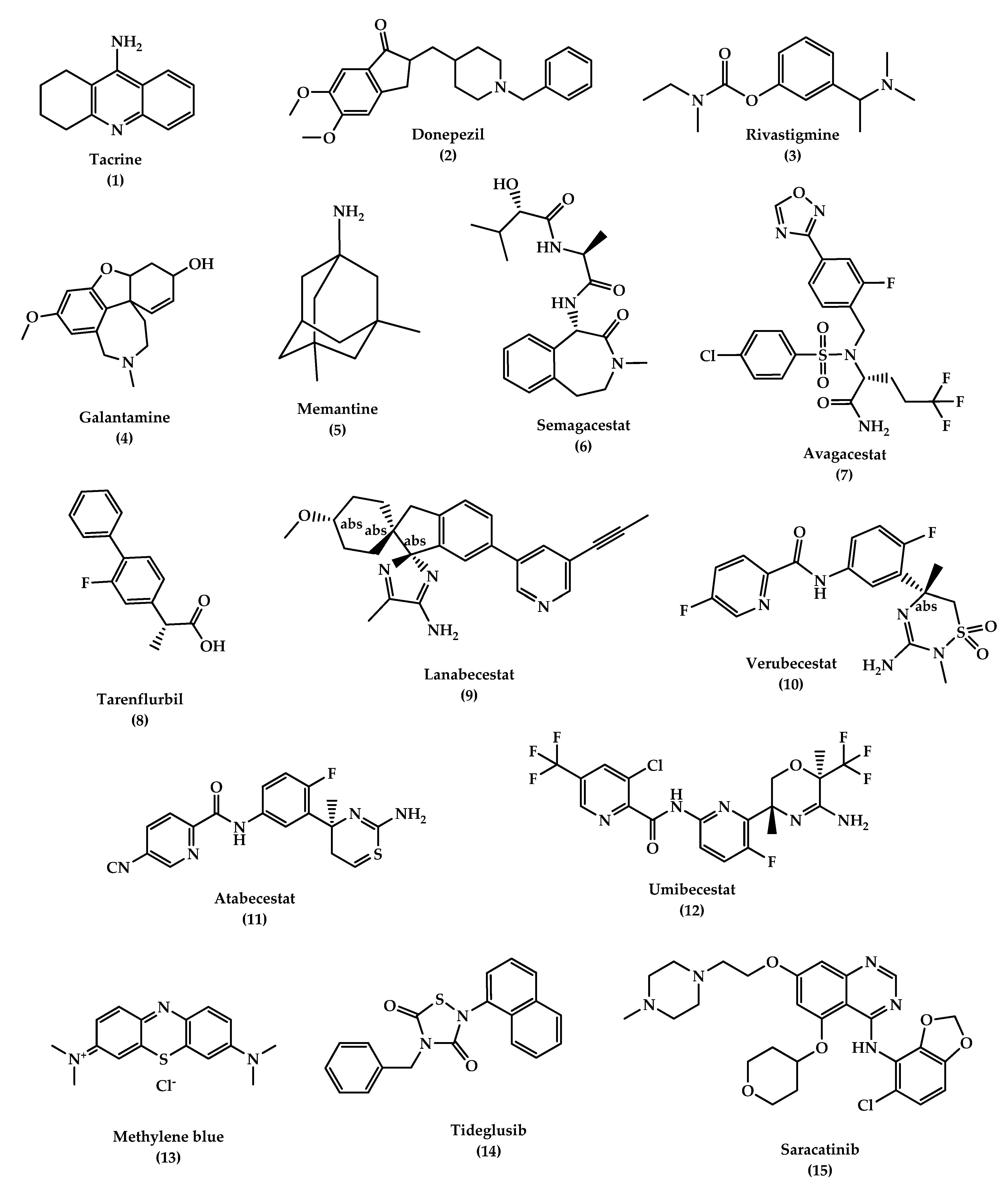
6. Treatment
6.1. Symptomatic Treatment of AD
6.1.1. Cholinesterase Inhibitors
- Donepezil
- Rivastigmine
- Galantamine (GAL)
6.1.2. N-methyl d-aspartate (NMDA) Antagonists
- Memantine
6.2. Promising Future Therapies
6.2.1. Disease-Modifying Therapeutics (DMT)
6.2.2. Chaperones
- Heat Shock Proteins (Hsps)
- Hsp60
- 2.
- Hsp70
- 3.
- Hsp90
- Vacuolar sorting protein 35 (VPS35)
- OT1001
6.2.3. Natural Extract
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- De-Paula, V.J.; Radanovic, M.; Diniz, B.S.; Forlenza, O.V. Alzheimer’s disease. Sub-Cell. Biochem. 2012, 65, 329–352. [Google Scholar] [CrossRef]
- Cipriani, G.; Dolciotti, C.; Picchi, L.; Bonuccelli, U. Alzheimer and his disease: A brief history. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2011, 32, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Blass, J.P. Alzheimer’s disease. Dis. A Mon. Dm 1985, 31, 1–69. [Google Scholar] [CrossRef]
- Terry, R.D.; Davies, P. Dementia of the Alzheimer type. Annu. Rev. Neurosci. 1980, 3, 77–95. [Google Scholar] [CrossRef] [PubMed]
- Rathmann, K.L.; Conner, C.S. Alzheimer’s disease: Clinical features, pathogenesis, and treatment. Drug Intell. Clin. Pharm. 1984, 18, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments in alzheimer disease: An update. J. Cent. Nerv. Syst. Dis. 2020, 12. [Google Scholar] [CrossRef] [Green Version]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef]
- Schachter, A.S.; Davis, K.L. Alzheimer’s disease. Dialogues Clin. Neurosci. 2000, 2, 91–100. [Google Scholar] [CrossRef]
- Jatoi, S.; Hafeez, A.; Riaz, S.U.; Ali, A.; Ghauri, M.I.; Zehra, M. Low Vitamin B12 levels: An underestimated cause of minimal cognitive impairment and dementia. Cureus 2020, 12, e6976. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.S.; Huang, L.K.; Lee, Y.T.; Chan, L.; Hong, C.T. Suboptimal baseline serum Vitamin B12 is associated with cognitive decline in people with Alzheimer’s disease undergoing cholinesterase inhibitor treatment. Front. Neurol. 2018, 9, 325. [Google Scholar] [CrossRef]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neugroschl, J.; Wang, S. Alzheimer’s disease: Diagnosis and treatment across the spectrum of disease severity. Mt. Sinai J. Med. N. Y. 2011, 78, 596–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayeux, R.; Stern, Y. Epidemiology of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaari, R.; Fleisher, A.S.; Tariot, P.N. Updates to diagnostic guidelines for Alzheimer’s disease. Prim. Care Companion Cns Disord. 2011, 13, 11f01262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Spires-Jones, T.L.; Hyman, B.T. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Srivastav, S.; Yadav, A.K.; Srikrishna, S.; Perry, G. Overview of Alzheimer’s disease and some therapeutic approaches targeting abeta by using several synthetic and herbal compounds. Oxidative Med. Cell. Longev. 2016, 2016, 7361613. [Google Scholar] [CrossRef] [Green Version]
- Cras, P.; Kawai, M.; Lowery, D.; Gonzalez-DeWhitt, P.; Greenberg, B.; Perry, G. Senile plaque neurites in Alzheimer disease accumulate amyloid precursor protein. Proc. Natl. Acad. Sci. USA 1991, 88, 7552–7556. [Google Scholar] [CrossRef] [Green Version]
- Perl, D.P. Neuropathology of Alzheimer’s disease. Mt. Sinai J. Med. N. Y. 2010, 77, 32–42. [Google Scholar] [CrossRef]
- Armstrong, R.A. The molecular biology of senile plaques and neurofibrillary tangles in Alzheimer’s disease. Folia Neuropathol. 2009, 47, 289–299. [Google Scholar] [PubMed]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- Tabaton, M.; Piccini, A. Role of water-soluble amyloid-beta in the pathogenesis of Alzheimer’s disease. Int. J. Exp. Pathol. 2005, 86, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Brion, J.P. Neurofibrillary tangles and Alzheimer’s disease. Eur. Neurol. 1998, 40, 130–140. [Google Scholar] [CrossRef]
- Metaxas, A.; Kempf, S.J. Neurofibrillary tangles in Alzheimer’s disease: Elucidation of the molecular mechanism by immunohistochemistry and tau protein phospho-proteomics. Neural Regen. Res. 2016, 11, 1579–1581. [Google Scholar] [CrossRef]
- Overk, C.R.; Masliah, E. Pathogenesis of synaptic degeneration in Alzheimer’s disease and Lewy body disease. Biochem Pharm. 2014, 88, 508–516. [Google Scholar] [CrossRef] [Green Version]
- Lleo, A.; Nunez-Llaves, R.; Alcolea, D.; Chiva, C.; Balateu-Panos, D.; Colom-Cadena, M.; Gomez-Giro, G.; Munoz, L.; Querol-Vilaseca, M.; Pegueroles, J.; et al. Changes in synaptic proteins precede neurodegeneration markers in preclinical Alzheimer’s disease cerebrospinal fluid. Mol. Cell. Proteom. Mcp 2019, 18, 546–560. [Google Scholar] [CrossRef] [Green Version]
- Tarawneh, R.; D’Angelo, G.; Crimmins, D.; Herries, E.; Griest, T.; Fagan, A.M.; Zipfel, G.J.; Ladenson, J.H.; Morris, J.C.; Holtzman, D.M. Diagnostic and prognostic utility of the synaptic marker neurogranin in Alzheimer Disease. JAMA Neurol. 2016, 73, 561–571. [Google Scholar] [CrossRef]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2016, 12, 292–323. [Google Scholar] [CrossRef]
- Kumar, A.; Sidhu, J.; Goyal, A. Alzheimer Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020; Available online: https://www.ncbi.nlm.nih.gov/books/NBK499922/ (accessed on 8 December 2020).
- Wattmo, C.; Minthon, L.; Wallin, A.K. Mild versus moderate stages of Alzheimer’s disease: Three-year outcomes in a routine clinical setting of cholinesterase inhibitor therapy. Alzheimer’s Res. Ther. 2016, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Apostolova, L.G. Alzheimer disease. Continuum 2016, 22, 419–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, R.A. Risk factors for Alzheimer’s disease. Folia Neuropathol. 2019, 57, 87–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, P.; Singh, B. A review on cholinesterase inhibitors for Alzheimer’s disease. Arch. Pharmacal Res. 2013, 36, 375–399. [Google Scholar] [CrossRef]
- Babic, T. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J. Neurol. Neurosurg. Psychiatry 1999, 67, 558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Monczor, M. Diagnosis and treatment of Alzheimer’s disease. Curr. Med. Chem. Cent. Nerv. Syst. Agents 2005, 5, 5–13. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain A J. Neurol. 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Paroni, G.; Bisceglia, P.; Seripa, D. Understanding the amyloid hypothesis in Alzheimer’s disease. J. Alzheimer’s Dis. Jad 2019, 68, 493–510. [Google Scholar] [CrossRef] [PubMed]
- Kametani, F.; Hasegawa, M. Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer’s disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef] [Green Version]
- Ricciarelli, R.; Fedele, E. The amyloid cascade hypothesis in Alzheimer’s disease: It’s time to change our mind. Curr. Neuropharmacol. 2017, 15, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Guerreiro, R.; Bras, J. The age factor in Alzheimer’s disease. Genome Med. 2015, 7, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedel, B.C.; Thompson, P.M.; Brinton, R.D. Age, APOE and sex: Triad of risk of Alzheimer’s disease. J. Steroid Biochem. Mol. Biol. 2016, 160, 134–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Bekris, L.M.; Yu, C.E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. Off. J. Am. Coll. Med Genet. 2016, 18, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Khanahmadi, M.; Farhud, D.D.; Malmir, M. Genetic of Alzheimer’s disease: A narrative review article. Iran. J. Public Health 2015, 44, 892–901. [Google Scholar]
- Li, N.M.; Liu, K.F.; Qiu, Y.J.; Zhang, H.H.; Nakanishi, H.; Qing, H. Mutations of beta-amyloid precursor protein alter the consequence of Alzheimer’s disease pathogenesis. Neural Regen. Res. 2019, 14, 658–665. [Google Scholar] [CrossRef]
- Tcw, J.; Goate, A.M. Genetics of beta-Amyloid precursor protein in Alzheimer’s disease. Cold Spring Harb. Perspect. Med. 2017, 7, a024539. [Google Scholar] [CrossRef]
- Bi, C.; Bi, S.; Li, B. Processing of mutant beta-amyloid precursor protein and the clinicopathological features of familial Alzheimer’s disease. Aging Dis. 2019, 10, 383–403. [Google Scholar] [CrossRef] [Green Version]
- Dai, M.H.; Zheng, H.; Zeng, L.D.; Zhang, Y. The genes associated with early-onset Alzheimer’s disease. Oncotarget 2018, 9, 15132–15143. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Liu, X.; Xia, W.; Zhang, Y.; Wang, C. Targeting amyloidogenic processing of APP in Alzheimer’s disease. Front. Mol. Neurosci. 2020, 13, 137. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; An, S.S.; Kim, S. Mutations in presenilin 2 and its implications in Alzheimer’s disease and other dementia-associated disorders. Clin. Interv. Aging 2015, 10, 1163–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanoiselee, H.M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Strooper, B. Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. Embo Rep. 2007, 8, 141–146. [Google Scholar] [CrossRef] [Green Version]
- Kelleher, R.J., 3rd; Shen, J. Presenilin-1 mutations and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 629–631. [Google Scholar] [CrossRef] [Green Version]
- Walker, E.S.; Martinez, M.; Brunkan, A.L.; Goate, A. Presenilin 2 familial Alzheimer’s disease mutations result in partial loss of function and dramatic changes in Abeta 42/40 ratios. J. Neurochem. 2005, 92, 294–301. [Google Scholar] [CrossRef]
- Kim, J.; Basak, J.M.; Holtzman, D.M. The role of apolipoprotein E in Alzheimer’s disease. Neuron 2009, 63, 287–303. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [Green Version]
- Giau, V.V.; Bagyinszky, E.; An, S.S.; Kim, S.Y. Role of apolipoprotein E in neurodegenerative diseases. Neuropsychiatr. Dis. Treat. 2015, 11, 1723–1737. [Google Scholar] [CrossRef] [Green Version]
- Koldamova, R.; Fitz, N.F.; Lefterov, I. ATP-binding cassette transporter A1: From metabolism to neurodegeneration. Neurobiol. Dis. 2014, 72 Pt A, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Nordestgaard, L.T.; Tybjaerg-Hansen, A.; Nordestgaard, B.G.; Frikke-Schmidt, R. Loss-of-function mutation in ABCA1 and risk of Alzheimer’s disease and cerebrovascular disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2015, 11, 1430–1438. [Google Scholar] [CrossRef] [PubMed]
- Foster, E.M.; Dangla-Valls, A.; Lovestone, S.; Ribe, E.M.; Buckley, N.J. Clusterin in Alzheimer’s disease: Mechanisms, genetics, and lessons from other pathologies. Front. Neurosci. 2019, 13, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holler, C.J.; Davis, P.R.; Beckett, T.L.; Platt, T.L.; Webb, R.L.; Head, E.; Murphy, M.P. Bridging integrator 1 (BIN1) protein expression increases in the Alzheimer’s disease brain and correlates with neurofibrillary tangle pathology. J. Alzheimer’s Dis. Jad 2014, 42, 1221–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrew, R.J.; De Rossi, P.; Nguyen, P.; Kowalski, H.R.; Recupero, A.J.; Guerbette, T.; Krause, S.V.; Rice, R.C.; Laury-Kleintop, L.; Wagner, S.L.; et al. Reduction of the expression of the late-onset Alzheimer’s disease (AD) risk-factor BIN1 does not affect amyloid pathology in an AD mouse model. J. Biol. Chem. 2019, 294, 4477–4487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soler-Lopez, M.; Badiola, N.; Zanzoni, A.; Aloy, P. Towards Alzheimer’s root cause: ECSIT as an integrating hub between oxidative stress, inflammation and mitochondrial dysfunction. Hypothetical role of the adapter protein ECSIT in familial and sporadic Alzheimer’s disease pathogenesis. Bioessays News Rev. Mol. Cell. Dev. Biol. 2012, 34, 532–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi Wi, S.; Park, J.; Shim, J.H.; Chun, E.; Lee, K.Y. Ubiquitination of ECSIT is crucial for the activation of p65/p50 NF-kappaBs in Toll-like receptor 4 signaling. Mol. Biol. Cell 2015, 26, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Soler-Lopez, M.; Zanzoni, A.; Lluis, R.; Stelzl, U.; Aloy, P. Interactome mapping suggests new mechanistic details underlying Alzheimer’s disease. Genome Res. 2011, 21, 364–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Woody, S.K.; Chhibber, A. Estrogen receptor beta in Alzheimer’s disease: From mechanisms to therapeutics. Ageing Res. Rev. 2015, 24, 178–190. [Google Scholar] [CrossRef] [Green Version]
- Sundermann, E.E.; Maki, P.M.; Bishop, J.R. A review of estrogen receptor alpha gene (ESR1) polymorphisms, mood, and cognition. Menopause 2010, 17, 874–886. [Google Scholar] [CrossRef] [Green Version]
- Yaffe, K.; Lindquist, K.; Sen, S.; Cauley, J.; Ferrell, R.; Penninx, B.; Harris, T.; Li, R.; Cummings, S.R. Estrogen receptor genotype and risk of cognitive impairment in elders: Findings from the Health ABC study. Neurobiol. Aging 2009, 30, 607–614. [Google Scholar] [CrossRef] [Green Version]
- Goumidi, L.; Dahlman-Wright, K.; Tapia-Paez, I.; Matsson, H.; Pasquier, F.; Amouyel, P.; Kere, J.; Lambert, J.C.; Meirhaeghe, A. Study of estrogen receptor-alpha and receptor-beta gene polymorphisms on Alzheimer’s disease. J. Alzheimer’s Dis. Jad 2011, 26, 431–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khorram Khorshid, H.R.; Gozalpour, E.; Saliminejad, K.; Karimloo, M.; Ohadi, M.; Kamali, K. Vitamin D Receptor (VDR) polymorphisms and late-onset Alzheimer’s disease: An association study. Iran. J. Public Health 2013, 42, 1253–1258. [Google Scholar] [PubMed]
- Liu, X.; Jiao, B.; Shen, L. The epigenetics of Alzheimer’s Disease: Factors and therapeutic implications. Front. Genet. 2018, 9, 579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wainaina, M.N.; Chen, Z.; Zhong, C. Environmental factors in the development and progression of late-onset Alzheimer’s disease. Neurosci. Bull. 2014, 30, 253–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, W.B.; Campbell, A.; Itzhaki, R.F.; Savory, J. The significance of environmental factors in the etiology of Alzheimer’s disease. J. Alzheimer’s Dis. Jad 2002, 4, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Moulton, P.V.; Yang, W. Air pollution, oxidative stress, and Alzheimer’s disease. J. Environ. Public Health 2012, 2012, 472751. [Google Scholar] [CrossRef] [PubMed]
- Croze, M.L.; Zimmer, L. Ozone atmospheric pollution and Alzheimer’s disease: From epidemiological facts to molecular mechanisms. J. Alzheimer’s Dis. Jad 2018, 62, 503–522. [Google Scholar] [CrossRef]
- Hu, N.; Yu, J.T.; Tan, L.; Wang, Y.L.; Sun, L.; Tan, L. Nutrition and the risk of Alzheimer’s disease. Biomed. Res. Int. 2013, 2013, 524820. [Google Scholar] [CrossRef] [Green Version]
- Abate, G.; Marziano, M.; Rungratanawanich, W.; Memo, M.; Uberti, D. Nutrition and AGE-ing: Focusing on Alzheimer’s disease. Oxidative Med. Cell. Longev. 2017, 2017, 7039816. [Google Scholar] [CrossRef]
- Koyama, A.; Hashimoto, M.; Tanaka, H.; Fujise, N.; Matsushita, M.; Miyagawa, Y.; Hatada, Y.; Fukuhara, R.; Hasegawa, N.; Todani, S.; et al. Malnutrition in Alzheimer’s disease, dementia with lewy bodies, and frontotemporal lobar degeneration: Comparison using serum albumin, total protein, and hemoglobin level. PLoS ONE 2016, 11, e0157053. [Google Scholar] [CrossRef]
- Adlard, P.A.; Bush, A.I. Metals and Alzheimer’s disease. J. Alzheimer’s Dis. Jad 2006, 10, 145–163. [Google Scholar] [CrossRef] [PubMed]
- Colomina, M.T.; Peris-Sampedro, F. Aluminum and Alzheimer’s disease. Adv. Neurobiol. 2017, 18, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Huat, T.J.; Camats-Perna, J.; Newcombe, E.A.; Valmas, N.; Kitazawa, M.; Medeiros, R. Metal toxicity links to Alzheimer’s disease and neuroinflammation. J. Mol. Biol. 2019, 431, 1843–1868. [Google Scholar] [CrossRef] [PubMed]
- Sochocka, M.; Zwolinska, K.; Leszek, J. The infectious etiology of Alzheimer’s disease. Curr. Neuropharmacol. 2017, 15, 996–1009. [Google Scholar] [CrossRef] [Green Version]
- Fulop, T.; Itzhaki, R.F.; Balin, B.J.; Miklossy, J.; Barron, A.E. Role of microbes in the development of Alzheimer’s disease: State of the art—An international symposium presented at the 2017 IAGG congress in San Francisco. Front. Genet. 2018, 9, 362. [Google Scholar] [CrossRef] [Green Version]
- Muzambi, R.; Bhaskaran, K.; Brayne, C.; Smeeth, L.; Warren-Gash, C. Common bacterial infections and risk of incident cognitive decline or dementia: A systematic review protocol. BMJ Open 2019, 9, e030874. [Google Scholar] [CrossRef]
- Stampfer, M.J. Cardiovascular disease and Alzheimer’s disease: Common links. J. Intern. Med. 2006, 260, 211–223. [Google Scholar] [CrossRef]
- Santos, C.Y.; Snyder, P.J.; Wu, W.C.; Zhang, M.; Echeverria, A.; Alber, J. Pathophysiologic relationship between Alzheimer’s disease, cerebrovascular disease, and cardiovascular risk: A review and synthesis. Alzheimer’s Dement. 2017, 7, 69–87. [Google Scholar] [CrossRef] [Green Version]
- De Bruijn, R.F.; Ikram, M.A. Cardiovascular risk factors and future risk of Alzheimer’s disease. BMC Med. 2014, 12, 130. [Google Scholar] [CrossRef] [Green Version]
- Alford, S.; Patel, D.; Perakakis, N.; Mantzoros, C.S. Obesity as a risk factor for Alzheimer’s disease: Weighing the evidence. Obes. Rev. Off. J. Int. Assoc. Study Obes. 2018, 19, 269–280. [Google Scholar] [CrossRef]
- Pegueroles, J.; Jimenez, A.; Vilaplana, E.; Montal, V.; Carmona-Iragui, M.; Pane, A.; Alcolea, D.; Videla, L.; Casajoana, A.; Clarimon, J.; et al. Obesity and Alzheimer’s disease, does the obesity paradox really exist? A magnetic resonance imaging study. Oncotarget 2018, 9, 34691–34698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anjum, I.; Fayyaz, M.; Wajid, A.; Sohail, W.; Ali, A. Does obesity increase the risk of dementia: A literature review. Cureus 2018, 10, e2660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Seo, H.I.; Cha, H.Y.; Yang, Y.J.; Kwon, S.H.; Yang, S.J. Diabetes and Alzheimer’s disease: Mechanisms and nutritional aspects. Clin. Nutr. Res. 2018, 7, 229–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Sadiq, N.M. Cholinesterase Inhibitors. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020; Available online: https://www.ncbi.nlm.nih.gov/books/NBK544336/ (accessed on 8 December 2020).
- Eldufani, J.; Blaise, G. The role of acetylcholinesterase inhibitors such as neostigmine and rivastigmine on chronic pain and cognitive function in aging: A review of recent clinical applications. Alzheimers Dement 2019, 5, 175–183. [Google Scholar] [CrossRef]
- Sharma, K. Cholinesterase inhibitors as Alzheimer’s therapeutics (Review). Mol. Med. Rep. 2019, 20, 1479–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Reddy, P.H. Role of glutamate and NMDA receptors in Alzheimer’s disease. J. Alzheimer’s Dis. Jad 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Kuns, B.; Rosani, A.; Varghese, D. Memantine. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020; Available online: https://www.ncbi.nlm.nih.gov/books/NBK500025/ (accessed on 8 December 2020).
- Briggs, R.; Kennelly, S.P.; O’Neill, D. Drug treatments in Alzheimer’s disease. Clin. Med. 2016, 16, 247–253. [Google Scholar] [CrossRef]
- Crous-Bou, M.; Minguillon, C.; Gramunt, N.; Molinuevo, J.L. Alzheimer’s disease prevention: From risk factors to early intervention. Alzheimer’s Res. Ther. 2017, 9, 71. [Google Scholar] [CrossRef]
- Crismon, M.L. Tacrine: First drug approved for Alzheimer’s disease. Ann. Pharmacother. 1994, 28, 744–751. [Google Scholar] [CrossRef]
- Qizilbash, N.; Birks, J.; Lopez Arrieta, J.; Lewington, S.; Szeto, S. Tacrine for Alzheimer’s disease. Cochrane Database Syst. Rev. 2000, CD000202. [Google Scholar] [CrossRef]
- Cacabelos, R. Donepezil in Alzheimer’s disease: From conventional trials to pharmacogenetics. Neuropsychiatr. Dis. Treat. 2007, 3, 303–333. [Google Scholar] [PubMed]
- Kumar, A.; Sharma, S. Donepezil. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020; Available online: https://www.ncbi.nlm.nih.gov/books/NBK513257/ (accessed on 8 December 2020).
- Dooley, M.; Lamb, H.M. Donepezil: A review of its use in Alzheimer’s disease. Drugs Aging 2000, 16, 199–226. [Google Scholar] [CrossRef] [PubMed]
- Annicchiarico, R.; Federici, A.; Pettenati, C.; Caltagirone, C. Rivastigmine in Alzheimer’s disease: Cognitive function and quality of life. Ther. Clin. Risk Manag. 2007, 3, 1113–1123. [Google Scholar] [PubMed]
- Muller, T. Rivastigmine in the treatment of patients with Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2007, 3, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Khoury, R.; Rajamanickam, J.; Grossberg, G.T. An update on the safety of current therapies for Alzheimer’s disease: Focus on rivastigmine. Ther. Adv. Drug Saf. 2018, 9, 171–178. [Google Scholar] [CrossRef]
- Birks, J.; Grimley Evans, J.; Iakovidou, V.; Tsolaki, M.; Holt, F.E. Rivastigmine for Alzheimer’s disease. Cochrane Database Syst. Rev. 2009, CD001191. [Google Scholar] [CrossRef]
- Scott, L.J.; Goa, K.L. Galantamine: A review of its use in Alzheimer’s disease. Drugs 2000, 60, 1095–1122. [Google Scholar] [CrossRef]
- Prvulovic, D.; Hampel, H.; Pantel, J. Galantamine for Alzheimer’s disease. Expert Opin. Drug Metab. Toxicol. 2010, 6, 345–354. [Google Scholar] [CrossRef]
- Kim, J.K.; Park, S.U. Pharmacological aspects of galantamine for the treatment of Alzheimer’s disease. Excli J. 2017, 16, 35–39. [Google Scholar] [CrossRef]
- Wahba, S.M.; Darwish, A.S.; Kamal, S.M. Ceria-containing uncoated and coated hydroxyapatite-based galantamine nanocomposites for formidable treatment of Alzheimer’s disease in ovariectomized albino-rat model. Mater. Sci. Eng. C Mater. Biol. Appl. 2016, 65, 151–163. [Google Scholar] [CrossRef]
- Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y. The role of NMDA receptors in Alzheimer’s disease. Front. Neurosci. 2019, 13, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.J.; Lin, C.H.; Lane, H.Y.; Tsai, G.E. NMDA Neurotransmission dysfunction in behavioral and psychological symptoms of Alzheimer’s disease. Curr. Neuropharmacol. 2012, 10, 272–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Companys-Alemany, J.; Turcu, A.L.; Bellver-Sanchis, A.; Loza, M.I.; Brea, J.M.; Canudas, A.M.; Leiva, R.; Vazquez, S.; Pallas, M.; Grinan-Ferre, C. A novel NMDA receptor antagonist protects against cognitive decline presented by senescent mice. Pharmaceutics 2020, 12, 284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folch, J.; Busquets, O.; Ettcheto, M.; Sanchez-Lopez, E.; Castro-Torres, R.D.; Verdaguer, E.; Garcia, M.L.; Olloquequi, J.; Casadesus, G.; Beas-Zarate, C.; et al. Memantine for the treatment of dementia: A Review on its current and future applications. J. Alzheimer’s Dis. Jad 2018, 62, 1223–1240. [Google Scholar] [CrossRef] [Green Version]
- Cummings, J.; Fox, N. Defining disease modifying therapy for Alzheimer’s Disease. J. Prev. Alzheimer’s Dis. 2017, 4, 109–115. [Google Scholar] [CrossRef]
- Huang, L.K.; Chao, S.P.; Hu, C.J. Clinical trials of new drugs for Alzheimer disease. J. Biomed. Sci. 2020, 27, 18. [Google Scholar] [CrossRef]
- Neumann, U.; Ufer, M.; Jacobson, L.H.; Rouzade-Dominguez, M.L.; Huledal, G.; Kolly, C.; Luond, R.M.; Machauer, R.; Veenstra, S.J.; Hurth, K.; et al. The BACE-1 inhibitor CNP520 for prevention trials in Alzheimer’s disease. Embo Mol. Med. 2018, 10, e9316. [Google Scholar] [CrossRef]
- Vandenberghe, R.; Riviere, M.E.; Caputo, A.; Sovago, J.; Maguire, R.P.; Farlow, M.; Marotta, G.; Sanchez-Valle, R.; Scheltens, P.; Ryan, J.M.; et al. Active Abeta immunotherapy CAD106 in Alzheimer’s disease: A phase 2b study. Alzheimers Dement 2017, 3, 10–22. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2020. Alzheimers Dement 2020, 6, e12050. [Google Scholar] [CrossRef]
- Tolar, M.; Abushakra, S.; Hey, J.A.; Porsteinsson, A.; Sabbagh, M. Aducanumab, gantenerumab, BAN2401, and ALZ-801-the first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimer’s Res. Ther. 2020, 12, 95. [Google Scholar] [CrossRef]
- Galimberti, D.; Scarpini, E. Disease-modifying treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2011, 4, 203–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghezzi, L.; Scarpini, E.; Galimberti, D. Disease-modifying drugs in Alzheimer’s disease. Drug Des. Dev. Ther. 2013, 7, 1471–1478. [Google Scholar] [CrossRef] [Green Version]
- Medina, M. An Overview on the clinical development of tau-based therapeutics. Int. J. Mol. Sci. 2018, 19, 1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidowitz, E.J.; Krishnamurthy, P.K.; Lopez, P.; Jimenez, H.; Adrien, L.; Davies, P.; Moe, J.G. In vivo validation of a small molecule inhibitor of tau self-association in htau mice. J. Alzheimer’s Dis. Jad 2020, 73, 147–161. [Google Scholar] [CrossRef] [Green Version]
- Cortez, L.; Sim, V. The therapeutic potential of chemical chaperones in protein folding diseases. Prion 2014, 8, 197–202. [Google Scholar] [CrossRef] [Green Version]
- Campanella, C.; Pace, A.; Caruso Bavisotto, C.; Marzullo, P.; Marino Gammazza, A.; Buscemi, S.; Palumbo Piccionello, A. Heat shock proteins in Alzheimer’s disease: Role and targeting. Int. J. Mol. Sci. 2018, 19, 2603. [Google Scholar] [CrossRef] [Green Version]
- Wilhelmus, M.M.; de Waal, R.M.; Verbeek, M.M. Heat shock proteins and amateur chaperones in amyloid-Beta accumulation and clearance in Alzheimer’s disease. Mol. Neurobiol. 2007, 35, 203–216. [Google Scholar] [CrossRef] [Green Version]
- Martin-Pena, A.; Rincon-Limas, D.E.; Fernandez-Funez, P. Engineered Hsp70 chaperones prevent Abeta42-induced memory impairments in a Drosophila model of Alzheimer’s disease. Sci. Rep. 2018, 8, 9915. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Murshid, A. Molecular chaperone accumulation in cancer and decrease in Alzheimer’s disease: The potential roles of HSF1. Front. Neurosci. 2017, 11, 192. [Google Scholar] [CrossRef] [Green Version]
- Repalli, J.; Meruelo, D. Screening strategies to identify HSP70 modulators to treat Alzheimer’s disease. Drug Des. Dev. Ther. 2015, 9, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Shao, H.; Taylor, I.R.; Gestwicki, J.E. Targeting allosteric control mechanisms in heat shock protein 70 (Hsp70). Curr. Top. Med. Chem. 2016, 16, 2729–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abisambra, J.; Jinwal, U.K.; Miyata, Y.; Rogers, J.; Blair, L.; Li, X.; Seguin, S.P.; Wang, L.; Jin, Y.; Bacon, J.; et al. Allosteric heat shock protein 70 inhibitors rapidly rescue synaptic plasticity deficits by reducing aberrant tau. Biol. Psychiatry 2013, 74, 367–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohush, A.; Bieganowski, P.; Filipek, A. Hsp90 and its co-chaperones in neurodegenerative diseases. Int. J. Mol. Sci. 2019, 20, 4976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, J.R.; Tan, M.S.; Xie, A.M.; Yu, J.T.; Tan, L. Heat shock protein 90 in Alzheimer’s disease. Biomed Res. Int. 2014, 2014, 796869. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Liu, Y.; Huang, L.; Chen, J.; Li, J.J.; Wang, R.; Kim, E.; Chen, Y.; Justicia, C.; Sakata, K.; et al. A CNS-permeable Hsp90 inhibitor rescues synaptic dysfunction and memory loss in APP-overexpressing Alzheimer’s mouse model via an HSF1-mediated mechanism. Mol. Psychiatry 2017, 22, 990–1001. [Google Scholar] [CrossRef] [Green Version]
- Li, J.G.; Chiu, J.; Ramanjulu, M.; Blass, B.E.; Pratico, D. A pharmacological chaperone improves memory by reducing Abeta and tau neuropathology in a mouse model with plaques and tangles. Mol. Neurodegener. 2020, 15, 1. [Google Scholar] [CrossRef]
- Vagnozzi, A.N.; Li, J.G.; Chiu, J.; Razmpour, R.; Warfield, R.; Ramirez, S.H.; Pratico, D. VPS35 regulates tau phosphorylation and neuropathology in tauopathy. Mol. Psychiatry 2019. [Google Scholar] [CrossRef]
- Li, J.G.; Chiu, J.; Pratico, D. Full recovery of the Alzheimer’s disease phenotype by gain of function of vacuolar protein sorting 35. Mol. Psychiatry 2020, 25, 2630–2640. [Google Scholar] [CrossRef]
- Mecozzi, V.J.; Berman, D.E.; Simoes, S.; Vetanovetz, C.; Awal, M.R.; Patel, V.M.; Schneider, R.T.; Petsko, G.A.; Ringe, D.; Small, S.A. Pharmacological chaperones stabilize retromer to limit APP processing. Nat. Chem. Biol. 2014, 10, 443–449. [Google Scholar] [CrossRef]
- Knight, E.M.; Williams, H.N.; Stevens, A.C.; Kim, S.H.; Kottwitz, J.C.; Morant, A.D.; Steele, J.W.; Klein, W.L.; Yanagisawa, K.; Boyd, R.E.; et al. Evidence that small molecule enhancement of beta-hexosaminidase activity corrects the behavioral phenotype in Dutch APP(E693Q) mice through reduction of ganglioside-bound Abeta. Mol. Psychiatry 2015, 20, 109–117. [Google Scholar] [CrossRef]
- Andrade, S.; Ramalho, M.J.; Loureiro, J.A.; Pereira, M.D.C. Natural compounds for Alzheimer’s disease therapy: A systematic review of preclinical and clinical studies. Int. J. Mol. Sci. 2019, 20, 2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Yang, M.W.; Li, X.W.; Yue, J.W.; Chen, J.Z.; Yang, M.W.; Huang, X.; Zhu, L.L.; Hong, F.F.; Yang, S.L. Therapeutic effects of natural drugs on Alzheimer’s disease. Front. Pharmacol. 2019, 10, 1355. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Modifying Agents | Mechanism of Action |
|---|---|
| Phase 3 Clinical Trials | |
| Aducanumab | Monoclonal antibody—targets β-amyloid and removes it. |
| Gantenerumab | Monoclonal antibody—binds and removes β-amyloid. |
| CAD106b | Amyloid vaccine—stimulates production of antibodies against β-amyloid. |
| BAN2401 | Monoclonal antibody—reduces protofibrillar β-amyloid. |
| TRx0237 (LMTX) | Tau protein aggregation inhibitor. |
| AGB101 | Low-dose levetiracetam—improves synaptic function and reduces amyloid-induced neuronal hyperactivity |
| ALZT-OP1 (cromolyn + ibuprofen) | Mast cell stabilizer and anti-inflammatory—promotes microglial clearance of amyloid |
| Azeliragon | RAGE (Receptor for Advanced Glycation End-products) antagonist—reduces inflammation and amyloid transport into the brain |
| BHV4157 (troriluzole) | Glutamate modulator—reduces synaptic levels of glutamate and improves synaptic functioning |
| Masitinib | Tyrosine kinase inhibitor—modulates inflammatory mast cell and reduces amyloid protein and tau phosphorylation |
| Phase 2 Clinical Trials | |
| Crenezumab | Monoclonal antibody—targets soluble oligomers and removes β-amyloid |
| ABBV-8E12 | Monoclonal antibody—prevents tau propagation |
| ABvac40 | Active immunotherapy—targets β-amyloid and removes it |
| BAN2401 | Monoclonal antibody—removes amyloid protofibrils and reduces amyloid plaques |
| BIIB092 | Monoclonal antibody—removes tau and reduces tau propagation |
| LY3002813 (donanemab) | Monoclonal antibody—removes amyloid by recognizing aggregated pyroglutamate form of Aβ |
| LY3303560 (zagotenemab) | Monoclonal antibody—neutralizes soluble tau aggregates |
| Semorinemab (RO7105705) | Monoclonal antibody—removes extracellular tau |
| APH-1105 | Alpha-secretase modulator—reduces amyloid |
| Daratumumab | Monoclonal antibody—immunomodulatory that targets CD38 and regulates microglial activity |
| Dasatinib + Quercetin | Tyrosine kinase inhibitor (dasatinib) + flavonoid (quercetin)—reduces senescent cells and tau aggregation |
| IONIS MAPTRx (BIIB080) | Epigenetic, Tau Antisense oligonucleotide—reduces tau production |
| Lithium | Neurotransmitter receptors ion channel modulator—improves neuropsychiatric symptoms |
| Nilotinib | Tyrosine kinase inhibitor—promotes clearance of amyloid and tau proteins |
| Posiphen | Selective inhibitor of APP—reduces amyloid, tau, and α-synuclein production |
| PTI-125 | Filamin A protein inhibitor—reduces tau hyperphosphorylation, synaptic dysfunction, and stabilizes soluble amyloid and the α7 nicotinic acetylcholine receptor interaction |
| PQ912 | Glutaminyl cyclase (QC) enzyme inhibitor—reduces amyloid plaques and pyroglutamates Aβ production |
| Riluzole | Glutamate receptor antagonist—reduces glutamate-mediated excitotoxicity |
| Thiethylperazine (TEP) | Activates ABCC1 (ATP binding cassette subfamily C member 1 transport protein)—removes amyloid |
| Phase 1 Clinical Trials | |
| BIIB076 | Monoclonal antibody—removes tau and reduces tau propagation |
| Lu AF87908 | Monoclonal antibody—removes tau |
| anle138b | Aggregation inhibitor—reduces tau aggregation |
| RO7126209 | Monoclonal antibody—removes amyloid |
| TPI-287 | Stabilizes tubulin-binding, microtubule, and reduces cellular damage mediated by tau |
| Natural Compounds | Mechanism of Action |
|---|---|
| Schisantherin A, Ginsenoside Rh2, and Angelica sinensis extracts | Aβ formation inhibitors |
| Shengmai (SM) formula, Uncarinic acid C, andTanshinone IIA (TIIA) extract | Reduction of Aβ accumulation |
| Onjisaponin B, Notoginsenoside R1, and delta-9-Tetrahydrocannabinol (THC)/cannabidiol (CBD) | Promotion of Aβ degradation |
| Rhynchophylline (RIN), INM-176 (ethanolic extract of Angelica gigas), Houttuyniacordata Thunb. (Saururaceae) water extracts, Huperzine A, and ethyl acetate extract from Diospyros kaki L.f | Inhibition of Aβ Neurotoxicity and reduce over-activation of microglial cells, neuroinflammation, oxidative stress, and disruption of calcium homeostasis, which lead to neuron loss |
| Tongmai Yizhi Decoction (TYD) (which includes six raw materials: safflower yellow (SY) from Carthamustinctorius L., geniposide from the fruit of G. jasminoides J. Ellis, ginsenoside Rd from Panax ginseng C. A. Mey, crocin from Crocus sativus L., and quinones) | Inhibition of hyperphosphorylated tau protein and its aggregation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. https://doi.org/10.3390/molecules25245789
Breijyeh Z, Karaman R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules. 2020; 25(24):5789. https://doi.org/10.3390/molecules25245789
Chicago/Turabian StyleBreijyeh, Zeinab, and Rafik Karaman. 2020. "Comprehensive Review on Alzheimer’s Disease: Causes and Treatment" Molecules 25, no. 24: 5789. https://doi.org/10.3390/molecules25245789