Synthesis and In Vitro Anticancer Evaluation of Novel Chrysin and 7-Aminochrysin Derivatives

 ,
,
Abstract
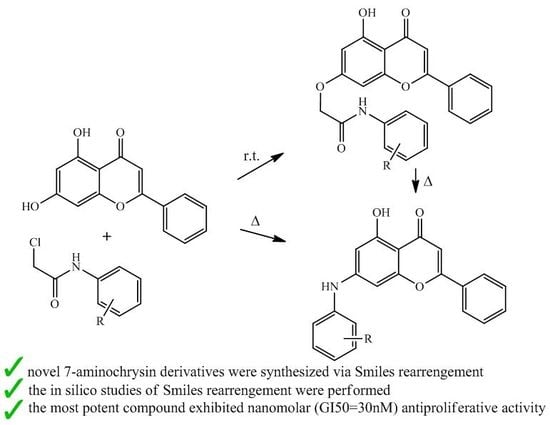
:
1. Introduction
2. Results and Discussion
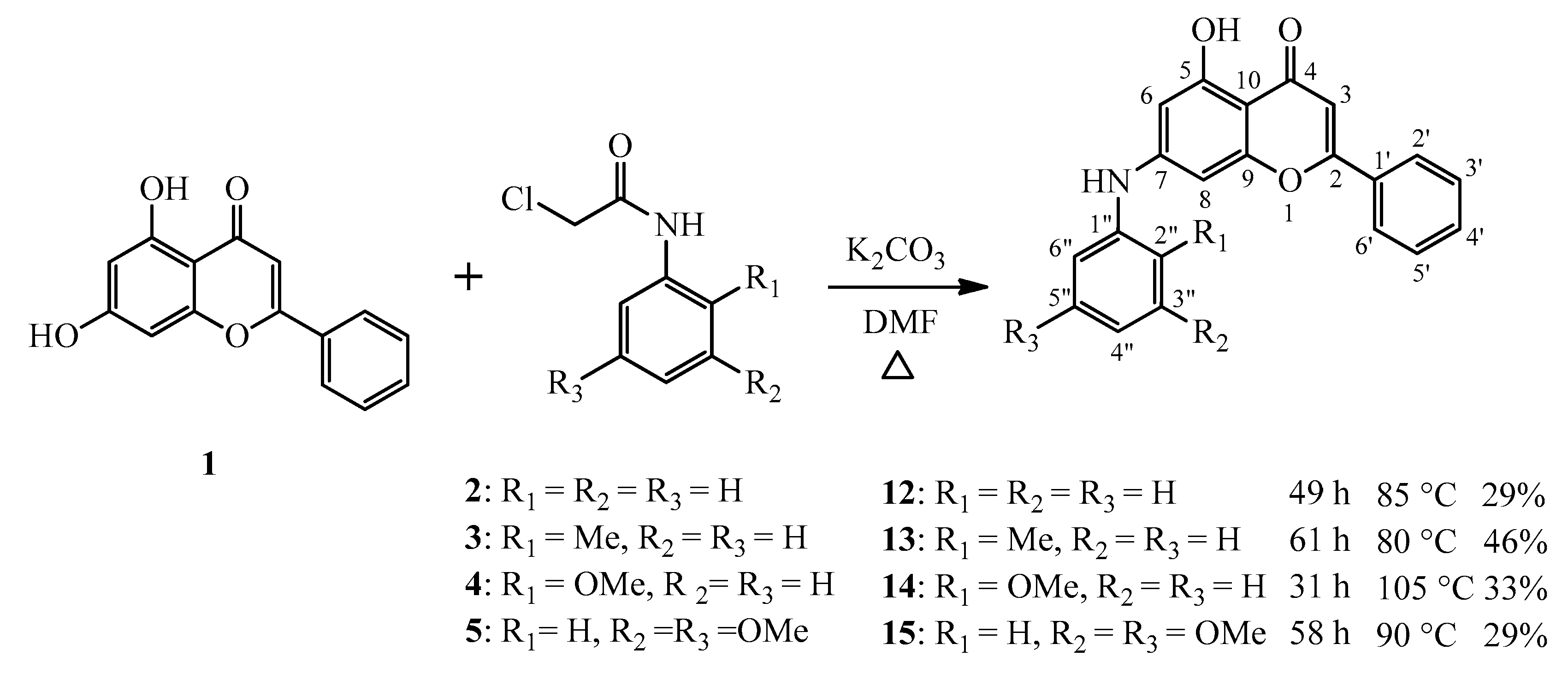
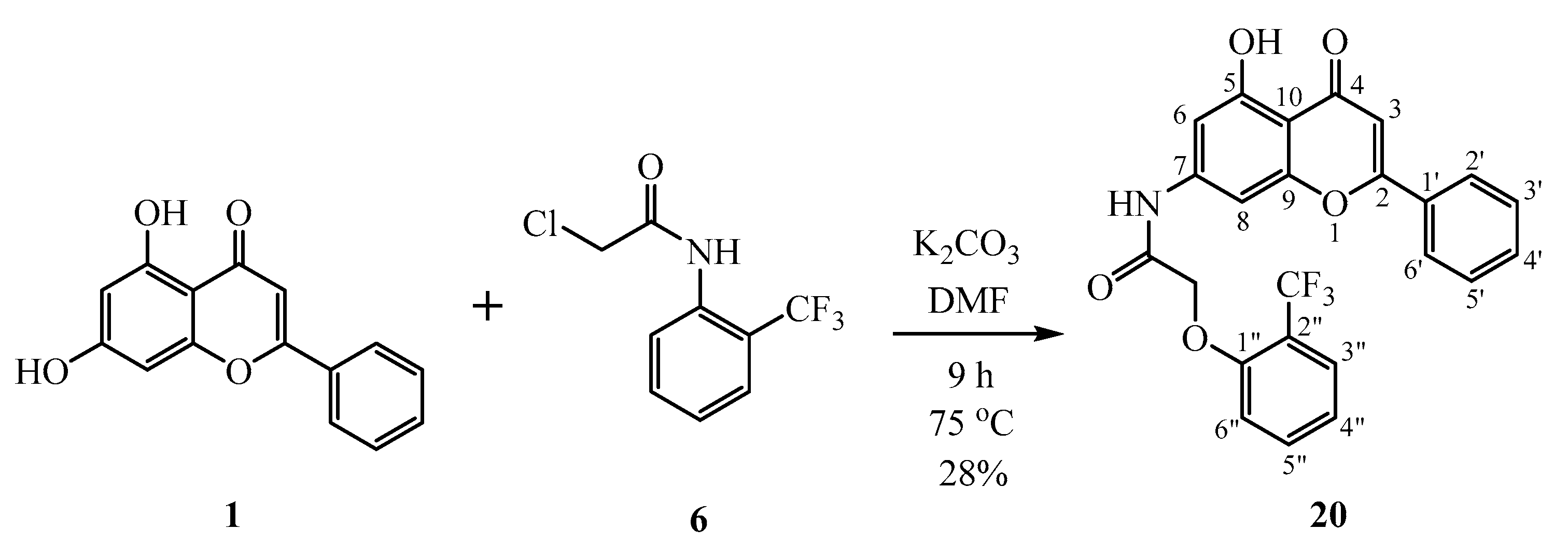
2.1. Chemistry
2.2. Kinetic and Mechanistic Investigation of the Reaction 1 + 4 → 9 + 14
2.3. Biological Evaluation
3. Materials and Methods
3.1. General Materials and Methods
3.2. In Silico Studies on the Smiles Rearrangement
3.3. Biological Evaluation
3.3.1. One-Dose Screen
3.3.2. Five-Dose Screen
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Raffa, D.; Maggio, B.; Riamondi, M.V.; Plescia, F.; Daidone, G. Recent discoveries of anticancer flavonoids. Eur. J. Med. Chem. 2017, 142, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Midelton, E., Jr.; Kandswami, C. The Flavonoids—Advences in Research Since 1986; Harborne, J.B., Ed.; Chapman and Hall: Cambridge, UK, 1993; pp. 619–652. ISBN 978-1-4899-2915-0. [Google Scholar]
- Read, M.A. Flavonoids: Naturally occurring anti-inflammatory agents. Am. J. Pathol. 1995, 147, 235–237. [Google Scholar] [PubMed]
- Mani, R.; Natesan, V. Chrysin: Sorurces, beneficial pharmacological activities, and molecular mechanism of action. Phytochemistry 2018, 145, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Boubakeur, B.; Tirtouil, A.; Meddah, B.; Khadem, H. The evaluation of the effect of synthetic flavonoids on growt of pathogenic and probiotic bacterica. J. Chem. Pharm. Res. 2015, 7, 228–236. [Google Scholar]
- Li, B.; Zhang, F.; Serrao, E.; Cheng, H.; Sanches, T.W.; Yang, L.; Nemati, N.; Zheng, Y.; Wang, H.; Long, Y. Design and discovery of flavonoid-based HIV-1 integrase inhibitors targeting both the active site and the interaction with LEDGF/p75. Bioorg. Med. Chem. 2014, 22, 3146–3158. [Google Scholar] [CrossRef]
- Brinkworth, R.I.; Stoermer, M.J.; Fairlie, D.P. Flavones are inhibitors of HIV-1 proteinase. Biochem. Biopshys. Res. Commun. 1992, 188, 631–637. [Google Scholar] [CrossRef]
- Catapano, A.L. Antioxidant effect of flavonoids. Angiology 1997, 48, 39–44. [Google Scholar] [CrossRef]
- Chen, Y.H.; Yang, Z.S.; Wen, C.C.; Chang, Y.S.; Wang, B.C.; Hsiao, C.A. Evaluation of the structure-activity relationship of flavonoids as antioxidants and toxicants of zebrafish larvae. Food Chem. 2012, 134, 717–724. [Google Scholar] [CrossRef]
- Medina, J.H.; Paladini, A.C.; Wolfman, C.; de Stein, M.L.; Calvo, D.; Diaz, L.E.; Peña, C. Chrysin (5,7-di-OH-flavone), a naturally-occurring ligand for benzodiazepine receptors, with anticonvulsant properties. Biochem. Pharmacol. 1990, 40, 2227–2231. [Google Scholar] [CrossRef]
- Dhawan, K.; Dhawan, S.; Sharma, A. Passiflora: A review update. J. Ethnopharmacol. 2004, 94, 1–23. [Google Scholar] [CrossRef]
- Ksaka, E.R.; Bodduluru, L.N.; Madana, R.M.; Athira, K.V.; Gogoi, R.; Barua, C.C. Chemopreventive and therapeutic potetntial of chrysin in cancer: Mechanistic perspectives. Toxicol. Lett. 2015, 233, 214–225. [Google Scholar] [CrossRef]
- Brechbul, H.M.; Kachadourian, R.; Min, E.; Chan, D.; Day, B.J. Chrysin enhances doxorubicin-induced cytotoxicity is human lung epithelial cancer cell lines: The role of gluthation. Toxicol. Appl. Pharmacol. 2012, 258, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, B.D.M.; Threadgill, M.D.; Groundwater, P.W.; Dale, I.L.; Hickaman, J.A. Synthesis and biological evaluation of a series of flavones designed as inhibitors of protein tyrosine kinases. Anticancer Drug Des. 1992, 7, 365–384. [Google Scholar] [PubMed]
- Cushman, M.; Zhu, H.; Geahlen, R.L.; Kraker, A.J. Synthesis and biochemical evaluation of a series of aminoflavones as potential inhibitors of protein-tyrosine kinases p56lck, EGFr, and p60v-src. J. Med. Chem. 1994, 37, 3353–3362. [Google Scholar] [CrossRef] [PubMed]
- Deka, N.; Hadjeri, M.; Lawson, M.; Beney, C.; Boumendjel, A. Acetylated dimethoxyaniline as a key intermediate for the synthesis of aminoflavones and quinolones. Heterocycles 2002, 57, 123–128. [Google Scholar]
- Deng, B.; Lepoivre, A.; Lemière, G. Synthesis of 7-Vinylflavone and 7-Aminoflavone by Palladium-Catalyzed Coupling Reactions. Eur. J. Org. Chem. 1999, 10, 2683–2688. [Google Scholar] [CrossRef]
- Choe, H.; Kim, J.; Hong, S. Structure-based design of flavone-based inhibitors of wild-type and T315I mutant of ABL. Bioorg. Med. Chem. Lett. 2013, 23, 4324–4327. [Google Scholar] [CrossRef]
- Warren, L.A.; Smiles, S. CXVII.—iso-β-Naphthol sulphide. J. Chem. Soc. 1930, 956–963. [Google Scholar] [CrossRef]
- Warren, L.A.; Smiles, S. CLXXI.—Dehydro-2-naphtholsulphone. J. Chem. Soc. 1930, 1327–1331. [Google Scholar] [CrossRef]
- Truce, W.E.; Kreider, E.M.; Brand, W.W. The Smiles and Related Rearrangements of Aromatic Systems. Org. React. 2011, 99–215. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.; Devlin, F.; Chabalowski, C.F.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Petersson, G.A.; Bennett, A.; Tensfeldt, T.G.; Al-Laham, M.A.; Shirley, W.A. A complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row elements. J. Chem. Phys. 1988, 89, 2193–2218. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. J. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.H.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A.; et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef]
- Alley, M.C.; Scudiero, D.A.; Monks, A.M.; Hursey, L.; Czerwinski, M.J.; Fine, D.L.; Abbott, B.J.; Mayo, J.G.; Shoemaker, R.H.; Boyd, M.R. Feasibility of Drug Screening with Panels of Human Tumor Cell Lines Using a Microculture Tetrazolium Assay. Cancer Res. 1988, 48, 589–601. [Google Scholar]
- Shoemaker, R.H.; Monks, A.; Alley, M.C.; Scudiero, D.A.; Fine, D.L.; McLemore, T.L.; Abbott, B.J.; Paull, K.D.; Mayo, J.G.; Boyd, M.R. Development of Human Tumor Cell Line Panels for Use in Disease-Oriented Drug Screening. Prog. Clin. Biol. Res. 1988, 276, 265–286. [Google Scholar]
- NCI-60 Screening Methodology. Available online: https://dtp.cancer.gov/discovery_development/nci-60/methodology.htm (accessed on 20 January 2020).
- Hratchian, H.P.; Schlegel, H.B. Accurate reaction paths using a Hessian based predictor-corrector integrator. J. Chem. Phys. 2004, 120, 9918–9924. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Time (h) | Conversion (MS, %) of Compounds with the Corresponding M + H+ in Brackets | |||||
|---|---|---|---|---|---|---|---|
| 1 (255) | 4 (200) | 9 (418) | 14 (360) | 16a (581) | 16b (523) | ||
| 1 | 0 | 100 | 100 | n.d. | n.d. | n.d. | n.d. |
| 2 | 0.25 | 29 | 27 | 61 | n.d. | n.d. | n.d. |
| 3 | 0.5 | 5 | 10 | 100 | n.d. | 4 | n.d. |
| 4 | 1 | n.d. | 7 | 96 | n.d. | 10 | n.d. |
| 5 | 2 | n.d. | 7 | 99 | n.d. | 10 | n.d. |
| 6 | 24 | n.d. | 5 | 61 | 19 | 9 | 2 |
| 7 | 48 | n.d. | 2 | 43 | 31 | 8 | 4 |
| Column | A | B | C | B | E | F | G | H | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | ΔG# TS2–6 | ΔG0 7–11 | ΔG# TS17 | ΔG0 18 | ΔG# TS18 | ΔG0 19 | ΔG# TS19 | ΔG0 12–16 |
| 1 | H | H | H | 73.3 | −67.2 | 31.0 | 16.1 | 34.9 | 9.9 | 140.7 | −7.0 |
| 2 | Me | H | H | 67.6 | −73.0 | 19.8 | 2.1 | 20.6 | −6.5 | 137.0 | −13.3 |
| 3 | OMe | H | H | 69.3 | −73.6 | 16.0 | −4.1 | 20.4 | −7.4 | 127.0 | −28.6 |
| 4 | H | OMe | OMe | 75.4 | −67.1 | 33.5 | 19.8 | 35.6 | 17.1 | 143.6 | −4.2 |
| 5 | CF3 | H | H | 73.3 | −71.0 | 32.7 | 25.6 | 42.9 | 17.6 | 155.8 | −9.0 |
| 1 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 20 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Leukemia | Living Cells, % | ||||||||||
| CCRF-CEM | 102.24 | 94.38 | 88.46 | 103.61 | 100.26 | 58.61 | 99.72 | 29.51 | 7.29 | 10.80 | 93.43 |
| HL-60(TB) | 116.20 | 74.53 | 80.72 | 96.19 | 86.01 | 55.03 | 95.29 | 8.58 | −6.07 | −35.66 | 96.64 |
| K-562 | 96.80 | 95.30 | 84.14 | 102.18 | 95.93 | 49.21 | 70.67 | 10.23 | 11.56 | 13.10 | 93.28 |
| MOLT-4 | 105.93 | 91.11 | 86.70 | 101.33 | 84.14 | 52.14 | 102.90 | 25.82 | 25.94 | 24.60 | 98.42 |
| RPMI-8226 | 101.04 | 93.25 | 100.27 | 103.24 | 101.95 | 54.84 | 103.51 | 29.70 | 16.42 | 17.06 | 90.20 |
| SR | 77.87 | n.d. | n.d. | 85.32 | n.d. | n.d. | 40.93 | 7.93 | 5.75 | 17.29 | n.d. |
| Non-Small-Cell Lung Cancer | |||||||||||
| A549/ATCC | 98.46 | 84.59 | 89.91 | 99.07 | 93.60 | 0.21 | 88.14 | 35.17 | 33.70 | 25.89 | 86.93 |
| EKVX | 89.35 | 102.66 | 95.38 | 100.69 | 104.69 | 51.40 | 95.16 | 35.54 | 57.37 | 38.16 | 100.07 |
| HOP-62 | 113.09 | 100.81 | 96.03 | 100.66 | 94.26 | 16.24 | 102.52 | 49.44 | 35.64 | 38.50 | 96.92 |
| HOP-92 | 77.93 | 89.87 | 94.70 | 122.42 | 90.77 | 51.61 | 97.26 | 86.73 | 62.17 | 79.55 | 92.47 |
| NCI-H226 | 86.84 | n.d. | n.d. | 96.59 | n.d. | n.d. | 82.23 | 57.82 | 76.84 | 67.67 | n.d. |
| NCI-H23 | 92.57 | 83.67 | 98.22 | 99.50 | 94.72 | 46.10 | 96.33 | 45.45 | 51.50 | 35.95 | 88.56 |
| NCI-H233M | 98.30 | 85.60 | 95.63 | 100.67 | 98.57 | 62.22 | 98.48 | 70.48 | 80.08 | 44.07 | 93.82 |
| NCI-H460 | 98.34 | 61.56 | 79.30 | 83.22 | 97.66 | −42.49 | 89.31 | 8.06 | 11.44 | 7.62 | 91.59 |
| NCI-H552 | 88.95 | 81.54 | 84.78 | 95.78 | 88.34 | 46.64 | 87.19 | 35.99 | 7.64 | −35.25 | 82.91 |
| Colon Cancer | |||||||||||
| COLO 205 | 104.94 | 108.40 | 108.85 | 117.93 | 109.55 | 60.11 | 117.09 | 61.77 | 76.85 | 48.45 | 106.47 |
| HCC-2998 | 102.88 | 99.66 | 104.54 | 102.54 | 97.53 | 89.85 | 98.67 | 76.72 | 71.93 | 32.39 | 106.49 |
| HCT-116 | 82.69 | 57.38 | 66.46 | 71.54 | 97.57 | 5.84 | 94.22 | 15.22 | 14.09 | 12.96 | 84.06 |
| HCT-15 | 90.99 | 96.02 | 97.82 | 110.39 | 101.89 | 53.37 | 88.34 | 17.62 | 29.25 | 25.00 | 102.90 |
| HT29 | 102.89 | 104.70 | 109.72 | 104.51 | 98.93 | 70.62 | 102.62 | 69.86 | 16.96 | 3.58 | 108.92 |
| KM12 | 92.93 | 86.55 | 100.16 | 104.94 | 103.34 | 68.50 | 98.53 | 36.41 | 36.18 | 20.97 | 93.03 |
| SW-620 | 101.60 | 87.53 | 91.85 | 101.53 | 96.14 | 21.80 | 92.66 | 21.04 | 20.80 | 24.89 | 92.57 |
| CNS Cancer | |||||||||||
| SF-268 | 101.55 | 90.96 | 92.88 | 103.44 | 95.94 | 63.49 | 108.82 | 61.47 | 44.88 | 48.20 | 93.54 |
| SF-295 | 99.86 | 100.68 | 105.53 | 107.28 | 101.39 | 34.55 | 94.93 | 27.12 | 25.87 | 33.51 | 99.41 |
| SF-539 | 92.17 | 90.97 | 95.00 | 94.46 | 95.80 | 35.24 | 88.56 | 23.08 | 28.35 | −35.20 | 101.29 |
| SNB-19 | 86.04 | 87.38 | 97.95 | 98.29 | 94.84 | 45.17 | 80.85 | 32.62 | 43.92 | 43.08 | 93.88 |
| SNB-75 | 88.98 | 88.03 | 89.86 | 98.11 | 85.65 | −4.06 | 85.97 | −5.66 | −4.79 | −9.05 | 84.74 |
| U251 | 80.67 | 93.73 | 99.75 | 95.07 | 99.90 | 4.09 | 103.19 | 36.19 | 22.12 | 22.78 | 105.43 |
| Melanoma | |||||||||||
| LOX IMVI | 85.08 | 89.59 | 91.33 | 97.12 | 94.23 | 51.99 | 98.19 | 30.95 | 39.93 | 41.67 | 91.07 |
| MALME-3M | 101.76 | 97.72 | 99.19 | 106.86 | 96.36 | 54.32 | 99.49 | 47.94 | 44.12 | 73.59 | 96.86 |
| M14 | 106.78 | 85.43 | 95.69 | 107.85 | 97.45 | 70.56 | 102.80 | 24.59 | 20.80 | 28.37 | 89.53 |
| MDA-MB-435 | 99.53 | 95.35 | 100.50 | 101.33 | 100.05 | 78.05 | 91.47 | 1.90 | −24.29 | 4.04 | 97.39 |
| SK-MEL-2 | 109.90 | 96.58 | 106.20 | 110.49 | 101.95 | 92.04 | 118.37 | 65.66 | 13.67 | 13.19 | 102.47 |
| SK-MEL-28 | 101.70 | 105.88 | 103.62 | 106.04 | 103.28 | 79.12 | 101.17 | 51.11 | 56.53 | 68.55 | 103.95 |
| SK-MEL-5 | 92.85 | 98.99 | 97.12 | 96.32 | n.d. | n.d. | 98.19 | 23.46 | 24.55 | 28.62 | n.d. |
| UACC-257 | 118.94 | 97.25 | 97.09 | 100.73 | 95.08 | 73.38 | 113.80 | 80.55 | 54.00 | 77.94 | 92.88 |
| UACC-62 | 82.24 | 93.67 | 94.23 | 100.84 | 93.14 | 77.53 | 94.55 | 43.38 | 35.81 | 42.50 | 90.77 |
| Ovarian Cancer | |||||||||||
| IGROV1 | 95.22 | 99.14 | 94.90 | 99.66 | 97.11 | 40.29 | 101.69 | 45.33 | 51.38 | 42.09 | 99.29 |
| OVCAR-3 | 97.60 | 105.55 | 112.68 | 117.24 | 116.11 | −2.06 | 119.29 | −4.27 | 5.90 | 0.37 | 107.74 |
| OVCAR-4 | 112.07 | 78.90 | 92.03 | 108.84 | 101.20 | −50.15 | 95.50 | 53.09 | 65.63 | 59.30 | 97.96 |
| OVCAR-5 | 99.07 | 98.07 | 98.20 | 106.13 | 98.13 | 62.88 | 107.85 | 89.20 | 78.14 | 45.48 | 101.59 |
| OVCAR-8 | 95.19 | 87.16 | 96.80 | 93.95 | 101.69 | 29.02 | 76.29 | 52.50 | 32.37 | 26.14 | 95.43 |
| NCI/ADR-RES | 92.84 | 88.88 | 98.42 | 98.91 | 96.78 | 51.21 | 84.49 | 4.90 | 11.48 | 22.21 | 93.45 |
| SK-OV-3 | 128.15 | 106.68 | 112.10 | 99.01 | 106.94 | 44.14 | 91.61 | 59.29 | 50.68 | 35.94 | 101.19 |
| Renal Cancer | |||||||||||
| 786-0 | 99.20 | 81.89 | 102.88 | 103.24 | 98.94 | 7.11 | 99.45 | 43.46 | 49.17 | 45.40 | 102.34 |
| A498 | 86.62 | 98.75 | 107.57 | 114.05 | 103.91 | 55.93 | 86.89 | 31.26 | 11.62 | −4.73 | 89.70 |
| ACHN | 85.03 | 57.89 | 89.02 | 95.66 | 100.57 | 17.85 | 88.95 | 34.50 | 50.84 | 48.27 | 90.18 |
| CAKI-1 | 83.56 | 77.85 | 85.07 | 94.67 | 83.96 | 22.26 | 78.85 | 39.00 | 41.29 | 47.20 | 84.52 |
| RXF 393 | 91.19 | 98.26 | 105.07 | 114.29 | 103.82 | 63.39 | 87.18 | 27.97 | n.d. | n.d. | 96.03 |
| SN12C | 85.77 | 89.89 | 100.73 | 93.53 | 96.93 | 27.73 | 89.88 | 31.78 | 36.59 | 44.31 | 94.21 |
| TK-10 | 107.21 | 102.52 | 101.40 | 107.48 | 96.05 | −13.91 | 115.18 | 110.08 | 66.29 | 43.97 | 104.00 |
| UO-31 | 89.42 | 80.20 | 80.65 | 81.08 | 84.04 | 17.07 | 113.09 | 42.28 | 46.60 | 40.33 | 88.09 |
| Prostate Cancer | |||||||||||
| PC-3 | 93.17 | 86.21 | 86.03 | 102.30 | 96.04 | 64.61 | 91.31 | 40.61 | 54.48 | 27.81 | 90.42 |
| DU-145 | 92.00 | 81.47 | 83.97 | 95.26 | 97.68 | 52.73 | 93.71 | 26.85 | 45.58 | 19.97 | 99.56 |
| Breast Cancer | |||||||||||
| MCF7 | 103.05 | 85.34 | 84.36 | 89.90 | 93.57 | 49.76 | 88.89 | 15.46 | 17.34 | 23.46 | 89.40 |
| MDA-MB-231/ATCC | 82.64 | 72.92 | 87.92 | 95.07 | 99.54 | 9.62 | 97.20 | 50.66 | 39.19 | 15.88 | 83.19 |
| HS 578T | 92.51 | 80.27 | 94.59 | 97.60 | 98.24 | 15.48 | 86.96 | 44.73 | 45.59 | 12.36 | 95.30 |
| BT-549 | 91.01 | n.d. | n.d. | 103.53 | n.d. | n.d. | 90.69 | 47.08 | 40.89 | 80.16 | n.d. |
| T-47D | 101.44 | 94.33 | 85.80 | 86.66 | 97.63 | 14.26 | 91.42 | 36.46 | 31.43 | 55.24 | 95.24 |
| MDA-MB-468 | 91.09 | 102.86 | 100.21 | 114.94 | 101.67 | 67.58 | 86.66 | −2.12 | −1.72 | 11.66 | 99.78 |
| Mean: | 96.10 | 90.32 | 95.08 | 100.69 | 97.53 | 40.61 | 94.75 | 38.33 | 34.57 | 29.48 | 95.38 |
| Delta: | 18.23 | 32.94 | 28.62 | 29.15 | 13.57 | 90.76 | 53.82 | 43.99 | 58.86 | 65.14 | 12.47 |
| Range: | 50.28 | 51.02 | 46.22 | 50.88 | 32.15 | 142.19 | 78.36 | 115.74 | 104.37 | 115.82 | 26.01 |
| 11 | 13 | 14 | 15 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Leukemia | GI50 (μM) | TGI (μM) | LC50 (μM) | GI50 (μM) | TGI (μM) | LC50 (μM) | GI50 (μM) | TGI (μM) | LC50 (μM) | GI50 (μM) | TGI (μM) | LC50 (μM) |
| CCRF-CEM | 19.6 | 64.1 | >100 | 2.25 | >100 | >100 | 3.57 | >100 | >100 | 0.4 | >100 | >100 |
| HL-60 (TB) | >100 | >100 | >100 | 2.16 | >100 | >100 | 2.79 | 34.9 | >100 | 0.32 | >100 | >100 |
| K-562 | 5.03 | >100 | >100 | 0.47 | >100 | >100 | 0.59 | >100 | >100 | 0.28 | >100 | >100 |
| MOLT-4 | 5.46 | >100 | >100 | 2.66 | >100 | >100 | 4.08 | >100 | >100 | 0.56 | >100 | >100 |
| RPMI-8226 | 7.03 | >100 | >100 | 2.87 | >100 | >100 | 4.51 | >100 | >100 | 0.39 | >100 | >100 |
| SR | 4.44 | >100 | >100 | 0.40 | >100 | >100 | 0.53 | >100 | >100 | 0.25 | >100 | >100 |
| Non-Small-Cell Lung Cancer | ||||||||||||
| A549/ATCC | 1.71 | 3.75 | 8.23 | 2.11 | >100 | >100 | 5.16 | >100 | >100 | 0.72 | 94.7 | >100 |
| EKVX | 9.37 | 62.4 | >100 | 3.35 | >100 | >100 | 4.83 | 5.6 | >100 | 0.64 | >100 | >100 |
| HOP-62 | 2.62 | 8.19 | 35.7 | 3.23 | >100 | >100 | 3.55 | >100 | >100 | 0.56 | 21.7 | >100 |
| HOP-92 | 2.13 | 6.71 | 90.1 | 3.51 | >100 | >100 | 3.76 | 23.7 | >100 | 1.95 | 21.3 | >100 |
| NCI-H226 | 2.28 | 6.48 | >100 | 4.11 | >100 | >100 | 4.38 | 28.8 | >100 | 1.25 | 71.2 | >100 |
| NCI-H23 | 3.07 | 11.6 | 49.6 | 4.75 | >100 | >100 | 4.22 | 52.6 | >100 | 0.93 | 28 | >100 |
| NCI-H233M | >100 | >100 | >100 | n.d. | >100 | >100 | 5.98 | 27.4 | >100 | 0.52 | 49.2 | >100 |
| NCI-H460 | 2.59 | 6.68 | >100 | 2.89 | >100 | >100 | 3.37 | 12 | >100 | 0.36 | 12.5 | 93.6 |
| NCI-H552 | 6.77 | >100 | >100 | 1.79 | >100 | >100 | 2.67 | 27.1 | >100 | 0.24 | 21.5 | >100 |
| Colon Cancer | ||||||||||||
| COLO 205 | 8.04 | >100 | >100 | n.d. | >100 | >100 | 9.92 | >100 | >100 | 5.13 | >100 | >100 |
| HCC-2998 | >100 | >100 | >100 | n.d. | >100 | >100 | 6.07 | >100 | >100 | 1.94 | 29.1 | >100 |
| HCT-116 | 1.83 | 4.06 | 8.99 | 3.36 | >100 | >100 | 3.31 | >100 | >100 | 0.42 | >100 | >100 |
| HCT-15 | 3.28 | 21.1 | >100 | 0.68 | >100 | >100 | 1.65 | >100 | >100 | 0.06 | >100 | >100 |
| HT29 | 6.48 | >100 | >100 | 3.25 | >100 | >100 | 3.64 | >100 | >100 | 3.18 | 13.6 | >100 |
| KM12 | 8.17 | >100 | >100 | 2.38 | >100 | >100 | 2.76 | 55.2 | >100 | 0.2 | 15.3 | >100 |
| SW-620 | 3.56 | >100 | >100 | 0.73 | >100 | >100 | 2.5 | >100 | >100 | 0.35 | >100 | >100 |
| CNS Cancer | ||||||||||||
| SF-268 | 4.94 | 26.1 | >100 | 4.49 | >100 | >100 | 4.71 | 48.2 | >100 | 0.6 | 31 | >100 |
| SF-295 | 2.94 | 9.13 | 32.6 | 2.36 | >100 | >100 | 2.98 | 13.2 | 43.2 | 0.3 | 3.79 | 99.3 |
| SF-539 | 2.21 | 5.50 | 20.5 | 3.10 | >100 | >100 | 2.73 | 10.4 | 36.3 | 0.28 | 1.25 | 18.2 |
| SNB-19 | 4.17 | 21.7 | >100 | 2.98 | >100 | >100 | 2.99 | 15.7 | >100 | 0.48 | 20.5 | >100 |
| SNB-75 | 1.65 | 4.41 | 14.9 | n.d. | n.d. | n.d. | 2.39 | 8.5 | 59.6 | 0.17 | 0.64 | 44.2 |
| U251 | 2.01 | 4.45 | 9.84 | 3.04 | >100 | >100 | 3.63 | >100 | >100 | 0.41 | 16 | >100 |
| Melanoma | ||||||||||||
| LOX IMVI | 4.74 | 22.6 | >100 | 2.75 | >100 | >100 | 3.41 | >100 | >100 | 0.51 | >100 | >100 |
| MALME-3M | 3.17 | >100 | >100 | 2.43 | >100 | >100 | 2.13 | 11.9 | 57.6 | 0.63 | >100 | >100 |
| M14 | 27.6 | >100 | >100 | 2.38 | >100 | >100 | n.d. | n.d. | n.d. | 0.5 | >100 | >100 |
| MDA-MB-435 | 6.03 | >100 | >100 | 0.31 | >100 | >100 | 0.36 | 2.03 | >100 | 0.1 | 0.33 | >100 |
| SK-MEL-2 | 15.1 | 38.0 | 95.7 | 2.44 | >100 | >100 | 5.62 | 34.2 | >100 | 0.79 | 78.5 | >100 |
| SK-MEL-28 | n.d. | >100 | >100 | n.d. | >100 | >100 | 3.64 | 25.8 | >100 | 2.78 | >100 | >100 |
| SK-MEL-5 | 4.10 | 25.4 | >100 | 2.39 | >100 | >100 | 3 | 9.58 | 42.4 | 0.17 | 3.25 | >100 |
| UACC-257 | >100 | >100 | >100 | n.d. | >100 | >100 | 6.72 | 63.5 | >100 | >100 | >100 | >100 |
| UACC-62 | 6.58 | >100 | >100 | 2.89 | >100 | >100 | 2.39 | 12.8 | 61.1 | 0.3 | 14 | >100 |
| Ovarian Cancer | ||||||||||||
| IGROV1 | 3.61 | 15.9 | 86.3 | 3.20 | >100 | >100 | 2.98 | 18.5 | 94.5 | 0.23 | 25.5 | >100 |
| OVCAR-3 | 2.12 | 4.12 | 7.99 | 1.72 | 4.91 | >100 | 2.05 | 5.2 | 21.5 | 0.22 | 1.59 | >100 |
| OVCAR-4 | 2.45 | 5.94 | >100 | 3.38 | >100 | >100 | 4.49 | 47.8 | >100 | 8.9 | 41.5 | >100 |
| OVCAR-5 | 7.18 | >100 | >100 | >100 | >100 | >100 | 4.82 | 27.3 | >100 | 0.55 | 22.6 | >100 |
| OVCAR-8 | 3.67 | 3.21 | >100 | 3.61 | >100 | >100 | 4.65 | >100 | >100 | 0.54 | 17.1 | >100 |
| NCI/ADR-RES | 3.45 | 22.5 | >100 | 1.28 | >100 | >100 | 1.99 | 18 | >100 | 0.45 | 26.7 | >100 |
| SK-OV-3 | 14.9 | 50.7 | >100 | 5.56 | >100 | >100 | 3.57 | 17.9 | 96.4 | 0.4 | 14.1 | 74.3 |
| Renal Cancer | ||||||||||||
| 786-0 | 2.01 | 4.15 | 8.56 | 4.42 | >100 | >100 | 5.15 | 27 | >100 | 1.04 | 20.7 | 91.4 |
| A498 | 4.05 | 17.8 | 65.2 | 2.48 | n.d. | >100 | 3.08 | 13.9 | 65.8 | 0.34 | 14 | 75.9 |
| ACHN | 2.91 | 9.63 | 60.5 | 4.25 | >100 | >100 | 3.38 | 13 | 41.5 | 0.59 | >100 | >100 |
| CAKI-1 | 3.74 | 18.7 | 78.2 | 2.77 | >100 | >100 | 4.24 | 20.3 | 73.5 | 0.28 | 15 | 80.3 |
| RXF 393 | 2.21 | 6.34 | 30.1 | 2.45 | 9.56 | >100 | 1.74 | 5.34 | 28.1 | 0.21 | 0.79 | 38.1 |
| SN12C | 4.02 | >100 | >100 | 4.05 | >100 | >100 | 3.5 | 17.6 | 94.5 | 0.79 | 60.4 | >100 |
| TK-10 | 1.88 | 3.71 | 7.30 | n.d. | >100 | >100 | 10.4 | 28.6 | 78.5 | 10.7 | 79.4 | >100 |
| UO-31 | 2.44 | n.d. | >100 | n.d. | >100 | >100 | 3.57 | 21.5 | >100 | 0.62 | 22.9 | 98.3 |
| Prostate Cancer | ||||||||||||
| PC-3 | 5.75 | >100 | >100 | 3.30 | >100 | >100 | 5.13 | 41.6 | >100 | 0.52 | 27.2 | >100 |
| DU-145 | 7.97 | >100 | >100 | 3.78 | >100 | >100 | 4.35 | 16.4 | 51.7 | 0.4 | 2.14 | >100 |
| Breast Cancer | ||||||||||||
| MCF7 | 7.40 | >100 | >100 | 0.82 | >100 | >100 | 1.63 | >100 | >100 | 0.03 | >100 | >100 |
| MDA-MB-231/ATCC | 2.04 | 5.26 | 22.6 | 3.58 | >100 | >100 | 3.96 | 18.4 | 68.2 | 1.05 | 22.4 | >100 |
| HS 578T | 2.60 | 7.80 | >100 | 2.46 | >100 | >100 | 3.61 | 32.6 | >100 | 0.3 | 2.45 | >100 |
| BT-549 | 5.19 | 38.9 | >100 | 4.03 | >100 | >100 | 4.96 | 37.2 | >100 | 0.77 | >100 | >100 |
| T-47D | 2.54 | n.d. | >100 | 2.96 | >100 | >100 | 4.36 | >100 | >100 | 0.21 | >100 | >100 |
| MDA-MB-468 | 4.95 | 35.8 | >100 | 0.38 | 3.03 | >100 | 1.5 | 6.42 | >100 | 0.13 | 0.64 | >100 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mayer, S.; Keglevich, P.; Ábrányi-Balogh, P.; Szigetvári, Á.; Dékány, M.; Szántay, C., Jr.; Hazai, L. Synthesis and In Vitro Anticancer Evaluation of Novel Chrysin and 7-Aminochrysin Derivatives. Molecules 2020, 25, 888. https://doi.org/10.3390/molecules25040888
Mayer S, Keglevich P, Ábrányi-Balogh P, Szigetvári Á, Dékány M, Szántay C Jr., Hazai L. Synthesis and In Vitro Anticancer Evaluation of Novel Chrysin and 7-Aminochrysin Derivatives. Molecules. 2020; 25(4):888. https://doi.org/10.3390/molecules25040888
Chicago/Turabian StyleMayer, Szabolcs, Péter Keglevich, Péter Ábrányi-Balogh, Áron Szigetvári, Miklós Dékány, Csaba Szántay, Jr., and László Hazai. 2020. "Synthesis and In Vitro Anticancer Evaluation of Novel Chrysin and 7-Aminochrysin Derivatives" Molecules 25, no. 4: 888. https://doi.org/10.3390/molecules25040888