Glycosaminoglycans as Tools to Decipher the Platelet Tumor Cell Interaction: A Focus on P-Selectin

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Breast Cancer Cell-Induced Platelet Activation and Secretion
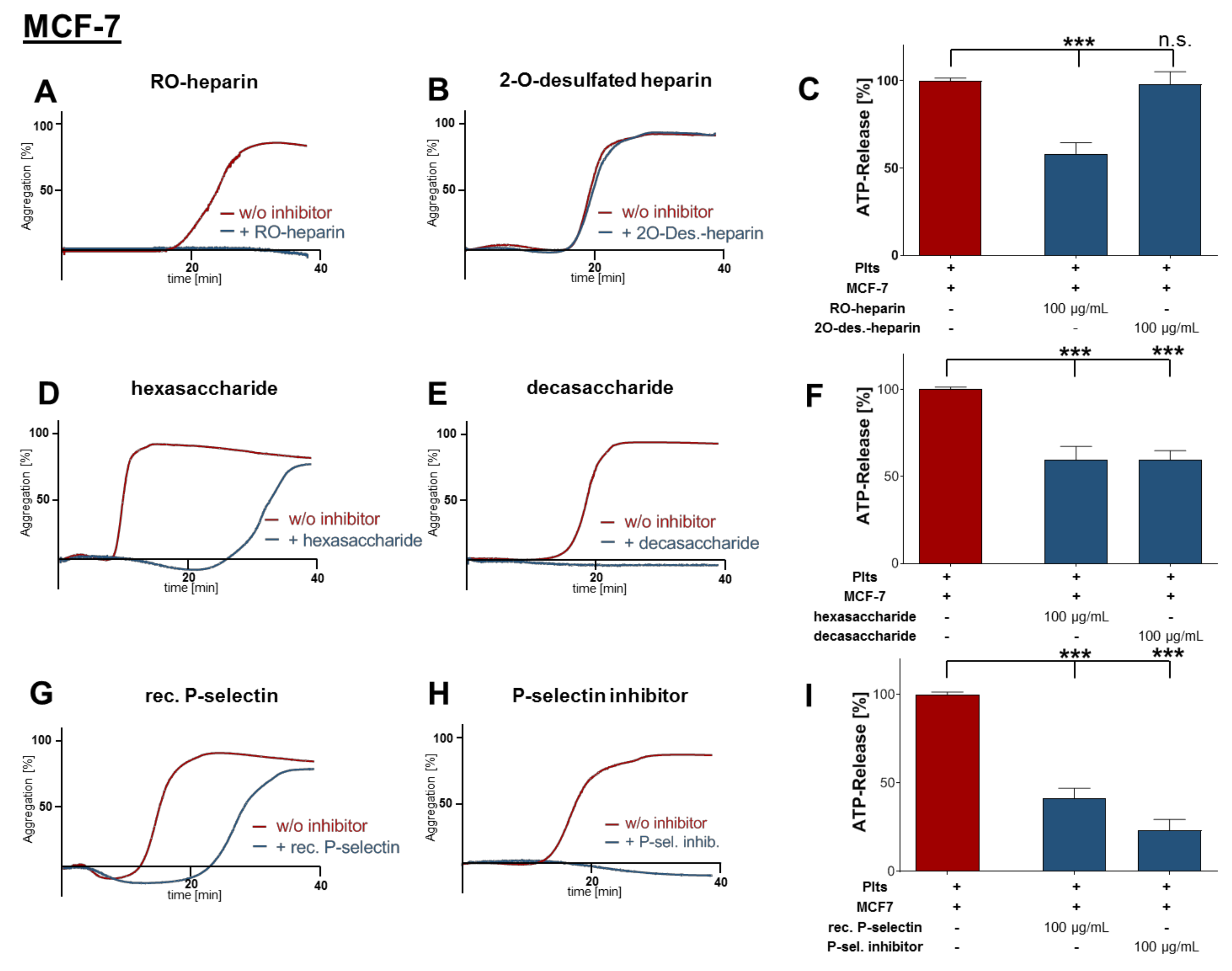
2.2. Inhibition of Platelet Aggregation and Dense Granule Secretion by Modified Heparin Derivatives
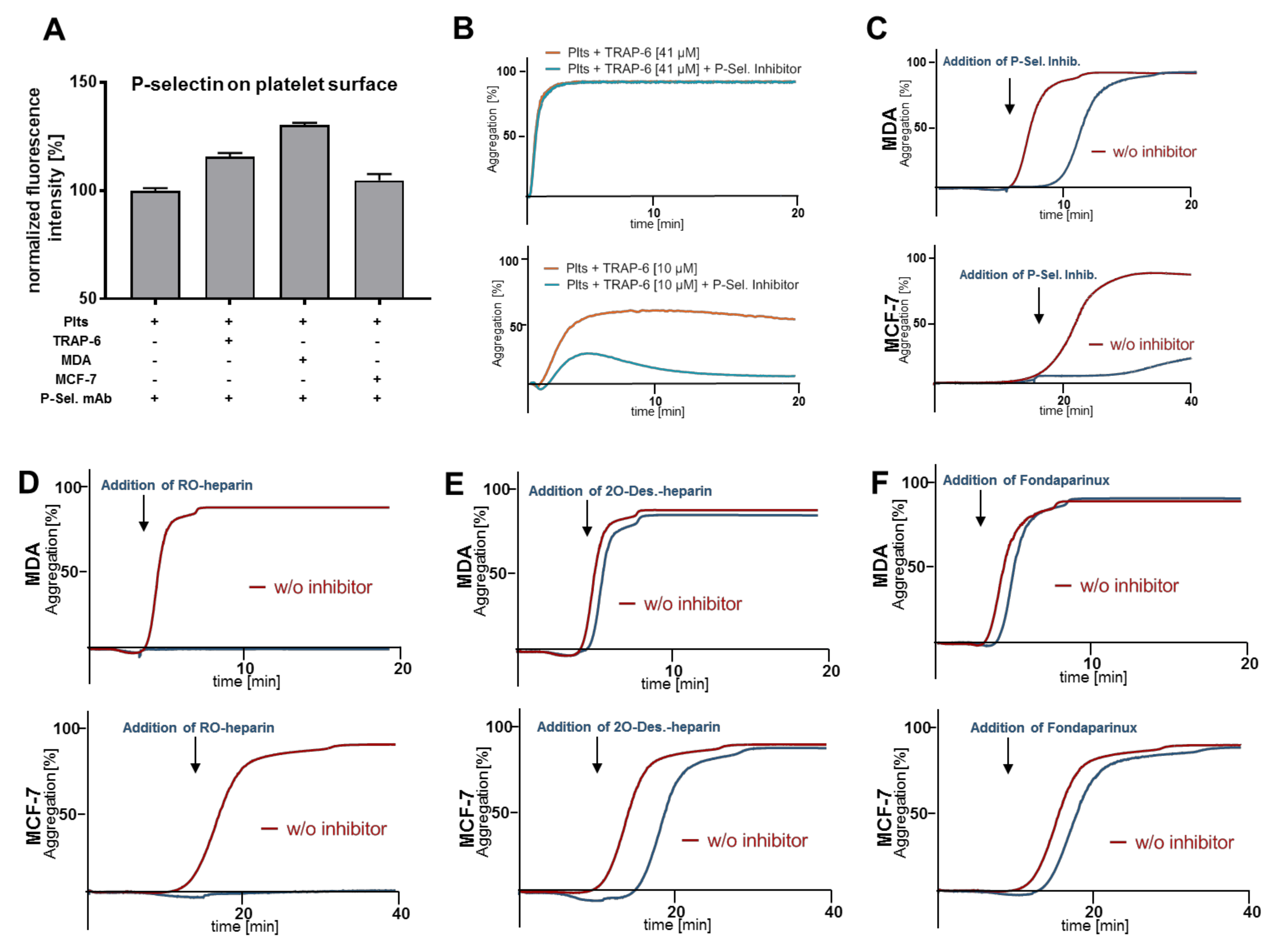
2.3. Heparin Mediated P-Selectin Blockade Subsequent to Tumor Cell Interaction
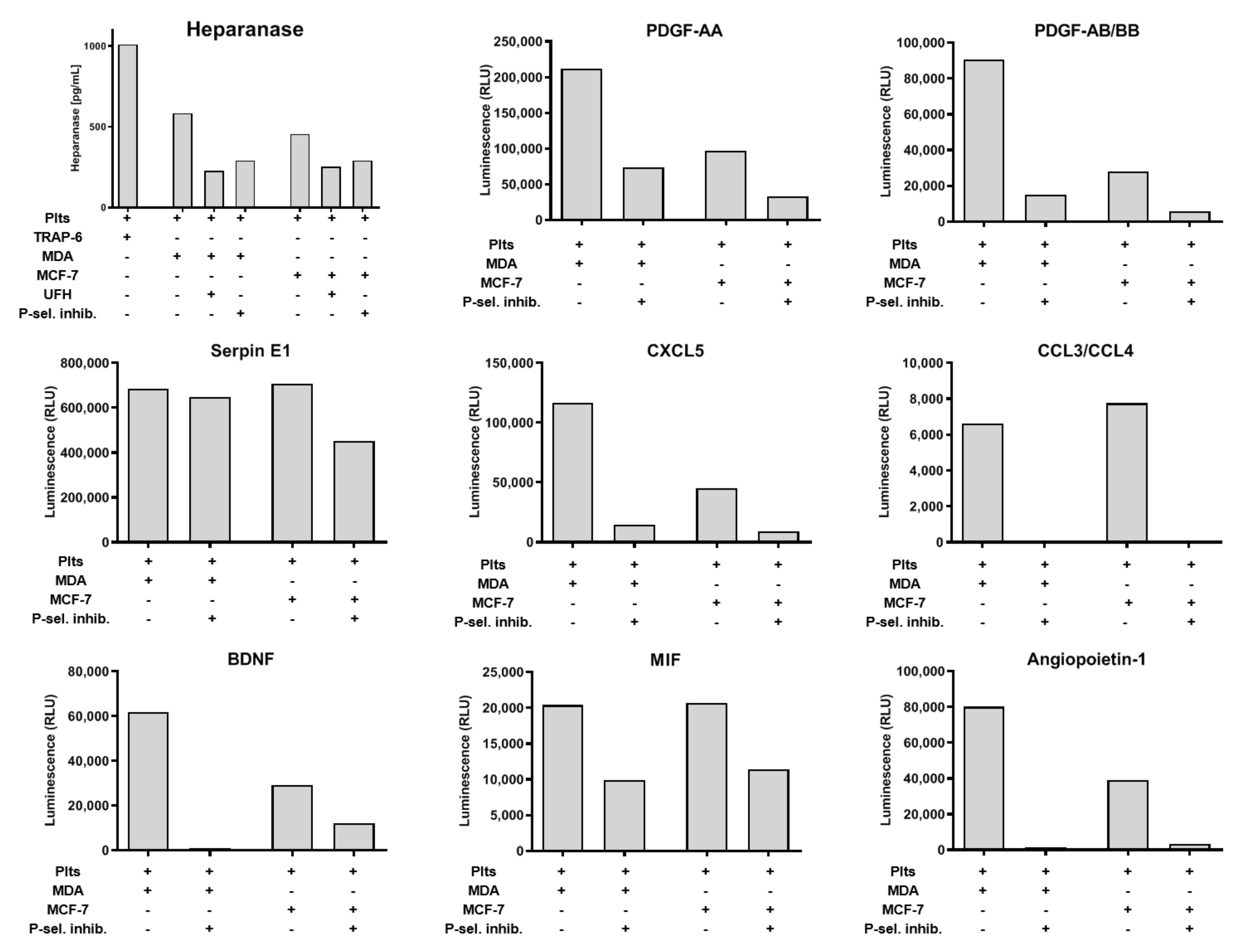
2.4. Impact of P-Selectin Inhibition on Platelet α-Granule Secretion
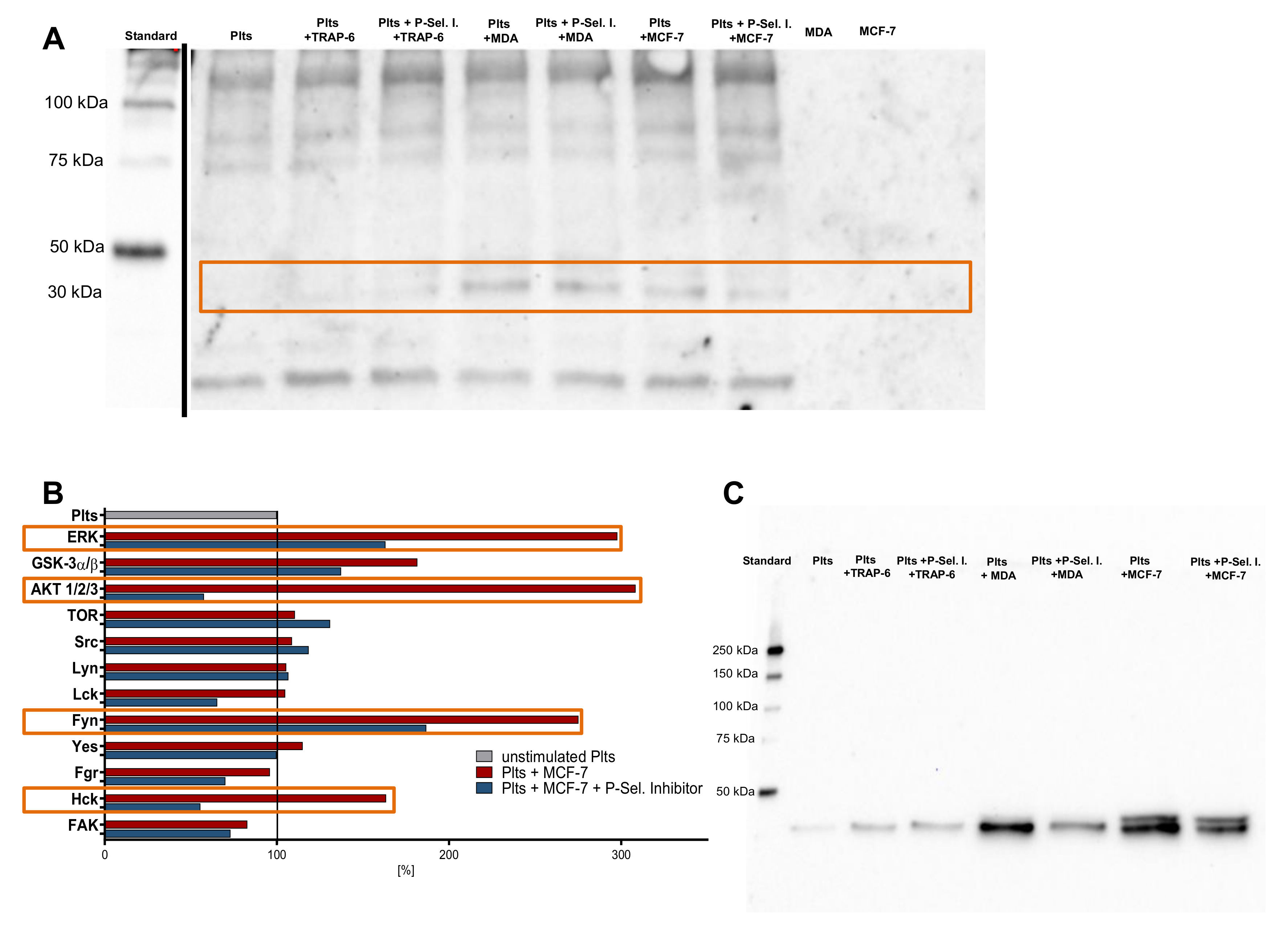
2.5. P-Selectin Signaling in Platelets
3. Discussion
4. Materials and Methods
4.1. Glycosaminogylcans and Sulfated Polysaccharides
4.2. Cell Lines
4.3. Preparation of Washed Platelets
4.4. Platelet Dense Granule Secretion Assay
4.5. Light Transmission Aggregometry
4.6. Western Blot
4.7. Cytokine and Phospho-Kinase Arrays
4.8. Heparanase ELISA
4.9. Flow Cytometry
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Labelle, M.; Hynes, R.O. The initial hours of metastasis: The importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discov. 2012, 2, 1091–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopp, H.-G.; Placke, T.; Salih, H.R. Platelet-derived transforming growth factor-beta down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res. 2009, 69, 7775–7783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, J.H.; Coupland, L.A.; Freeman, C.; Chong, B.H.; Parish, C.R. Activation of tumour cell ECM degradation by thrombin-activated platelet membranes: Potentially a P-selectin and GPIIb/IIIa-dependent process. Clin. Exp. Metastasis 2015, 32, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Coupland, L.A.; Chong, B.H.; Parish, C.R. Platelets and P-selectin control tumor cell metastasis in an organ-specific manner and independently of NK cells. Cancer Res. 2012, 72, 4662–4671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyslop, S.R.; Josefsson, E.C. Undercover Agents: Targeting Tumours with Modified Platelets. Trends Cancer 2017, 3, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Zarà, M.; Canobbio, I.; Visconte, C.; Canino, J.; Torti, M.; Guidetti, G.F. Molecular mechanisms of platelet activation and aggregation induced by breast cancer cells. Cell. Signal. 2018, 48, 45–53. [Google Scholar] [CrossRef]
- Ueno, T.; Toi, M.; Koike, M.; Nakamura, S.; Tominaga, T. Tissue factor expression in breast cancer tissues: Its correlation with prognosis and plasma concentration. Br. J. Cancer 2000, 83, 164–170. [Google Scholar] [CrossRef]
- Adams, G.N.; Rosenfeldt, L.; Frederick, M.; Miller, W.; Waltz, D.; Kombrinck, K.; McElhinney, K.E.; Flick, M.J.; Monia, B.P.; Revenko, A.S.; et al. Colon Cancer Growth and Dissemination Relies upon Thrombin, Stromal PAR-1, and Fibrinogen. Cancer Res. 2015, 75, 4235–4243. [Google Scholar] [CrossRef] [Green Version]
- Gockel, L.M.; Ponert, J.M.; Schwarz, S.; Schlesinger, M.; Bendas, G. The Low Molecular Weight Heparin Tinzaparin Attenuates Platelet Activation in Terms of Metastatic Niche Formation by Coagulation-Dependent and Independent Pathways. Molecules 2018, 23, 2753. [Google Scholar] [CrossRef] [Green Version]
- Zucchella, M.; Dezza, L.; Pacchiarini, L.; Meloni, F.; Tacconi, F.; Bonomi, E.; Grignani, G.; Notario, A. Human tumor cells cultured “in vitro” activate platelet function by producing ADP or thrombin. Haematologica 1989, 74, 541–545. [Google Scholar] [PubMed]
- Aitokallio-Tallberg, A.M.; Viinikka, L.U.; Ylikorkala, R.O. Increased synthesis of prostacyclin and thromboxane in human ovarian malignancy. Cancer Res. 1988, 48, 2396–2398. [Google Scholar] [PubMed]
- Yu, L.-X.; Yan, L.; Yang, W.; Wu, F.-Q.; Ling, Y.; Chen, S.-Z.; Tang, L.; Tan, Y.-X.; Cao, D.; Wu, M.-C.; et al. Platelets promote tumour metastasis via interaction between TLR4 and tumour cell-released high-mobility group box1 protein. Nat. Commun. 2014, 5, 5256. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, M. Role of platelets and platelet receptors in cancer metastasis. J. Hematol. Oncol. 2018, 11, 125. [Google Scholar] [CrossRef] [PubMed]
- Mammadova-Bach, E.; Zigrino, P.; Brucker, C.; Bourdon, C.; Freund, M.; De Arcangelis, A.; Abrams, S.I.; Orend, G.; Gachet, C.; Mangin, P.H. Platelet integrin α6β1 controls lung metastasis through direct binding to cancer cell-derived ADAM9. JCI Insight 2016, 1, e88245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dovizio, M.; Maier, T.J.; Alberti, S.; Di Francesco, L.; Marcantoni, E.; Münch, G.; John, C.M.; Suess, B.; Sgambato, A.; Steinhilber, D.; et al. Pharmacological inhibition of platelet-tumor cell cross-talk prevents platelet-induced overexpression of cyclooxygenase-2 in HT29 human colon carcinoma cells. Mol. Pharmacol. 2013, 84, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Connors, J.M. Prophylaxis against venous thromboembolism in ambulatory patients with cancer. N. Engl. J. Med. 2014, 370, 2515–2519. [Google Scholar] [CrossRef] [Green Version]
- Spek, C.A.; Versteeg, H.H.; Borensztajn, K.S. Anticoagulant therapy of cancer patients: Will patient selection increase overall survival? Thromb. Haemost. 2015, 114, 530–536. [Google Scholar]
- Kakkar, A.K.; Levine, M.N.; Kadziola, Z.; Lemoine, N.R.; Low, V.; Patel, H.K.; Rustin, G.; Thomas, M.; Quigley, M.; Williamson, R.C.N. Low molecular weight heparin, therapy with dalteparin, and survival in advanced cancer: The fragmin advanced malignancy outcome study (FAMOUS). J. Clin. Oncol 2004, 22, 1944–1948. [Google Scholar] [CrossRef]
- Klerk, C.P.W.; Smorenburg, S.M.; Otten, H.-M.; Lensing, A.W.A.; Prins, M.H.; Piovella, F.; Prandoni, P.; Bos, M.M.E.M.; Richel, D.J.; van Tienhoven, G.; et al. The effect of low molecular weight heparin on survival in patients with advanced malignancy. J. Clin. Oncol 2005, 23, 2130–2135. [Google Scholar] [CrossRef]
- Macbeth, F.; Noble, S.; Evans, J.; Ahmed, S.; Cohen, D.; Hood, K.; Knoyle, D.; Linnane, S.; Longo, M.; Moore, B.; et al. Randomized Phase III Trial of Standard Therapy Plus Low Molecular Weight Heparin in Patients With Lung Cancer: FRAGMATIC Trial. J. Clin. Oncol. 2016, 34, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Ek, L.; Gezelius, E.; Bergman, B.; Bendahl, P.O.; Anderson, H.; Sundberg, J.; Wallberg, M.; Falkmer, U.; Verma, S.; Belting, M.; et al. Randomized phase III trial of low-molecular-weight heparin enoxaparin in addition to standard treatment in small-cell lung cancer: The RASTEN trial. Ann. Oncol. 2018, 29, 398–404. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Singh, P.; Boyango, I.; Gutter-Kapon, L.; Elkin, M.; Sanderson, R.D.; Ilan, N. Heparanase: From basic research to therapeutic applications in cancer and inflammation. Drug Resist. Updat. 2016, 29, 54–75. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, M.; Roblek, M.; Ortmann, K.; Naggi, A.; Torri, G.; Borsig, L.; Bendas, G. The role of VLA-4 binding for experimental melanoma metastasis and its inhibition by heparin. Thromb. Res. 2014, 133, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Hostettler, N.; Naggi, A.; Torri, G.; Ishai-Michaeli, R.; Casu, B.; Vlodavsky, I.; Borsig, L. P-selectin- and heparanase-dependent antimetastatic activity of non-anticoagulant heparins. FASEB J. 2007, 21, 3562–3572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, R.M.; Cecconi, O.; Roberts, W.G.; Aruffo, A.; Linhardt, R.J.; Bevilacqua, M.P. Heparin oligosaccharides bind L- and P-selectin and inhibit acute inflammation. Blood 1993, 82, 3253–3258. [Google Scholar] [CrossRef] [Green Version]
- Stevenson, J.L.; Choi, S.H.; Varki, A. Differential metastasis inhibition by clinically relevant levels of heparins--correlation with selectin inhibition, not antithrombotic activity. Clin. Cancer Res. 2005, 11, 7003–7011. [Google Scholar] [CrossRef] [Green Version]
- Watz, H.; Bock, D.; Meyer, M.; Schierhorn, K.; Vollhardt, K.; Woischwill, C.; Pedersen, F.; Kirsten, A.; Beeh, K.-M.; Meyer-Sabellek, W.; et al. Inhaled pan-selectin antagonist Bimosiamose attenuates airway inflammation in COPD. Pulm. Pharmacol. Ther. 2013, 26, 265–270. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Elkin, M.; Abboud-Jarrous, G.; Levi-Adam, F.; Fuks, L.; Shafat, I.; Ilan, N. Heparanase: One molecule with multiple functions in cancer progression. Connect. Tissue Res. 2008, 49, 207–210. [Google Scholar] [CrossRef]
- Haemmerle, M.; Stone, R.L.; Menter, D.G.; Afshar-Kharghan, V.; Sood, A.K. The Platelet Lifeline to Cancer: Challenges and Opportunities. Cancer Cell 2018, 33, 965–983. [Google Scholar] [CrossRef]
- Li, N. Platelets in cancer metastasis: To help the “villain” to do evil. Int. J. Cancer 2016, 138, 2078–2087. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.S.; Noh, K.; Haemmerle, M.; Li, D.; Park, H.; Hu, Q.; Hisamatsu, T.; Mitamura, T.; Mak, S.L.C.; Kunapuli, S.; et al. Role of ADP receptors on platelets in the growth of ovarian cancer. Blood 2017, 130, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, Y.W.; Osanto, S.; Reitsma, P.H.; Versteeg, H.H. The relationship between tissue factor and cancer progression: Insights from bench and bedside. Blood 2012, 119, 924–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, S.; Sato, S.; Oh-hara, T.; Takami, M.; Koike, S.; Mishima, Y.; Hatake, K.; Fujita, N. Platelets promote tumor growth and metastasis via direct interaction between Aggrus/podoplanin and CLEC-2. PLoS ONE 2013, 8, e73609. [Google Scholar] [CrossRef]
- Dovizio, M.; Alberti, S.; Guillem-Llobat, P.; Patrignani, P. Role of platelets in inflammation and cancer: Novel therapeutic strategies. Basic Clin. Pharmacol. Toxicol. 2014, 114, 118–127. [Google Scholar] [CrossRef]
- Lian, L.; Li, W.; Li, Z.-Y.; Mao, Y.-X.; Zhang, Y.-T.; Zhao, Y.-M.; Chen, K.; Duan, W.-M.; Tao, M. Inhibition of MCF-7 breast cancer cell-induced platelet aggregation using a combination of antiplatelet drugs. Oncol. Lett. 2013, 5, 675–680. [Google Scholar] [CrossRef] [Green Version]
- Varki, N.M.; Varki, A. Heparin inhibition of selectin-mediated interactions during the hematogenous phase of carcinoma metastasis: Rationale for clinical studies in humans. Semin. Thromb. Hemost. 2002, 28, 53–66. [Google Scholar] [CrossRef] [Green Version]
- Borsig, L.; Wong, R.; Feramisco, J.; Nadeau, D.R.; Varki, N.M.; Varki, A. Heparin and cancer revisited: Mechanistic connections involving platelets, P-selectin, carcinoma mucins, and tumor metastasis. Proc. Natl. Acad. Sci. USA 2001, 98, 3352–3357. [Google Scholar] [CrossRef] [Green Version]
- Ornstein, D.L.; Zacharski, L.R. The use of heparin for treating human malignancies. Haemostasis 1999, 29 (Suppl. S1), 48–60. [Google Scholar] [CrossRef]
- Stevenson, J.L.; Varki, A.; Borsig, L. Heparin attenuates metastasis mainly due to inhibition of P- and L-selectin, but non-anticoagulant heparins can have additional effects. Thromb. Res 2007, 120 (Suppl. S2), S107–S111. [Google Scholar] [CrossRef]
- Pfankuchen, D.B.; Baltes, F.; Batool, T.; Li, J.-P.; Schlesinger, M.; Bendas, G. Heparin antagonizes cisplatin resistance of A2780 ovarian cancer cells by affecting the Wnt signaling pathway. Oncotarget 2017, 8, 67553–67566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raskob, G.E.; van Es, N.; Verhamme, P.; Carrier, M.; Di Nisio, M.; Garcia, D.; Grosso, M.A.; Kakkar, A.K.; Kovacs, M.J.; Mercuri, M.F.; et al. Edoxaban for the Treatment of Cancer-Associated Venous Thromboembolism. N. Engl. J. Med. 2018, 378, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Young, A.M.; Marshall, A.; Thirlwall, J.; Chapman, O.; Lokare, A.; Hill, C.; Hale, D.; Dunn, J.A.; Lyman, G.H.; Hutchinson, C.; et al. Comparison of an Oral Factor Xa Inhibitor With Low Molecular Weight Heparin in Patients With Cancer With Venous Thromboembolism: Results of a Randomized Trial (SELECT-D). J. Clin. Oncol. 2018, 36, 2017–2023. [Google Scholar] [CrossRef] [PubMed]
- Voigtlaender, M.; Langer, F. Low-Molecular-Weight Heparin in Cancer Patients: Overview and Indications. Hamostaseologie 2019, 39, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Kahale, L.A.; Hakoum, M.B.; Tsolakian, I.G.; Alturki, F.; Matar, C.F.; Terrenato, I.; Sperati, F.; Barba, M.; Yosuico, V.E.; Schünemann, H.; et al. Anticoagulation for the long-term treatment of venous thromboembolism in people with cancer. Cochrane Database Syst. Rev. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, M.; Frenette, P.S.; Saffaripour, S.; Johnson, R.C.; Hynes, R.O.; Wagner, D.D. Defects in hemostasis in P-selectin-deficient mice. Blood 1996, 87, 1238–1242. [Google Scholar] [CrossRef] [Green Version]
- Qi, C.-L.; Wei, B.; Ye, J.; Yang, Y.; Li, B.; Zhang, Q.-Q.; Li, J.-C.; He, X.-D.; Lan, T.; Wang, L.-J. P-selectin-mediated platelet adhesion promotes the metastasis of murine melanoma cells. PLoS ONE 2014, 9, e91320. [Google Scholar] [CrossRef]
- Mannori, G.; Crottet, P.; Cecconi, O.; Hanasaki, K.; Aruffo, A.; Nelson, R.M.; Varki, A.; Bevilacqua, M.P. Differential colon cancer cell adhesion to E-, P-, and L-selectin: Role of mucin-type glycoproteins. Cancer Res. 1995, 55, 4425–4431. [Google Scholar]
- Läubli, H.; Borsig, L. Selectins as mediators of lung metastasis. Cancer Microenviron 2010, 3, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Théorêt, J.-F.; Yacoub, D.; Hachem, A.; Gillis, M.-A.; Merhi, Y. P-selectin ligation induces platelet activation and enhances microaggregate and thrombus formation. Thromb. Res. 2011, 128, 243–250. [Google Scholar] [CrossRef]
- Merten, M.; Beythien, C.; Gutensohn, K.; Kühnl, P.; Meinertz, T.; Thiagarajan, P. Sulfatides Activate Platelets Through P-Selectin and Enhance Platelet and Platelet–Leukocyte Aggregation. Arter. Thromb. Vasc. Biol. 2005, 25, 258–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sathish, J.G.; Falati, S.; Croce, K.; Crump, C.; Furie, B.C.; Furie, B.; Poole, A.W. Antibody cross-linking of human platelet P-selectin induces calcium entry by a mechanism dependent upon Fcgamma receptor IIA. Thromb. Haemost. 2004, 92, 598–605. [Google Scholar] [PubMed]
- Fujimoto, T.; McEver, R.P. The cytoplasmic domain of P-selectin is phosphorylated on serine and threonine residues. Blood 1993, 82, 1758–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crovello, C.S.; Furie, B.C.; Furie, B. Histidine phosphorylation of P-selectin upon stimulation of human platelets: A novel pathway for activation-dependent signal transduction. Cell 1995, 82, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Nolo, R.; Herbrich, S.; Rao, A.; Zweidler-McKay, P.; Kannan, S.; Gopalakrishnan, V. Targeting P-selectin blocks neuroblastoma growth. Oncotarget 2017, 8, 86657–86670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flevaris, P.; Li, Z.; Zhang, G.; Zheng, Y.; Liu, J.; Du, X. Two distinct roles of mitogen-activated protein kinases in platelets and a novel Rac1-MAPK-dependent integrin outside-in retractile signaling pathway. Blood 2009, 113, 893–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Xi, X.; Du, X. A mitogen-activated protein kinase-dependent signaling pathway in the activation of platelet integrin alpha IIbbeta3. J. Biol. Chem. 2001, 276, 42226–42232. [Google Scholar] [CrossRef] [Green Version]
- Estevez, B.; Du, X. New Concepts and Mechanisms of Platelet Activation Signaling. Physiology (Bethesda) 2017, 32, 162–177. [Google Scholar] [CrossRef] [Green Version]
- Crockett-Torabi, E. Selectins and mechanisms of signal transduction. J. Leukoc. Biol. 1998, 63, 1–14. [Google Scholar] [CrossRef]
- Becker, K.A.; Beckmann, N.; Adams, C.; Hessler, G.; Kramer, M.; Gulbins, E.; Carpinteiro, A. Melanoma cell metastasis via P-selectin-mediated activation of acid sphingomyelinase in platelets. Clin. Exp. Metastasis 2017, 34, 25–35. [Google Scholar] [CrossRef]
- Matsuo, Y.; Amano, S.; Furuya, M.; Namiki, K.; Sakurai, K.; Nishiyama, M.; Sudo, T.; Tatsumi, K.; Kuriyama, T.; Kimura, S.; et al. Involvement of p38alpha mitogen-activated protein kinase in lung metastasis of tumor cells. J. Biol. Chem. 2006, 281, 36767–36775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esumi, N.; Todo, S.; Imashuku, S. Platelet aggregating activity mediated by thrombin generation in the NCG human neuroblastoma cell line. Cancer Res. 1987, 47, 2129–2135. [Google Scholar] [PubMed]
- Bradley, C.J.; Dauer, R.J.; Thurlow, P.J.; Connellan, J.M. Characterization of platelet aggregation induced by the human carcinosarcoma Colo 526: Role of platelet activation, tumor cell cytoskeleton and tumor cell plasma membrane. Pathology 1997, 29, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Wei, B.; Zhou, W.; Yang, Y.; Li, B.; Guo, S.; Li, J.; Ye, J.; Li, J.; Zhang, Q.; et al. P-selectin-mediated platelet adhesion promotes tumor growth. Oncotarget 2015, 6, 6584–6596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battinelli, E.M.; Markens, B.A.; Kulenthirarajan, R.A.; Machlus, K.R.; Flaumenhaft, R.; Italiano, J.E. Anticoagulation inhibits tumor cell-mediated release of platelet angiogenic proteins and diminishes platelet angiogenic response. Blood 2014, 123, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, C.G.; Michelson, A.D.; Flaumenhaft, R. Granule exocytosis is required for platelet spreading: Differential sorting of α-granules expressing VAMP-7. Blood 2012, 120, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Naggi, A.; Casu, B.; Perez, M.; Torri, G.; Cassinelli, G.; Penco, S.; Pisano, C.; Giannini, G.; Ishai-Michaeli, R.; Vlodavsky, I. Modulation of the heparanase-inhibiting activity of heparin through selective desulfation, graded N-acetylation, and glycol splitting. J. Biol. Chem. 2005, 280, 12103–12113. [Google Scholar] [CrossRef] [Green Version]
- Bisio, A.; Vecchietti, D.; Citterio, L.; Guerrini, M.; Raman, R.; Bertini, S.; Eisele, G.; Naggi, A.; Sasisekharan, R.; Torri, G. Structural features of low-molecular-weight heparins affecting their affinity to antithrombin. Thromb. Haemost. 2009, 102, 865–873. [Google Scholar] [CrossRef] [Green Version]
- Ross, T.; Jakubzig, B.; Grundmann, M.; Massing, U.; Kostenis, E.; Schlesinger, M.; Bendas, G. The molecular mechanism by which saturated lysophosphatidylcholine attenuates the metastatic capacity of melanoma cells. FEBS Open Bio. 2016, 6, 1297–1309. [Google Scholar] [CrossRef]
- Shafat, I.; Zcharia, E.; Nisman, B.; Nadir, Y.; Nakhoul, F.; Vlodavsky, I.; Ilan, N. An ELISA method for the detection and quantification of human heparanase. Biochem. Biophys. Res. Commun. 2006, 341, 958–963. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the different heparins are available from the authors. |

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schwarz, S.; Gockel, L.M.; Naggi, A.; Barash, U.; Gobec, M.; Bendas, G.; Schlesinger, M. Glycosaminoglycans as Tools to Decipher the Platelet Tumor Cell Interaction: A Focus on P-Selectin. Molecules 2020, 25, 1039. https://doi.org/10.3390/molecules25051039
Schwarz S, Gockel LM, Naggi A, Barash U, Gobec M, Bendas G, Schlesinger M. Glycosaminoglycans as Tools to Decipher the Platelet Tumor Cell Interaction: A Focus on P-Selectin. Molecules. 2020; 25(5):1039. https://doi.org/10.3390/molecules25051039
Chicago/Turabian StyleSchwarz, Svenja, Lukas Maria Gockel, Annamaria Naggi, Uri Barash, Martina Gobec, Gerd Bendas, and Martin Schlesinger. 2020. "Glycosaminoglycans as Tools to Decipher the Platelet Tumor Cell Interaction: A Focus on P-Selectin" Molecules 25, no. 5: 1039. https://doi.org/10.3390/molecules25051039