Enantioselective HPLC Analysis to Assist the Chemical Exploration of Chiral Imidazolines

 ,
, 
 , , and
, , and
Abstract
:1. Introduction
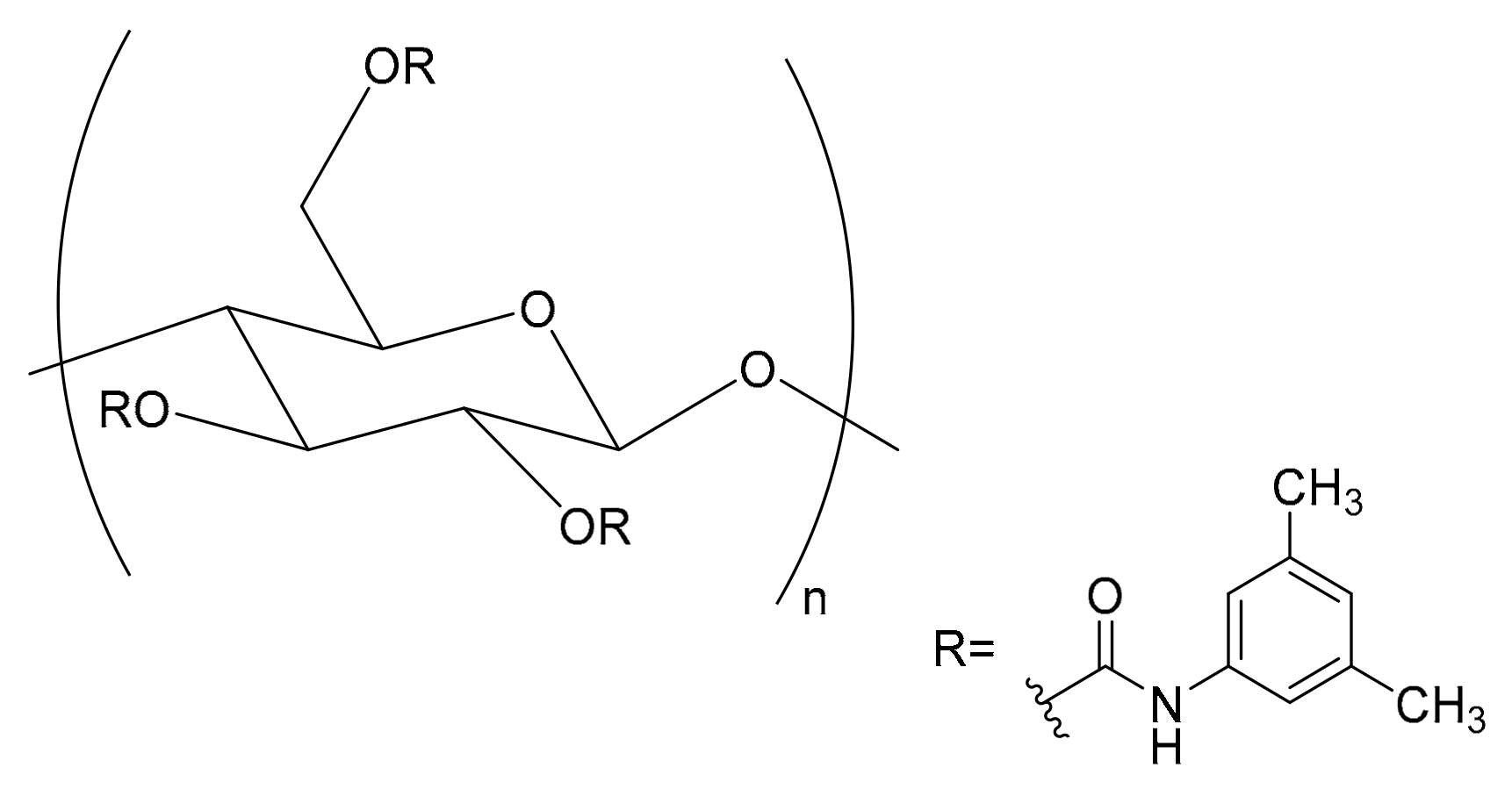
2. Results and Discussion
2.1. Optimization of the Enantioselective HPLC Analysis
2.2. Mechanism of Retention and Enantiorecognition Under RP Conditions
3. Material and Methods
3.1. Chemicals
3.2. Instrumentations
3.3. Molecular Modelling and Statistical Analysis.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References and Notes
- Gumustas, M.; Ozkan, S.A.; Chankvetadze, B. Analytical and preparative scale separation of enantiomers of chiral drugs by chromatography and related methods. Curr. Med. Chem. 2018, 25, 4152–4188. [Google Scholar] [CrossRef] [PubMed]
- Ilisz, I.; Bajtai, A.; Lindner, W.; Péter, A. Liquid chromatographic enantiomer separations applying chiral ion-exchangers based on Cinchona alkaloids. J. Pharm. Biomed. Anal. 2018, 159, 127–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chankvetadze, B. Recent developments on polysaccharide-based chiral stationary phases for liquid-phase separation of enantiomers. J. Chromatogr. A 2012, 1269, 26–51. [Google Scholar] [CrossRef] [PubMed]
- Ward, T.J.; Ward, K.D. Chiral separations: A review of current topics and trends. Anal. Chem. 2012, 84, 626–635. [Google Scholar] [CrossRef]
- Okamoto, Y.; Ikai, T. Chiral HPLC for efficient resolution of enantiomers. Chem. Soc. Rev. 2008, 37, 2593–2608. [Google Scholar] [CrossRef]
- Shen, J.; Okamoto, Y. Efficient separation of enantiomers using stereoregular chiral polymers. Chem. Rev. 2016, 116, 1094–1138. [Google Scholar] [CrossRef]
- Shen, J.; Ikai, T.; Okamoto, Y. Synthesis and application of immobilized polysaccharide-based chiral stationary phases for enantioseparation by high-performance liquid chromatography. J. Chromatogr. A 2014, 1363, 51–61. [Google Scholar] [CrossRef]
- Zhang, T.; Nguyen, D.; Franco, P.; Isobe, Y.; Michishita, T.; Murakami, T. Cellulose tris(3,5-dichlorophenylcarbamate) immobilised on silica: A novel chiral stationary phase for resolution of enantiomers. J. Pharm. Biomed. Anal. 2008, 46, 882–891. [Google Scholar] [CrossRef]
- Ali, I.; Aboul-Enein, H.Y. Impact of immobilized polysaccharide chiral stationary phases on enantiomeric separations. J. Sep. Sci. 2006, 6, 762–769. [Google Scholar] [CrossRef]
- Peluso, P.; Dessì, A.; Dallocchio, R.; Mamane, V.; Cossu, S. Recent studies of docking and molecular dynamics simulation for liquid-phase enantioseparations. Electrophoresis 2019, 40, 1881–1896. [Google Scholar] [CrossRef]
- Natalini, B.; Sardella, R.; Massari, S.; Ianni, F.; Tabarrini, O.; Cecchetti, V. Synthesis and chromatographic enantioresolution of anti-HIV quinolone derivatives. Talanta 2011, 85, 1392–1397. [Google Scholar] [CrossRef]
- Sardella, R.; Levent, S.; Ianni, F.; Çalişkan, B.; Gerstmeier, J.; Pergola, C.; Werz, O.; Banoglu, E.; Natalini, B. Chromatographic separation and biological evaluation of benzimidazole derivative enantiomers as inhibitors of leukotriene biosynthesis. J. Pharm. Biomed. Anal. 2014, 89, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, A.; Wang, C. Enantioselective separation of racemates using CHIRALPAK IG amylose-based chiral stationary phase under normal standard, non-standard and reversed phase high performance liquid chromatography. J. Chromatogr. A 2018, 1532, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Vojtylová, T.; Hamplová, V.; Galewski, Z.; Korbecka, I.; Sýkora, D. Chiral separation of novel diazenes on a polysaccharide-based stationary phase in the reversed-phase mode. J. Sep. Sci. 2017, 40, 1465–1469. [Google Scholar] [CrossRef] [PubMed]
- Geryk, R.; Kalíková, K.; Vozka, J.; Tesařová, E. Immobilized polysaccharide-based stationary phases for enantioseparation in normal versus reversed phase HPLC. Chromatographia 2015, 78, 909–915. [Google Scholar] [CrossRef]
- Geryk, R.; Kalíková, K.; Vozka, J.; Plecitá, D.; Schmid, M.G.; Tesařová, E. Enantioselective potential of chiral stationary phases based on immobilized polysaccharides in reversed phase mode. J. Chromatogr. A 2014, 1363, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Jayapalan, S.; Chankvetadze, B.; Farkas, T. Reversed-phase chiral HPLC and LC/MS analysis with tris(chloromethylphenylcarbamate) derivatives of cellulose and amylose as chiral stationary phases. J. Chromatogr. A 2010, 1217, 6942–6955. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Franco, P.; Nguyen, D.; Hamasaki, R.; Miyamoto, S.; Ohnishi, A.; Murakami, T. Complementary enantiorecognition patterns and specific method optimization aspects on immobilized polysaccharide-derived chiral stationary phases. J. Chromatogr. A 2012, 1269, 178–188. [Google Scholar] [CrossRef]
- Gioiello, A. Unpublished work, manuscript in preparation.
- Zhang, T.; Nguyen, D.; Franco, P. Reversed-phase screening strategies for liquid chromatography on polysaccharide-derived. J. Chromatogr. A 2010, 1217, 1048–1055. [Google Scholar] [CrossRef]
- Ghanem, A.; Naim, L. Immobilized versus coated amylose tris(3,5-dimethylphenylcarbamate) chiral stationary phases for the enantioselective separation of cyclopropane derivatives by liquid chromatography. J. Chromatogr. A 2006, 1101, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Thunberg, L.; Hashemi, J.; Andersson, S. Comparative study of coated and immobilized polysaccharide-based chiral stationary phases and their applicability in the resolution of enantiomers. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2008, 875, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Katz, E.D.; Lochmuller, C.H.; Scott, P.W. Methanol-water association and its effect on solute retention in liquid chromatography. Anal. Chem. 1989, 61, 349–355. [Google Scholar] [CrossRef]
- Lämmerhofer, M. Chiral recognition by enantioselective liquid chromatography: Mechanisms and modern chiral stationary phases. J. Chromatogr. A 2010, 1217, 814–856. [Google Scholar] [CrossRef] [PubMed]
- Lipka, E.; Descamps, C.; Vaccher, C.; Bonte, J.P. Reversed-phase liquid chromatographic separation, on cellulose chiral stationary phases, of the stereoisomers of methoxytetra-hydronaphthalene derivatives, new agonist and antagonist ligands for melatonin receptors. Chromatographia 2005, 61, 143–149. [Google Scholar] [CrossRef]
- Kubota, T.; Yamamoto, C.; Okamoto, Y. Reversed-phase liquid chromatographic enantioseparation by cycloalkylcarboxylates of cellulose and amylose. Chirality 2004, 16, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Yashima, E. Polysaccharide-based chiral stationary phases for high-performance liquid chromatographic enantioseparation. J. Chromatogr. A 2001, 906, 105–125. [Google Scholar] [CrossRef]
- Aboul-Enein, H.Y.; Ali, I.; Gübitz, G.; Simons, C.; Nicholls, P.J. HPLC enantiomeric resolution of novel aromatase inhibitors on cellulose- and amylose-based chiral stationary phases under reversed phase mode. Chirality 2000, 12, 727–733. [Google Scholar] [CrossRef]
- Hou, J.G.; Wang, Y.L.; Li, C.X.; Han, X.Q.; Gao, J.Z.; Kang, J.W. Reversed-phase separations on cellulose tris(3,5-dimethylphenylcarbamate) chiral stationary phase. Chromatographia 1999, 50, 89–95. [Google Scholar] [CrossRef]
- Aboul-Enein, H.Y.; Abou-Basha, L.I.; Bakr, S.A. Direct enantioselective separation of some propranolol analogs by HPLC on normal and reversed cellulose chiral stationary phases. Chirality 1996, 8, 153–156. [Google Scholar] [CrossRef]
- Eiji, Y.; Yoshio, O. Chiral discrimination on polysaccharides derivatives. Bull. Chem. Soc. Jpn. 1995, 68, 3289–3307. [Google Scholar]
- Yang, M.; Fazio, S.; Munch, D.; Drumm, P. Impact of methanol and acetonitrile on separations based on pi-pi interactions with a reversed-phase phenyl column. J. Chromatogr. A 2005, 1097, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Booth, T.D.; Wainer, I.W. Mechanistic investigation into the enantioselective separation of mexiletine and related compounds, chromatographed on an amylose tris(3,5-dimethylphenylcarbamate) chiral stationary phase. J. Chromatogr. A 1996, 741, 205–211. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The R Project for Statistical Computing. Available online: http://www.R-project.org (accessed on 19 December 2019).
Sample Availability: Samples of the compounds 1–10 are not available. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | % MeOH (v) | Selected Chromatographic Parameters | |||
|---|---|---|---|---|---|
| k1 | k2 | RS | α | ||
| 1 | 25 | 4.99 | 5.50 | 1.94 | 1.10 |
| 2 | 25 | 11.28 | 11.97 | 1.21 | 1.06 |
| 3 | 30 | 6.85 | 7.48 | 1.90 | 1.09 |
| 4 | 30 | 5.51 | 5.97 | 1.48 | 1.08 |
| 5 | 25 | 6.98 | 7.67 | 2.06 | 1.10 |
| 6 | 30 | 9.55 | 10.43 | 1.96 | 1.09 |
| 7 | 20 | 3.49 | 3.94 | 2.28 | 1.13 |
| 8 | 15 | 4.06 | 4.54 | 2.31 | 1.12 |
| 9 | 15 | 9.84 | 10.74 | 2.16 | 1.09 |
| 10 | Co-elution with all the tested conditions | ||||
| Descriptor | r - logk1st | r - logk2nd |
|---|---|---|
| AlogP | 0.45 (p = 0.20) | 0.50 (p = 0.14) |
| QlogPo/w | 0.41 (p = 0.23) | 0.48 (p = 0.16) |
| ilogP | 0.23 (p = 0.53) | 0.25 (p = 0.49) |
| XlogP3 | 0.46 (p = 0.18) | 0.50 (p = 0.14) |
| WlogP | 0.70 (p = 0.03) | 0.75 (p = 0.01) |
| MlogP | 0.53 (p = 0.11) | 0.58 (p = 0.08) |
| SilicosIT-logP | 0.39 (p = 0.26) | 0.46 (p = 0.18) |
| ESOL-logS | −0.80 (p = 0.005) | −0.83 (p = 0.003) |
| Ali-logS | −0.93 (p < 0.001) | −0.94 (p < 0.001) |
| SilicosIT-logSw | −0.39 (p = 0.27) | −0.45 (p = 0.19) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cerra, B.; Macchiarulo, A.; Carotti, A.; Camaioni, E.; Varfaj, I.; Sardella, R.; Gioiello, A. Enantioselective HPLC Analysis to Assist the Chemical Exploration of Chiral Imidazolines. Molecules 2020, 25, 640. https://doi.org/10.3390/molecules25030640
Cerra B, Macchiarulo A, Carotti A, Camaioni E, Varfaj I, Sardella R, Gioiello A. Enantioselective HPLC Analysis to Assist the Chemical Exploration of Chiral Imidazolines. Molecules. 2020; 25(3):640. https://doi.org/10.3390/molecules25030640
Chicago/Turabian StyleCerra, Bruno, Antonio Macchiarulo, Andrea Carotti, Emidio Camaioni, Ina Varfaj, Roccaldo Sardella, and Antimo Gioiello. 2020. "Enantioselective HPLC Analysis to Assist the Chemical Exploration of Chiral Imidazolines" Molecules 25, no. 3: 640. https://doi.org/10.3390/molecules25030640