Unexpected Reaction Pathway of the Alpha-Aminoalkyl Radical Derived from One-Electron Oxidation of S-Alkylglutathiones


Abstract
:1. Introduction
2. Results
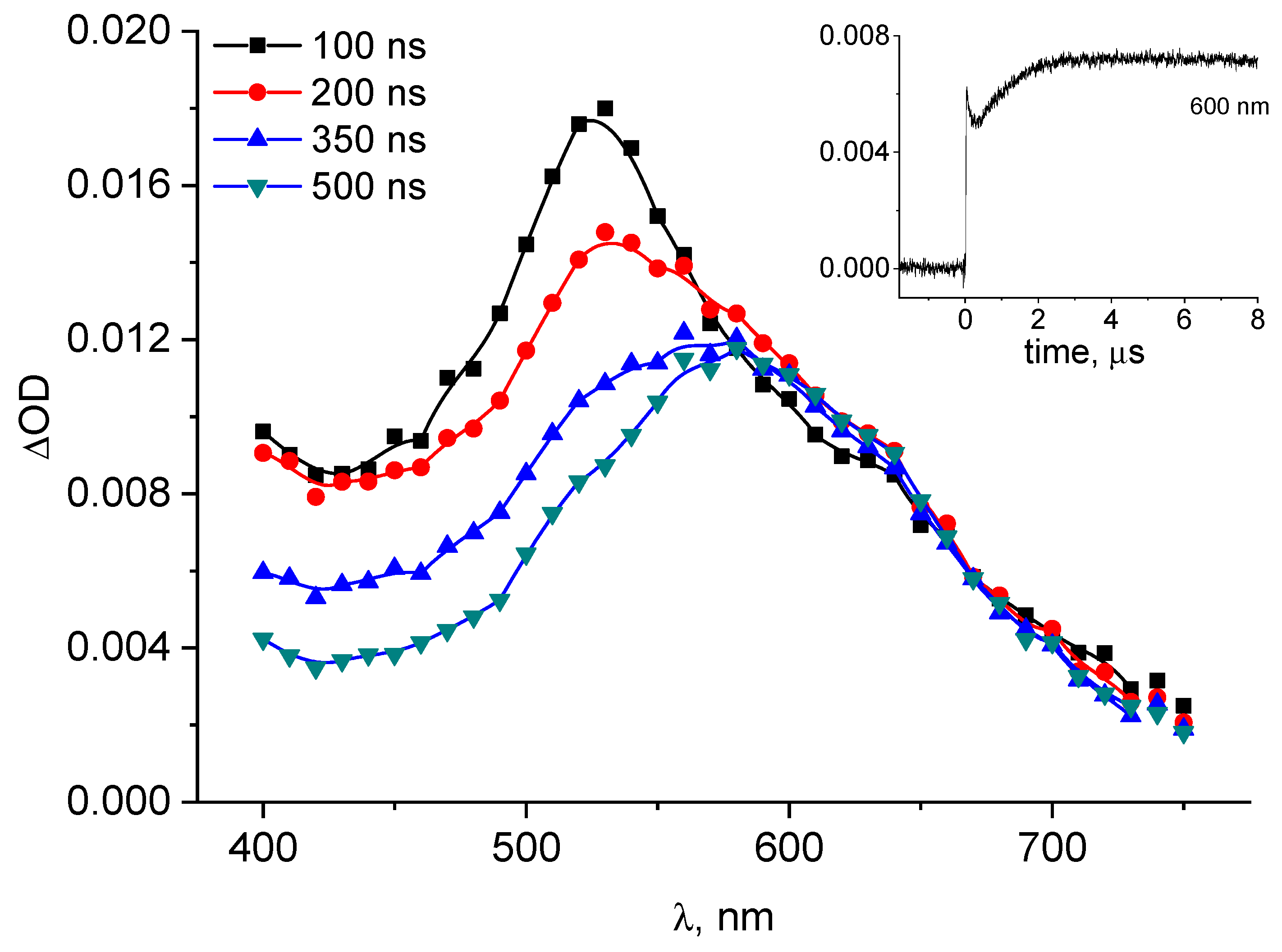
2.1. Laser Flash Photolysis
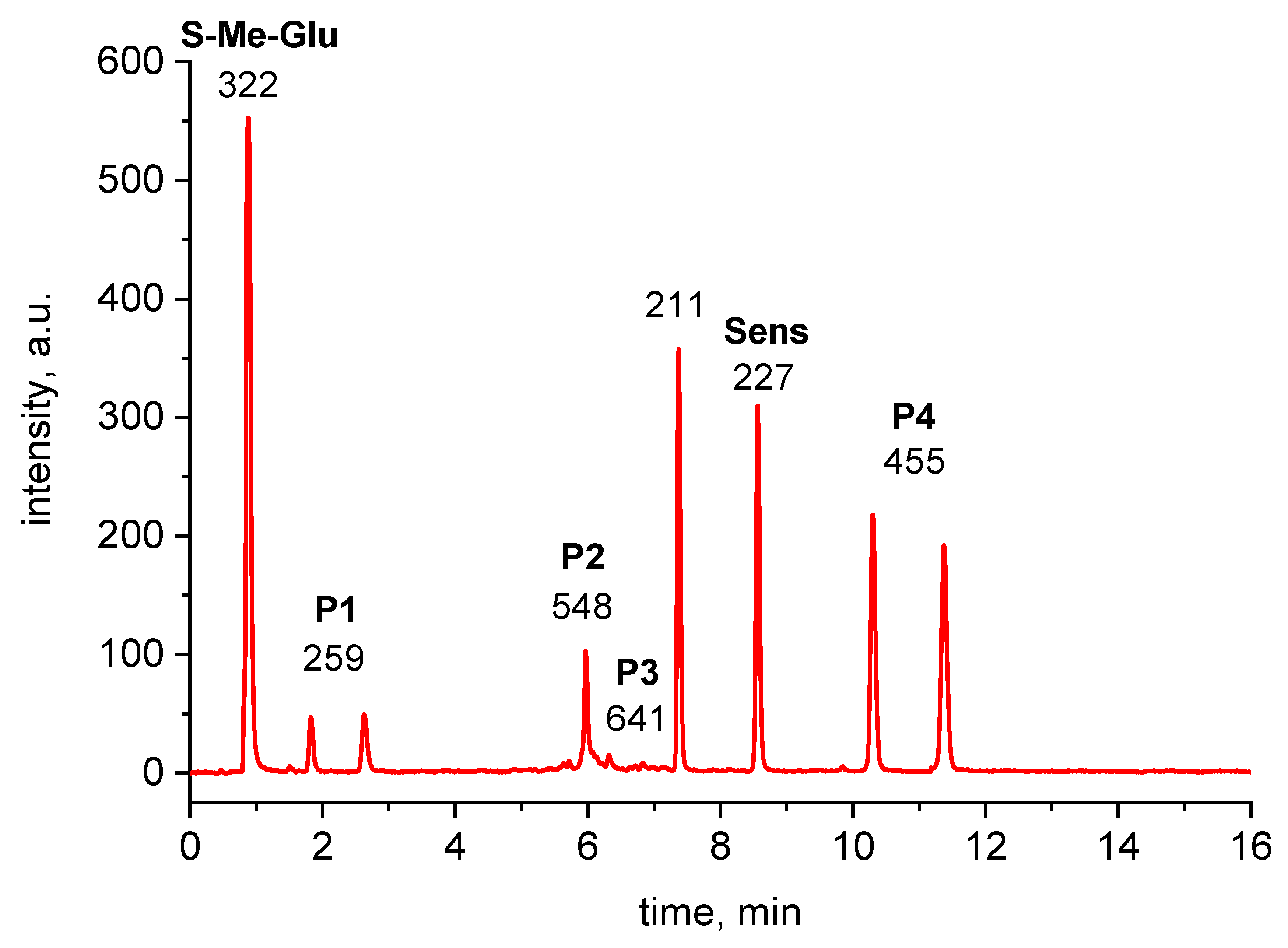
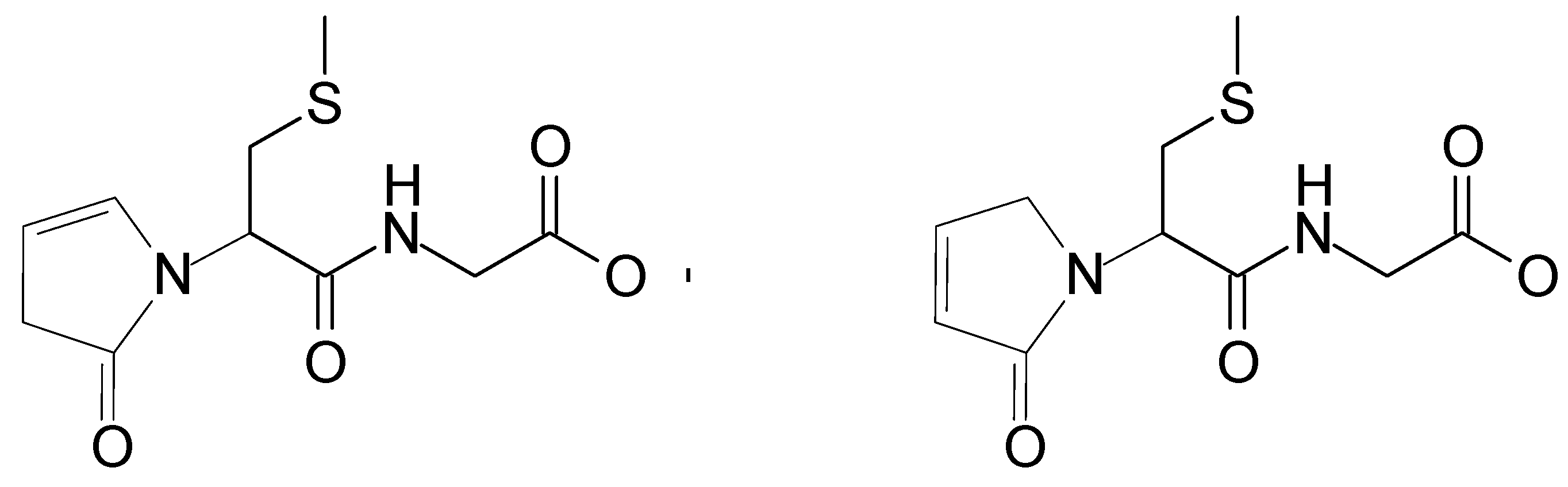
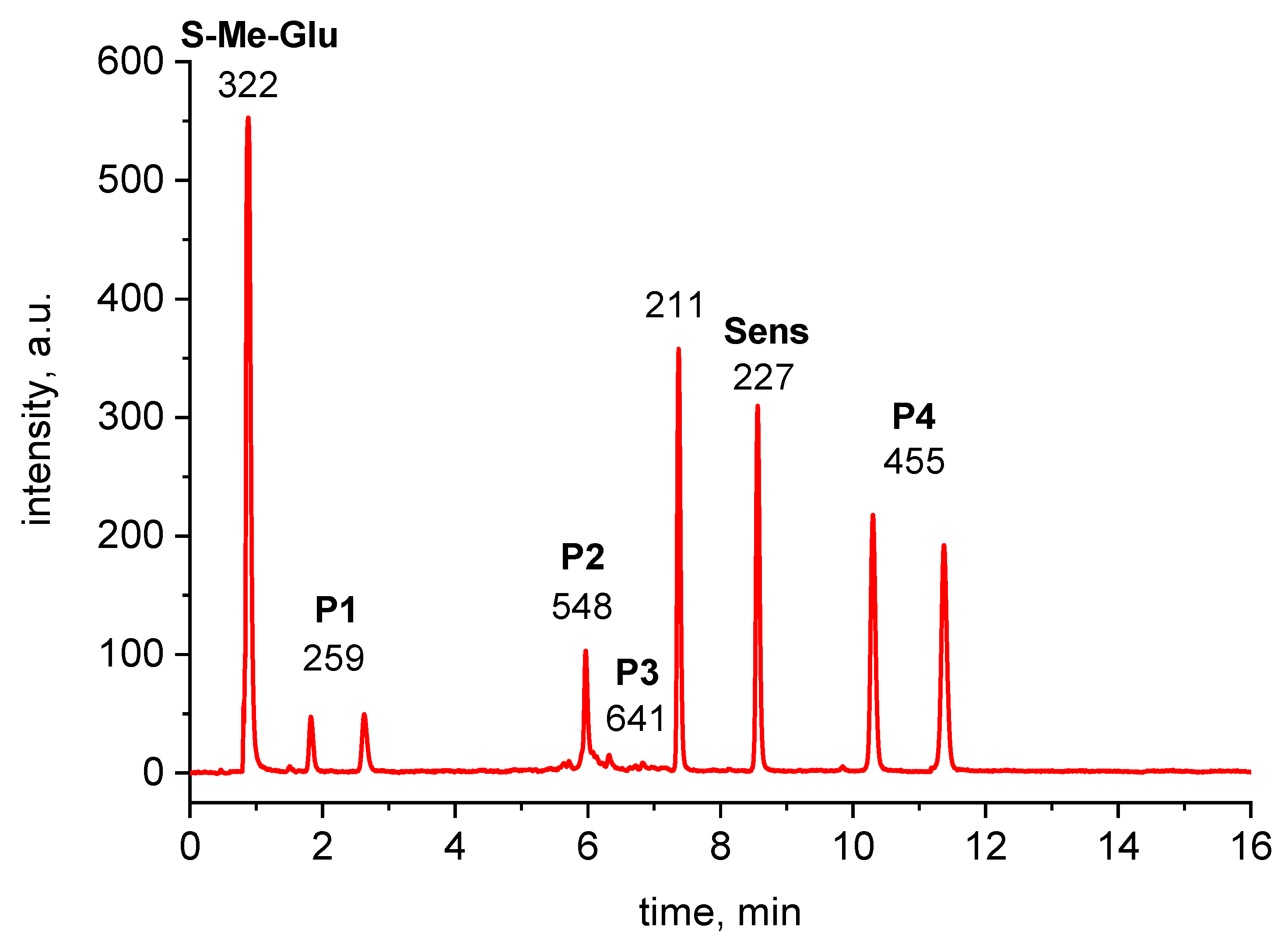
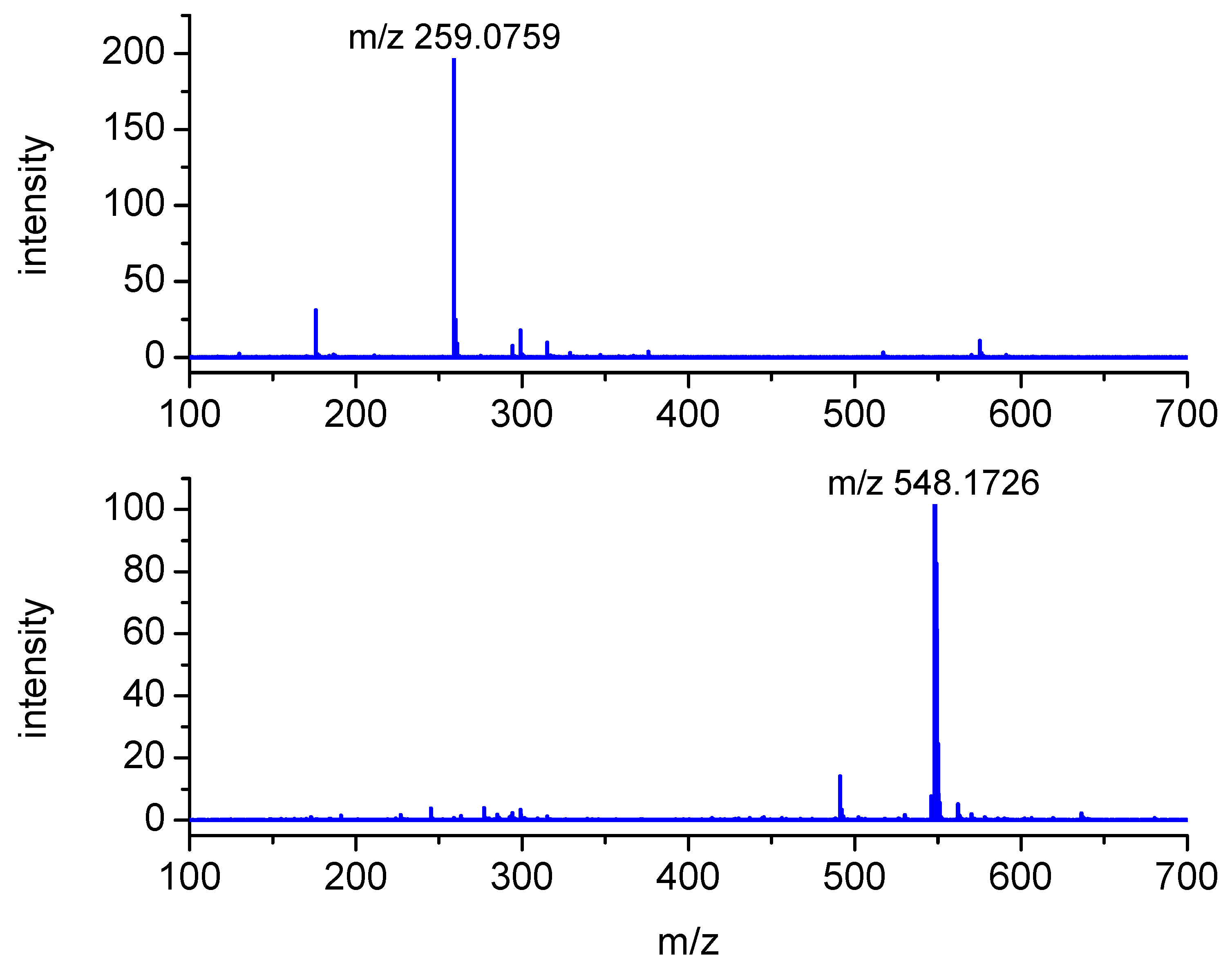
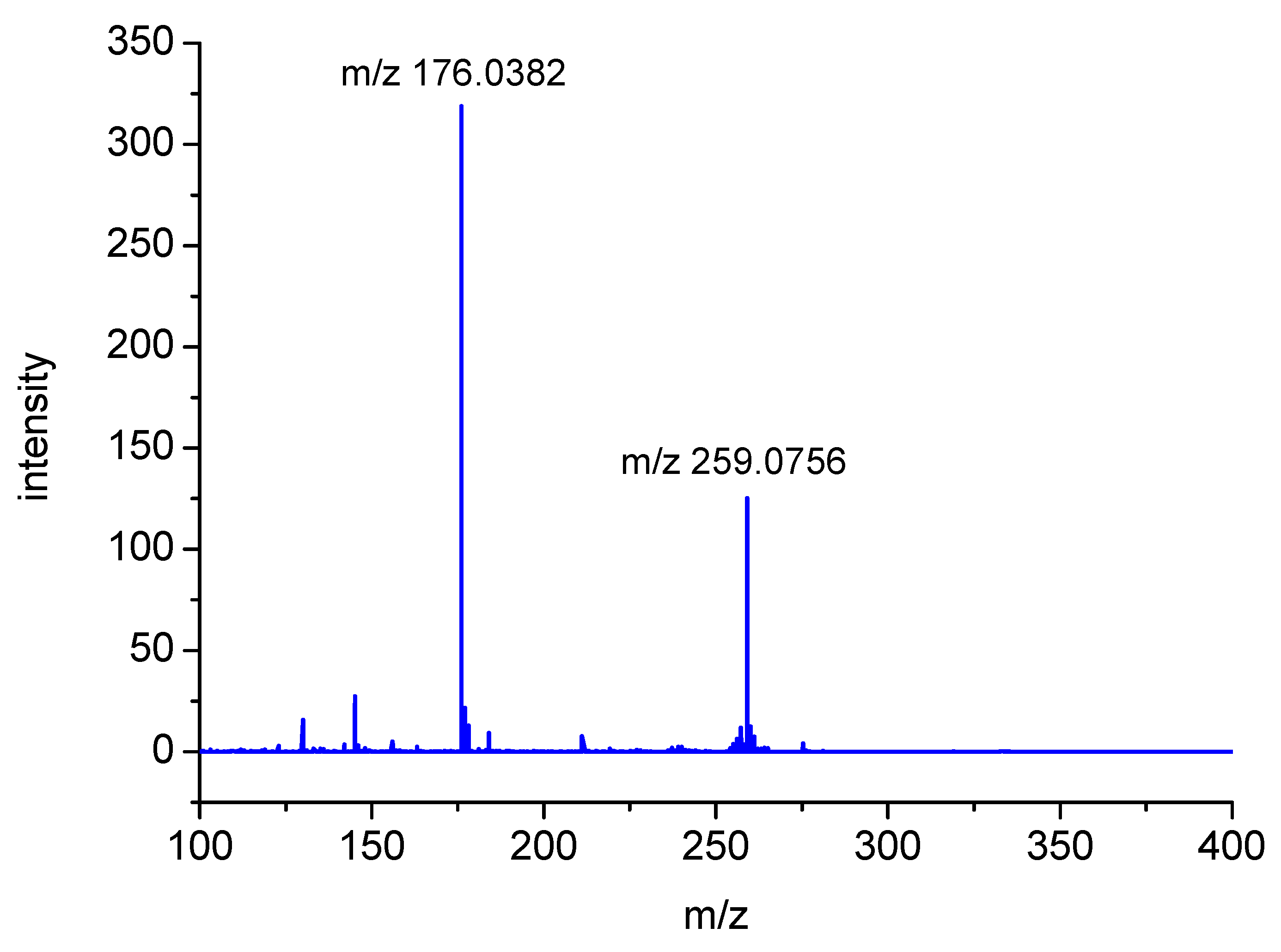
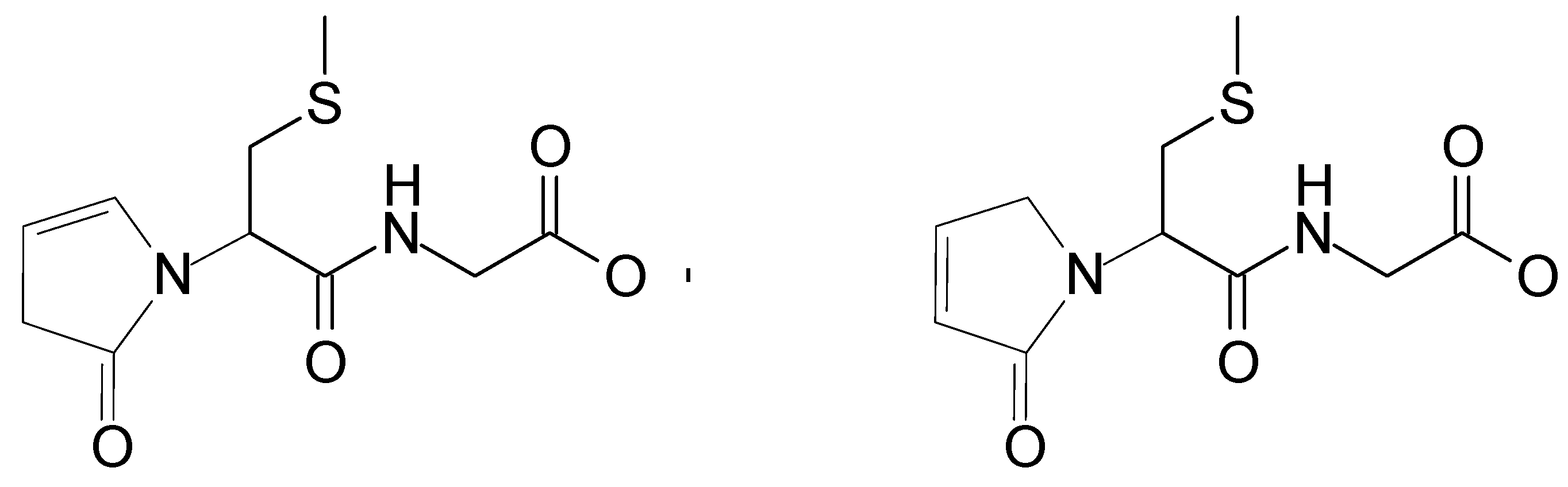
2.2. Analysis of High-Resolution Mass Spectra
3. Discussion
Mechanism
4. Materials and Methods
4.1. Nanosecond Laser Flash Photolysis (LFP)
4.2. Preparation of Solutions
4.3. Steady-State Photolysis
4.4. Chemicals
4.5. LC-MS Measurements
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maw, G.A. Biochemistry of S-methyl-L-cysteine and its principal derivatives. Sulfur Rep. 1982, 2, 1–26. [Google Scholar] [CrossRef]
- Kanazawa, A.; Kakimoto, Y.; Nakajima, T.; Sano, I. Identification of γ-glutamylserine, γ-glutamylalanine, γ-glutamylvaline and S-methylglutathione of bovine brain. Biochim. Et Biophys. Acta (Bba) - Gen. Subj. 1965, 111, 90–95. [Google Scholar] [CrossRef]
- Maw, G.A.; Coyne, C.M. The metabolism of S-methylcysteine in yeasts. Arch. Biochem. Biophys. 1968, 127, 241–251. [Google Scholar] [CrossRef]
- Terwilliger, T.C.; Bollag, G.E.; Sternberg, D.W.; Koshland, D.E. S-methyl glutathione synthesis is catalyzed by the cheR methyltransferase in Escherichia coli. J. Bacteriol. 1986, 165, 958–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallier, E.; Deutschmann, S.; Reichel, C.; Bolt, H.M.; Peter, H. A comparative investigation of the metabolism of methyl bromide and methyl iodide in human erythrocytes. Int. Arch. Occup. Environ. Health 1990, 62, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Peter, H.; Deutschmann, S.; Reichel, C.; Hallier, E. Metabolism of methyl chloride by human erythrocytes. Arch. Toxicol. 1989, 63, 351–355. [Google Scholar] [CrossRef]
- Redford-Ellis, M.; Gowenlock, A.H. Studies on the reaction of chloromethane with preparations of liver, brain and kidney. Acta Pharmacol. Et Toxicol. 1971, 30, 49–58. [Google Scholar] [CrossRef]
- Medinsky, M.A.; Bond, J.A.; Dutcher, J.S.; Birnbaum, L.S. Disposition of [14C] methyl bromide in fischer-344 rats after oral or intraperitoneal administration. Toxicology 1984, 32, 187–196. [Google Scholar] [CrossRef]
- Johnson, M.K. Studies on glutathione S-alkyltransferase of the rat. Biochem. J. 1966, 98, 44–56. [Google Scholar] [CrossRef] [Green Version]
- Jenei, Z.; Janáky, R.; Varga, V.; Saransaari, P.; Oja, S.S. Interference of S-alkyl derivatives of glutathione with brain ionotropic glutamate receptors. Neurochem. Res. 1998, 23, 1085–1091. [Google Scholar] [CrossRef]
- Elango, N.; Janaki, S.; Rao, A.R. Two affinity chromatography methods for the purification of glyoxalase I from rabbit liver. Biochem. Biophys. Res. Commun. 1978, 83, 1388–1395. [Google Scholar] [CrossRef]
- Vince, R.; Daluge, S.; Wadd, W.B. Inhibition of glyoxalase I by S-substituted glutathiones. J. Med. Chem. 1971, 14, 402–404. [Google Scholar] [CrossRef] [PubMed]
- Di Ilio, C.; Sacchetta, P.; Angelucci, S.; Bucciarelli, T.; Pennelli, A.; Mazzetti, A.P.; Lo Bello, M.; Aceto, A. Interaction of glutathione transferase P1-1 with captan and captafol. Biochem. Pharmacol. 1996, 52, 43–48. [Google Scholar] [CrossRef]
- Staab, C.A.; Hellgren, M.; Grafström, R.C.; Höög, J.-O. Medium-chain fatty acids and glutathione derivatives as inhibitors of S-nitrosoglutathione reduction mediated by alcohol dehydrogenase 3. Chem. -Biol. Interact. 2009, 180, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Lakomkin, V.L.; Lukoshkova, E.V.; Abramov, A.A.; Ermishkin, V.V.; Kapelko, V.I. Protective action of ethylglutathione at hypoxia-reoxygenation of the heart: Role of glucose. Kardiologiya 2013, 53, 49–54. [Google Scholar]
- Regan, R.F. Modulation of N-methyl-d-aspartate receptor responses with S-substituted derivatives of glutathione. US6329430B1, 17 August 2001. [Google Scholar]
- Anderson, E.I.; Wright, D.D. Effects of S-methyl glutathione, S-methyl cysteine, and the concentration of oxidized glutathione on transendothelial fluid transport. Investig. Ophthalmol. Vis. Sci. 1980, 19, 684–686. [Google Scholar]
- Spear, N.; Aust, S.D. Hydroxylation of deoxyguanosine in DNA by copper and thiols. Arch. Biochem. Biophys. 1995, 317, 142–148. [Google Scholar] [CrossRef]
- Cilento, G.; Nascimento, A.L.T.O. Generation of electronically excited triplet species at the cellular level: A potential source of genotoxicity. Toxicol. Lett. 1993, 67, 17–28. [Google Scholar] [CrossRef]
- Cilento, G. Generation of electronically excited triplet species in biochemical systems. In Pure and Applied Chemistry; Burrows, H., Stohner, J., Eds.; De Gruyter: Berlin, Germany, 1984; Volume 56, p. 1179. [Google Scholar]
- Glass, R.S. Sulfur-Centered Reactive Intermediates in Chemistry and Biology; Plenum Press: New York, NY, USA, 1990; Volume 97, pp. 213–226. [Google Scholar]
- Bobrowski, K.; Hug, G.L.; Pogocki, D.; Marciniak, B.; Schöneich, C. Sulfur radical cation−peptide bond complex in the one-electron oxidation of S-Methylglutathione. J. Am. Chem Soc. 2007, 129, 9236–9245. [Google Scholar] [CrossRef]
- Filipiak, P.; Bobrowski, K.; Hug, G.L.; Pogocki, D.; Schoneich, C.; Marciniak, B. Formation of a three-electron sulfur-sulfur bond as a probe for interaction between side chains of methionine residues. J. Phys. Chem. B 2016, 120, 9732–9744. [Google Scholar] [CrossRef]
- Bobrowski, K.; Holcman, J. Formation and stability of intramolecular three-electron SN, SS, and SO bonds in one-electron-oxidized simple methionine peptides. Pulse radiolysis. J. Phys. Chem. 1989, 93, 6381–6387. [Google Scholar] [CrossRef]
- Schoneich, C.; Zhao, F.; Madden, K.P.; Bobrowski, K. Side chain fragmentation of N-terminal threonine or serine residue induced through intramolecular proton transfer to hydroxy sulfuranyl radical formed at neighboring methionine in dipeptides. J. Am. Chem Soc. 1994, 116, 4641–4652. [Google Scholar] [CrossRef]
- Hug, G.L.; Bobrowski, K.; Pogocki, D.; Hoerner, G.; Marciniak, B. Conformational influence on the type of stabilization of sulfur radical cations in cyclic peptides. Chemphyschem 2007, 8, 2202–2210. [Google Scholar] [CrossRef] [PubMed]
- Bobrowski, K.; Hug, G.L.; Pogocki, D.; Marciniak, B.; Schoeneich, C. Stabilization of sulfide radical cations through complexation with the peptide bond: Mechanisms relevant to oxidation of proteins containing multiple methionine residues. J. Phys. Chem. B 2007, 111, 9608–9620. [Google Scholar] [CrossRef] [PubMed]
- Bobrowski, K.; Houée-Levin, C.; Marciniak, B. Stabilization and reactions of sulfur radical cations: Relevance to one-electron oxidation of methionine in peptides and proteins. Chim. Int. J. Chem. 2008, 62, 728–734. [Google Scholar] [CrossRef]
- Ignasiak, M.T.; Pedzinski, T.; Rusconi, F.; Filipiak, P.; Bobrowski, K.; Houee-Leyin, C.; Marciniak, B. Photosensitized oxidation of methionine-containing dipeptides. From the transients to the final products. J. Phys. Chem. B 2014, 118, 8549–8558. [Google Scholar] [CrossRef]
- Filipiak, P.; Bobrowski, K.; Hug, G.L.; Pogocki, D.; Schoneich, C.; Marciniak, B. New insights into the reaction paths of 4-carboxybenzophenone triplet with oligopeptides containing N- and C-terminal methionine residues. J. Phys. Chem. B 2017, 121, 5247–5258. [Google Scholar] [CrossRef]
- Filipiak, P.; Hug, G.L.; Bobrowski, K.; Pedzinski, T.; Kozubek, H.; Marciniak, B. Sensitized photooxidation of S-methylglutathione in aqueous solution: Intramolecular (SO) and (SN) bonded species. J. Phys. Chem. B 2013, 117, 2359–2368. [Google Scholar] [CrossRef]
- Morozova, O.B.; Panov, M.S.; Vieth, H.M.; Yurkovskaya, A.V. CIDNP study of sensitized photooxidation of S-methylcysteine and S-methylglutathione in aqueous solution. J. Photochem. Photobiol. A: Chem. 2016, 321, 90–98. [Google Scholar] [CrossRef]
- Hiller, K.O.; Masloch, B.; Goebl, M.; Asmus, K.D. Mechanism of the hydroxyl radical induced oxidation of methionine in aqueous solution. J. Am. Chem Soc. 1981, 103, 2734–2743. [Google Scholar] [CrossRef]
- Marciniak, B.; Hug, G.L.; Kozubek, H.; Bobrowski, K. Mechanism of 4-carboxybenzophenone-sensitized photooxidation of methionine-containing dipeptides and tripeptides in aqueous solution. J. Phys. Chem. 1995, 99, 13560–13568. [Google Scholar] [CrossRef]
- Hug, G.L.; Marciniak, B.; Bobrowski, K. Sensitized photo-oxidation of sulfur-containing amino acids and peptides in aqueous solution. J. Photochem. Photobiol. A: Chem. 1996, 95, 81–88. [Google Scholar] [CrossRef]
- Hug, G.L.; Bobrowski, K.; Kozubek, H.; Marciniak, B. Photo-oxidation of methionine-containing peptides by the 4-carboxybenzophenone triplet state in aqueous solution. Competition between intramolecular two-centered three-electron bonded (SS)+ and (SN)+ formation. Photochem. Photobiol. 2000, 72, 1–9. [Google Scholar] [CrossRef]
- Pedzinski, T.; Bobrowski, K.; Ignasiak, M.; Kciuk, G.; Hug, G.L. Lewandowska-Andralojc, A.; Marciniak, B., 3-Carboxybenzophenone (3-CB) as an efficient sensitizer in the photooxidation of methionyl-leucine in aqueous solutions: Spectral, kinetic and acid-base properties of 3-CB derived transients. J. Photochem. Photobiol. A: Chem. 2014, 287, 1–7. [Google Scholar] [CrossRef]
- Pedzinski, T.; Markiewicz, A.; Marciniak, B. Photosensitized oxidation of methionine derivatives. Laser flash photolysis studies. Res. Chem. Intermed. 2009, 35, 497–506. [Google Scholar] [CrossRef]
- Hug, G.L.; Marciniak, B.; Bobrowski, K. Acid-base equilibria involved in secondary reactions following the 4-carboxybenzophenone sensitized photooxidation of methionylglycine in aqueous solution. Spectral and time resolution of the decaying (S therefore N)(+) radical cation. J. Phys. Chem. 1996, 100, 14914–14921. [Google Scholar] [CrossRef]
- Hiller, K.O.; Asmus, K.D. Oxidation of methionine by X in aqueous solution and characterization of some three-electron bonded intermediates. A pulse radiolysis study. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1981, 40, 583–595. [Google Scholar] [CrossRef]
- Bobrowski, K.; Marciniak, B.; Hug, G.L. 4-Carboxybenzophenone-sensitized photooxidation of sulfur-containing amino acids. Nanosecond laser flash photolysis and pulse radiolysis studies. J. Am. Chem Soc. 1992, 114, 10279–10288. [Google Scholar] [CrossRef]
- Hiller, K.O.; Asmus, K.D. Formation and reduction reactions of alpha-amino radicals derived from methionine and its derivatives in aqueous solutions. J. Phys. Chem. 1983, 87, 3682–3688. [Google Scholar] [CrossRef]
- Das, P.K. Transient carbocations and carbanions generated by laser flash photolysis and pulse radiolysis. Chem. Rev. 1993, 93, 119–144. [Google Scholar] [CrossRef]
- Bobrowski, K.; Pogocki, D.; Schöneich, C. Oxidation of (carboxyalkyl)thiopropionic acid derivatives by hydroxyl radicals. Mechanisms and kinetics of competitive inter- and intramolecular formation of σ- and σ*-type sulfuranyl radicals. J. Phys. Chem. A 1998, 102, 10512–10521. [Google Scholar] [CrossRef]
- Cilento, G. Photobiochemistry without light. Experientia 1988, 44, 572–576. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MH+ Exact Mass (Measured) | MH+ Exact Mass (Calculated) | Most Abundant Fragment (MSMS) | Molecular Composition (MH+) | Product |
|---|---|---|---|---|
| 259.0759 | 259.0753 | 176.0382 | C10H15N2O4S | P1 |
| 548.1726 | 548.1703 | - | C25H30N3O9S | P2 (αS-Sens) |
| 273.1003 | 273.0909 | 190.0540 | C11H17N2O4S | P’1 |
| 562.1864 | 562.1859 | - | C26H32N3O9S | P’2 (αS-Sens) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pedzinski, T.; Bobrowski, K.; Marciniak, B.; Filipiak, P. Unexpected Reaction Pathway of the Alpha-Aminoalkyl Radical Derived from One-Electron Oxidation of S-Alkylglutathiones. Molecules 2020, 25, 877. https://doi.org/10.3390/molecules25040877
Pedzinski T, Bobrowski K, Marciniak B, Filipiak P. Unexpected Reaction Pathway of the Alpha-Aminoalkyl Radical Derived from One-Electron Oxidation of S-Alkylglutathiones. Molecules. 2020; 25(4):877. https://doi.org/10.3390/molecules25040877
Chicago/Turabian StylePedzinski, Tomasz, Krzysztof Bobrowski, Bronislaw Marciniak, and Piotr Filipiak. 2020. "Unexpected Reaction Pathway of the Alpha-Aminoalkyl Radical Derived from One-Electron Oxidation of S-Alkylglutathiones" Molecules 25, no. 4: 877. https://doi.org/10.3390/molecules25040877