
Synthesis, Structure and In Vitro Anti-Trypanosomal Activity of Non-Toxic Arylpyrrole-Based Chalcone Derivatives

 , , , , and
, , , , and
Abstract
:
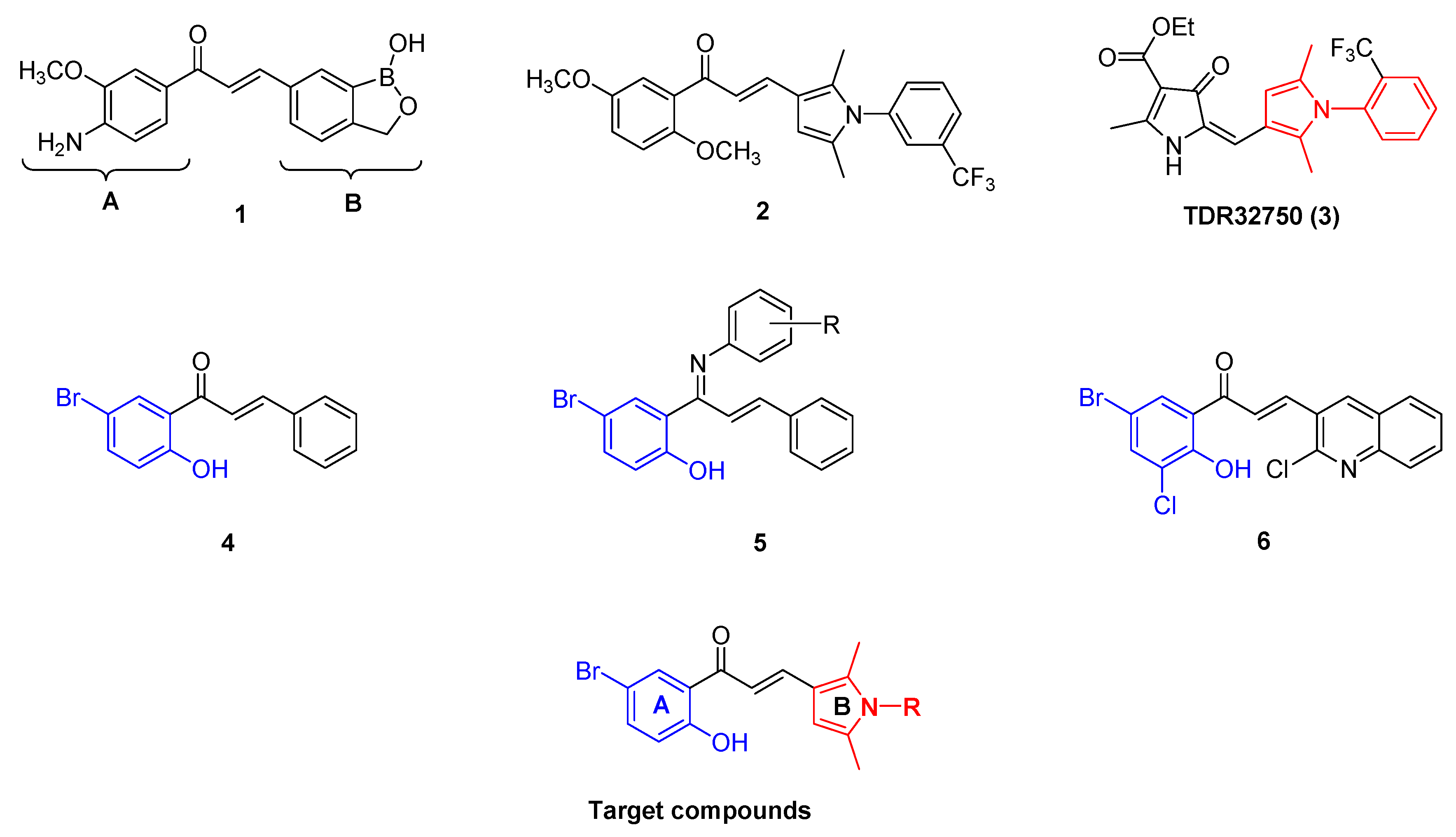
1. Introduction
2. Results and Discussion
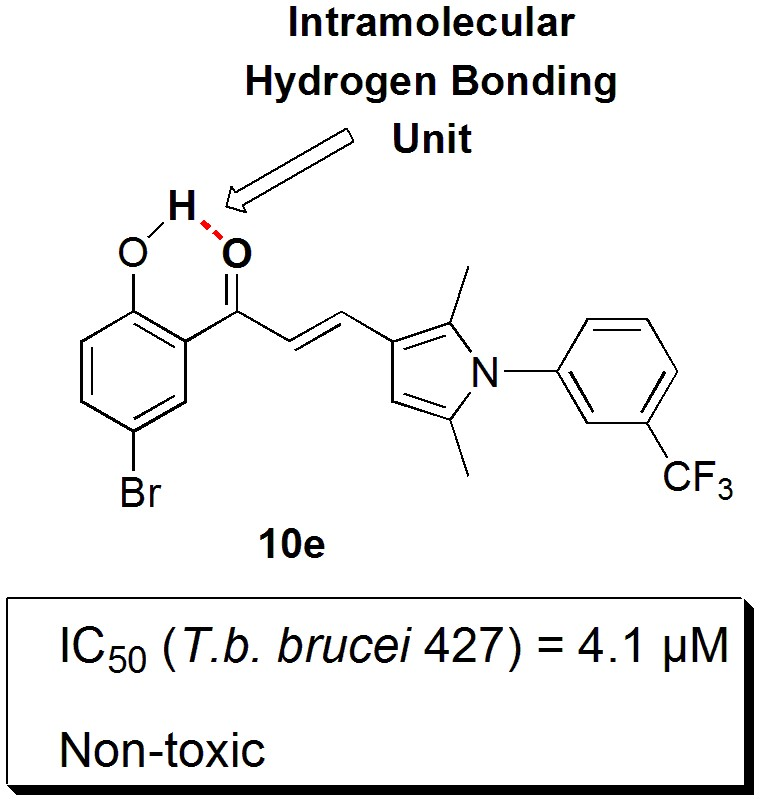
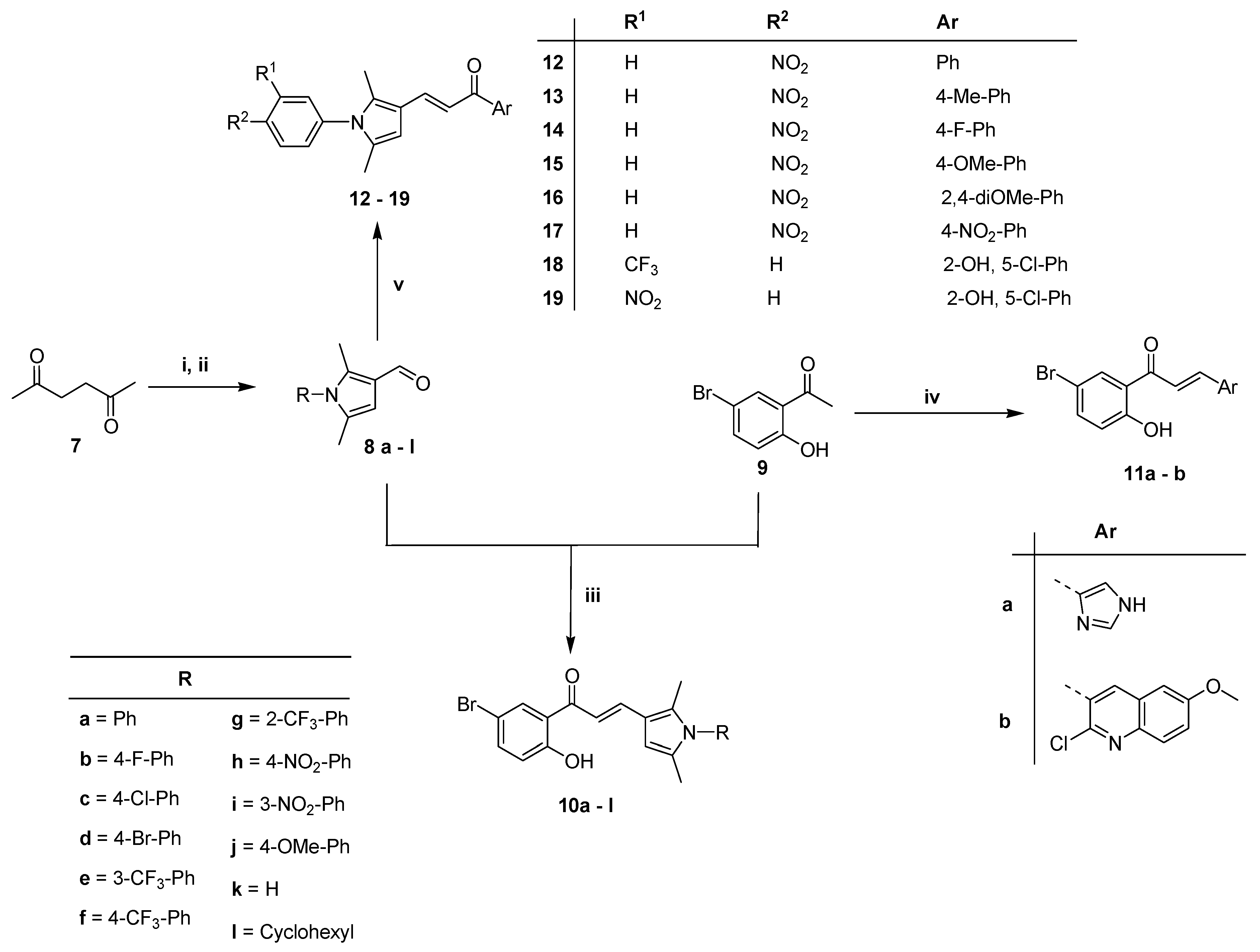
2.1. Chemical Synthesis
2.2. In Vitro Biological Assessment
3. Materials and Methods
3.1. General Information
3.2. General Procedure for the Synthesis of Arylpyrrole-Chalcone Hybrids
3.3. X-ray Crystallographic Analysis
3.4. In Vitro Antitrypanosomal Assay
3.5. In Vitro Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Deeks, E.D.; Lyseng-Williamson, K.A. Fexinidazole in human African trypanosomiasis: A profile of its use. Drugs Ther. Perspect. 2019, 35, 529–535. [Google Scholar] [CrossRef]
- Berninger, M.; Schunidt, I.; Ponte-Sucre, A.; Holzgrabe, U. Novel lead compounds in pre-clinical development against African sleeping sickness. Med. Chem. Commun. 2017, 8, 1872–1890. [Google Scholar] [CrossRef] [PubMed]
- Njogore, M.; Njuguna, N.M.; Mutai, P.; Ongarora, D.S.B.; Smith, P.W.; Chibale, K. Recent approaches to chemical discovery and development against malaria and the neglected tropical diseases human African trypanosomiasis and schistosomiasis. Chem. Rev. 2014, 114, 11138–11163. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Acosta, V.; Ruiz-Pérez, L.; Etxebarria, J.; Reichardt, N.; Navarro, M.; Igarashi, Y.; Liekens, S.; Balzarini, J.; González-Pacanowska, D. Open source drug discovery with the malaria box compound collection for neglected diseases and beyond. PLoS Pathog. 2016, 12, e1005851. [Google Scholar]
- Burrows, J.N.; Elliott, R.L.; Kaneko, T.; Mowbray, C.E.; Waterson, D. The role of modern drug discovery in the fight against neglected and tropical diseases. Med. Chem. Comm. 2014, 5, 688–700. [Google Scholar] [CrossRef]
- Kwofie, K.D.; Tung, N.H.; Suzuki-Ohashi, M.; Amoa-Bosompem, M.; Adegle, R.; Sakyiamah, M.M.; Ayertey, F.; Owusu, K.B.-A.; Tuffour, I.; Atchoglo, P. Antitrypanosomal activities and mechanisms of action of novel tetracyclic iridoids from Morinda lucida benth. Antimicrob. Agents Chemother. 2016, 60, 3283–3290. [Google Scholar] [CrossRef] [Green Version]
- WHO. Trypanosomiasis, Human African (Sleeping Sickness). Available online: http://www.who.int/mediacentre/factsheets/fs259/en (accessed on 1 December 2019).
- Klung, D.M.; Gelb, M.H.; Pollastri, M.P. Repurposing strategies for tropical disease drug discovery. Bioorg. Med. Chem. Lett. 2016, 26, 2569–2576. [Google Scholar] [CrossRef] [Green Version]
- CDC. Parasites African Trypanosomiasis (Also Known as Sleeping Sickness). Available online: https://www.cdc.gov/parasites/sleepingsickness/ (accessed on 1 December 2019).
- Franco, J.R.; Simarro, P.P.; Diarra, A.; Jannin, J.G. Epidemiology of human African trypanosomiasis. Clin. Epidemiol. 2014, 6, 257–275. [Google Scholar]
- Bouteille, B.; Buguet, A. The detection and treatment of human African trypanosomiasis. Res. Rep. Trop. Med. 2012, 3, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, M.; Bray, M.A.; Cal, M.; Trunz, B.; Torreele, E.; Brun, R. Anti-trypanosomal activity of fexinidazole, a new oral nitroimidazole drug candidate for treatment of sleeping sickness. Antimicrob. Agents Chemother. 2011, 55, 5602–5608. [Google Scholar] [CrossRef] [Green Version]
- Mishina, Y.V.; Krishna, S.; Haynes, R.K.; Meade, J.C. Artemisinins inhibit Trypanosoma cruzi and Trypanosoma brucei rhodisiense in vitro growth. Antimicrob. Agents Chemother. 2007, 51, 1852–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willyard, C. Putting sleeping sickness to bed. Nat. Med. 2011, 17, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Balasegana, M.; Young, H.; Chappuis, F.; Priotto, G.; Raguenaud, M.E.; Checchi, F. Effectiveness of melarsoprol and eflornithine as first-line regimens for gambiense sleeping sickness in nine Médecins Sans Frontières programmes. Trans. R. Soc. Trop. Med. Hyg. 2009, 103, 280–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, P.Y.; Wang, M.; Li, L.; Wu, H.; He, C.Y.; Yao, S.Q. Design, synthesis and biological evaluation of potent azadipeptide nitrile inhibitors and activity-based probes as promising anti-trypanosoma brucei agents. Chem. Eur. J. 2012, 18, 6528–6541. [Google Scholar] [CrossRef]
- Giordani, F.; Morrison, L.J.; Rowan, T.G.; De Koning, H.P.; Barrett, M.P. The animal trypanosomiases and their chemotherapy: A review. Parasitology 2016, 143, 1862–1889. [Google Scholar] [CrossRef]
- Babokhov, P.; Sanyaolu, A.O.; Oyibo, W.A.; Fagbeno-Beioku, A.F.; Iriemenam, N.C. A current analysis of chemotherapy strategies for the treatment of human African trypanosomiasis: A review. Pathog. Glob. Health 2013, 107, 242–252. [Google Scholar] [CrossRef] [Green Version]
- Priotto, G.; Kasparian, S.; Mutombo, W.; Ngovama, D.; Ghorashian, S.; Arnold, U.; Ghabri, S.; Baudin, E.; Buard, V.; Kazadi-Kyanza, S.; et al. Nifurtimox-eflornithine combination therapy for second-stage African Trypanosoma brucei gambiense trypanosomiasis: A multicentre, randomised, phase III, non-inferiority trial. Lancet 2000, 374, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Deeks, E.D. Fexinidazole: First global approval. Drugs 2019, 79, 215–220. [Google Scholar] [CrossRef]
- Veale, C.G.L.; Laming, D.; Swart, T.; Chibale, K.; Hoppe, H.C. Exploring the antiplasmodial 2-aminopyrridines as potential anti-trypanosomal agents. ChemMedChem 2019, 14, 2034–2041. [Google Scholar] [CrossRef]
- Veale, C.G.L.; Hoppe, C. Screening of the Pathogen Box reveals new starting points for anti-trypanosomal drug discovery. MedChemComm 2019, 9, 2037–2044. [Google Scholar] [CrossRef] [Green Version]
- Pedron, J.; Boudo, C.; Bourgeade-Delmas, S.; Sourin-Saquet, A.; Paloque, L.; Rastegari, M.; Abdoulaye, M.; El-Kashef, H.; Bonduelle, C.; Pratviel, G.; et al. Anti-trypanosomatid pharmacomodulation at position 3 of the 8-nitroquinolin-2(1H)-one scaffold using palladium-catalysed cross-coupling reactions. ChemMedChem 2018, 13, 2217–2228. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, D.K.; Bharti, S.K.; Asati, V. Chalcone scaffolds as anti-infective agents: Structural and molecular target perspectives. Eur. J. Med. Chem. 2015, 101, 496–524. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C. Chalcone: A privileged structure in medicinal chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef] [PubMed]
- Rohrmann, E.; Jones, R.G.; Shonle, H.A. The use of chalcones in the synthesis of medicinal intermediates. Am. Chem. Soc. 1944, 66, 1856–1857. [Google Scholar] [CrossRef]
- Bano, S.; Javed, K.; Ahmad, S.; Rathish, I.G.; Singh, S.; Chaitanya, M.; Arunasree, K.M. Synthesis of some novel chalcones, flavanones and flavones and evaluation of their anti-inflammatory activity. Eur. J. Med. Chem. 2013, 65, 51–59. [Google Scholar] [CrossRef]
- Anandam, R.; Jadav, S.S.; Ala, V.B.; Ahsan, M.J.; Bollikolla, H.B. Synthesis of new C-dimethylated chalcones as potent antitubercular agents. Med. Chem. Res. 2018, 27, 1690–1704. [Google Scholar] [CrossRef]
- Ventura, T.L.B.; Calixto, S.D.; Abrahim-Vieira, B.A.; de Souza, A.M.T.; Mello, M.V.P.; Rodrigues, C.R.; Miranda, L.S.M.; de Souza, R.O.C.; Leal, I.C.R.; Lasunskaia, E.B.; et al. Antimycobacterial and anti-inflammatory activities of substituted chalcones focusing on an anti-tuberculosis dual treatment approach. Molecules 2015, 20, 8072–8093. [Google Scholar] [CrossRef] [Green Version]
- Syahri, J.; Yuanita, E.; Nurohmah, B.A.; Armunanto, R.; Purwono, B. Chalcone analogue as potent anti-malarial compounds against Plasmodium falciparum: Synthesis, biological evaluation, and docking simulation study. Asian Pac. J. Trop. Biomed. 2017, 7, 675–679. [Google Scholar] [CrossRef]
- Sinha, S.; Batovska, D.I.; Medhi, B.; Radotra, B.D.; Bhalla, A.; Markova, N.; Sehgal, R. In vitro anti-malarial efficacy of chalcones: Cytotoxicity profile, mechanism of action and their effect on erythrocytes. Malar. J. 2019, 18, 421–431. [Google Scholar] [CrossRef]
- Wang, M.; Qin, H.L.; Leng, J.; Amjad, M.W.; Raja, M.A.G.; Hussain, M.A.; Bukhari, S.N.A. Synthesis and biological evaluation of new tetramethylpyrazine-based chalcone derivatives as potential anti-Alzheimer agents. Chem. Biol. Drug Des. 2018, 92, 1859–1866. [Google Scholar] [CrossRef]
- Karthikeyan, C.; Narayana Moorthy, N.S.H.; Ramasamy, S.; Vanam, U.; Manivannan, E.; Karunagaran, D.; Trivedi, P. Advances in chalcones with anticancer activities. Recent Pat. Anti-Cancer Drug Discov. 2015, 10, 97–115. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Z.; Wang, Q.; Zhang, F.; Bowling, T.; Nare, B.; Jacobs, R.T.; Zhang, J.; Ding, D.; Liu, Y.; Zhou, H. Chalcone-benzoxaborole hybrid molecules as potent antitrypanosomal agents. J. Med. Chem. 2012, 55, 3553–3557. [Google Scholar] [CrossRef] [PubMed]
- Osório, T.M.; Monache, F.D.; Chiaradia, L.D.; Mascarello, A.; Stumpf, T.R.; Zanetti, C.R.; Silveira, D.B.; Barardi, C.R.M.; de Fatima Albino Smânia, E.; Viancelli, A.; et al. Antibacterial activity of chalcones, hydrazones and oxadiazoles against methicillin-resistant Stapphylococcus aureus. Bioorg. Med. Chem. Lett. 2012, 22, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Murugesan, D.; Kaiser, M.; White, K.L.; Norval, S.; Riley, J.; Wyatt, P.G.; Charman, S.A.; Read, K.D.; Yeates, C.; Gilbert, I.H. Structure-activity relationship studies of pyrrolone antimalarial agents. ChemMedChem 2013, 8, 1537–1544. [Google Scholar] [CrossRef] [Green Version]
- Patil, S.; Utale, P.; Gholse, S.; Pande, S.; Thakur, S. Synthesis, characterization and antimicrobial activity of 2-hydroxy-5-bromo-4-methoxy-N-(substituted phenyl) chalconeimine. Int. J. Pharm. Res. Sch. 2013, 2, 129–135. [Google Scholar]
- Dave, S.S.; Ghatole, A.M.; Rahatgaonkar, A.M.; Chorghade, M.S.; Chauhan, P.; Srivastava, K. Experimental and computational evaluation of new quinolinyl chalcones as potent antiplasmodial agents. Indian J. Chem. Sect. B 2009, 48B, 1780–1793. [Google Scholar]
- Cardona, G.W.; Yepes, A.F.; Herrera, R.A. Hybrid molecules: Promising compounds for the development of new treatment against leishmaniasis and Chagas disease. Curr. Med. Chem. 2018, 25, 1–43. [Google Scholar] [CrossRef]
- Kerru, N.; Singh, P.; Koorbanally, N.; Raj, R.; Kumar, V. Recent advances (2015–2016) in anticancer hybrids. Eur. J. Med. Chem. 2017, 142, 179–212. [Google Scholar] [CrossRef]
- Mishra, S.; Singh, P. Hybrid molecules: The privileged scaffolds for various pharmaceuticals. Eur. J. Med. Chem. 2016, 124, 500–536. [Google Scholar]
- Darrell, O.T.; Hulushe, S.T.; Mtshare, T.E.; Beteck, R.M.; Isaacs, M.; Laming, D.; Hoppe, H.C.; Krause, R.W.M.; Khanye, S.D. Synthesis, antiplasmodial and antitrypanosomal evaluation of a series of novel 2-oxoquinoline-based thiosemicarbazone derivatives. S. Afr. J. Chem. 2018, 71, 174–181. [Google Scholar] [CrossRef]
- Gumbo, M.; Beteck, R.M.; Mandizvo, T.; Seldon, R.; Warner, D.F.; Hoppe, H.C.; Isaacs, M.; Laming, D.; Tam, C.C.; Cheng, L.W.; et al. Cinnamoyl-Oxaborole Amides: Synthesis and Their in Vitro Biological Activity. Molecules 2018, 23, 2038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beteck, R.M.; Isaacs, M.; Hoppe, H.C.; Khanye, S.D. Synthesis, in vitro cytotoxicity and trypanocidal evaluation of novel 1,3,6-substituted non-fluoroquinolones. S. Afr. J. Chem. 2018, 71, 188–195. [Google Scholar] [CrossRef]
- Amarnath, V.; Anthony, D.C.; Amarnath, K.; Valentine, W.M.; Wetterau, L.A.; Graham, D.G. Intermediates in the Paal-Knorr synthesis of pyrroles. J. Org. Chem. 1991, 56, 6924–6931. [Google Scholar] [CrossRef]
- OpenWetWare Contributors, Todd: Synthesis of Analogs of Arylpyrrole Antimalarial Drug Leads. Available online: http://www.openwetware.org/index.php?title=Todd:Synthesisof__Analogs_of_Arylpyrrole_Antimalarial_Drug_Leads&oldid=567645 (accessed on 20 February 2020).
- Petrov, V.; Stanimirov, S.; Petrov, I.K.; Fernandes, A.; de freitas, V.; Pina, F. Emptying the β-Cyclodextrin Cavity by Light: Photochemical Removal of the trans-Chalcone of 4′,7-Dihydroxyflavylium. J. Phys. Chem. A 2013, 117, 10692–10701. [Google Scholar] [CrossRef]
- Vazquez-Vuelvas, O.F.; Enriquez-Figueroa, R.A.; Garcia-Ortega, H.; Flores-Alamo, M.; Pineda-Contreras, A. Crystal structure of the chalcone(E)-3-(furan-2-yl)-1-phenylprop-2-en-1-one. Acta Cryst. 2015, E71, 161–164. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, B.; Mohr, P.; Stahl, M. Intramolecular hydrogen bonding in medicinal chemistry. J. Med. Chem. 2010, 53, 2601–2611. [Google Scholar] [CrossRef]
- Attram, H.D.; Wittlin, S.; Chibale, K. Incorporation of an intramolecular hydrogen bonding motif in the side chain of antimalarial benzimidazoles. Med. Chem. Commun. 2019, 10, 450–455. [Google Scholar] [CrossRef]
- Elemental Composition Calculator. Available online: https://webapp.scs.illlinois.edu/microanalysis/calc/ (accessed on 16 December 2019).
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Barbour, L.J. X-Seed–A software tool for supramolecular crystallography. J. Supram. Chem. 2001, 1, 189–191. [Google Scholar] [CrossRef]
- Hirymi, H.; Hirumi, K. Continous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 1989, 75, 985–989. [Google Scholar] [CrossRef] [Green Version]
- Oderinlo, O.O.; Tukulula, M.; Isaacs, M.; Hoppe, H.C.; Taylor, D.; Smith, V.J.; Khanye, S.D. New thiazolidine-2,4-dione derivatives combined with organometallic ferrocene: Synthesis, structure and antiparasitic activity. Appl. Organomet. Chem. 2018, 32, e4385. [Google Scholar] [CrossRef]
- Beteck, R.M.; Legoabe, L.J.; Isaacs, M.; Khanye, S.D.; Laming, D.; Hoppe, H.C. Anti-trypanosomal and antimalarial properties of tetralone derivatives and structurally related benzocycloalkanones. Medicina 2019, 55, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of the compounds are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp | T. b. brucei | Comp | T. b. brucei |
|---|---|---|---|
| IC50/µM | IC50/µM | ||
| 10a | 11.5 | 10l | 11.5 |
| 10b | 6.9 | 11a | na |
| 10c | 40.0 | 11b | 9.1 |
| 10d | 10.5 | 12 | na |
| 10e | 4.1 | 13 | 44.3 |
| 10f | 11.4 | 14 | 36.5 |
| 10g | 10.0 | 15 | 12.3 |
| 10h | 5.1 | 16 | 10.6 |
| 10i | 9.3 | 17 | na |
| 10j | na | 18 | 9.3 |
| 10k | na | 19 | 11.7 |
| PMD | 0.68 nM | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zulu, A.I.; Oderinlo, O.O.; Kruger, C.; Isaacs, M.; Hoppe, H.C.; Smith, V.J.; Veale, C.G.L.; Khanye, S.D. Synthesis, Structure and In Vitro Anti-Trypanosomal Activity of Non-Toxic Arylpyrrole-Based Chalcone Derivatives. Molecules 2020, 25, 1668. https://doi.org/10.3390/molecules25071668
Zulu AI, Oderinlo OO, Kruger C, Isaacs M, Hoppe HC, Smith VJ, Veale CGL, Khanye SD. Synthesis, Structure and In Vitro Anti-Trypanosomal Activity of Non-Toxic Arylpyrrole-Based Chalcone Derivatives. Molecules. 2020; 25(7):1668. https://doi.org/10.3390/molecules25071668
Chicago/Turabian StyleZulu, Ayanda I., Ogunyemi O. Oderinlo, Cuan Kruger, Michelle Isaacs, Heinrich C. Hoppe, Vincent J. Smith, Clinton G. L. Veale, and Setshaba D. Khanye. 2020. "Synthesis, Structure and In Vitro Anti-Trypanosomal Activity of Non-Toxic Arylpyrrole-Based Chalcone Derivatives" Molecules 25, no. 7: 1668. https://doi.org/10.3390/molecules25071668