Experimental and Computational Studies Unraveling the Peculiarity of Enolizable Oxoesters in the Organocatalyzed Mannich-Type Addition to Cyclic N-Acyl Iminium Ions

 , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Metal-Organocatalyzed Mannich Reactions with Quinoline N,O-Acetals
2.2. Metal-Organocatalyzed Mannich Reactions with Isoquinoline N,O-Acetals
2.3. Reductive Cyclization to Heteroaryl Lactones
2.4. Computational Data
3. Materials and Methods
3.1. Computational Section
3.1.1. General Procedure
3.1.2. General Procedure for Lactonization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Harris, S.A.; Wolf, D.E.; Mozingo, R.; Arth, G.E.; Anderson, R.C.; Easton, N.R.; Folkers, K.; Biotin, V. Synthesis of d,l-Biotin, d,l-Allobiotin and d,l-epiAllobiotin. J. Am. Chem. Soc. 1945, 67, 2096–2100. [Google Scholar] [CrossRef]
- Towada, R.; Kuwahara, S. Synthesis of Topsentolides A2 and C2, and non-Enzymatic Conversion of the Former to the Latter. Tetrahedron 2014, 70, 3774–3781. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Morita, M.; Ogawa, N.; Kondo, D.; Tojo, T. Asymmetric Synthesis of 12-Hydroxyheptadecatrienoic Acid and its 5,6-Dihydro- and 14,15-Dehydro-derivatives. Org. Biomol. Chem. 2016, 14, 10667–10673. [Google Scholar] [CrossRef] [PubMed]
- Mackai, E.G.; Nörret, M.; Wong, L.S.-M.; Louis, I.; Lawrence, A.L.; Willis, A.C.; Sherburn, M.S. A domino Diels-Alder Approach toward the Tetracyclic Nicandrenone Framework. Org. Lett. 2015, 17, 5517–5519. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhang, W.; Nam, S.; Horne, D.A.; Jove, R.; Carter, R.G. Amphidinolide B: Total Synthesis, Structural Investigation, and Biological Evaluation. J. Org. Chem. 2013, 78, 2213–2247. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Chen, J.; Xie, Y.; Zhang, H. Enantioselective Total Synthesis of (-)-Stenine. Angew. Chem. Int. Ed. 2012, 51, 1024–1027. [Google Scholar] [CrossRef]
- Hötling, S.; Haberlag, B.; Tamm, M.; Collatz, J.; Mack, P.; Steidle, J.L.M.; Vences, M.; Schulz, S. Identification and Synthesis of Macrolide Pheromones of the Grain Beetle Oryzaephilus Surinamensis and the Frog Spinomantis Aglavei. Chem. Eur. J. 2014, 20, 3183–3191. [Google Scholar] [CrossRef]
- Ciufolini, M.; Zhu, S. Practical Synthesis of (±)-Chlorovulone II. J. Org. Chem. 1998, 63, 1668–1675. [Google Scholar] [CrossRef]
- Egger, J.; Bretscher, P.; Freignag, S.; Kopf, M.; Carreira, E.M. Discovery of Highly Potent Anti-inflammatory Epoxyisoprostane-Derived Lactone. J. Am. Chem. Soc. 2014, 136, 17382–17385. [Google Scholar] [CrossRef]
- Jiménez, J.; Landa, A.; Lizarraga, A.; Maestro, M.; Mielgo, A.; Oiarbide, M.; Velilla, I.; Palomo, C. Enantio- and Diastereoselective Organocatalytic α-Alkylation of Aldehydes with 3-Substituted 2-(Bromomethyl)acrylates. J. Org. Chem. 2012, 77, 747–753. [Google Scholar] [CrossRef]
- Chi, Y.; Guo, L.; Kopf, N.A.; Gellman, S.H. Enantioselective Organocatalytic Michael Addition of Aldehydes to Nitroethylene: Efficient Access to γ2-Amino Acids. J. Am. Chem. Soc. 2008, 130, 5608–5609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesner, M.; Revell, J.D.; Tonazzi, S.; Wennemers, H. Peptide Catalyzed Asymmetric Conjugate Addition Reactions of Aldehydes to Nitroethylene-A Convenient Entry into γ2-Amino Acids. J. Am. Chem. Soc. 2008, 130, 5610–5611. [Google Scholar] [CrossRef] [PubMed]
- Kano, T.; Shirozu, F.; Akakura, M.; Maruoka, K. Powerful Amino Diol Catalyst for Effecting the Direct Asymmetric Conjugate Addition of Aldehydes to Acrylates. J. Am. Chem. Soc. 2012, 134, 16068–16073. [Google Scholar] [CrossRef] [PubMed]
- Sarkale, A.M.; Kumar, A.; Appayee, C. Organocatalytic Approach for Short Asymmetric Synthesis of (R)-Paraconyl Alchol:Application to the Total Synthesis of IM-2,SCB2, and A-factor γ-Butyrolactone Autoregulators. J. Org. Chem. 2018, 83, 4167–4172. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Korotkov, A.; Chapman, C.W.; Eastman, A.; Wu, J. Enantioselective Formal Syntheses of 11 Nuphar Alkaloids and Discovery of Potent Apoptotic Monomeric Analogues. Angew. Chem. Int. Ed. 2016, 55, 3509–3513. [Google Scholar] [CrossRef]
- Li, H.; Cooke, T.J.; Korotkov, A.; Chapman, C.W.; Eastman, A.; Wu, J. Stereoselective Synthesis and Biological Evaluation of C1-Epimeric and Desmethyl Monomeric Nuphar Analogues. J. Org. Chem. 2017, 82, 2648–2655. [Google Scholar] [CrossRef]
- Lindemann, C.; Schneider, C. Quinolizidine-Based Alkaloids: A General Catalytic, Highly Enantio- and Diastereoselective Synthetic Approach. Synthesis 2016, 48, 828–844. [Google Scholar] [CrossRef]
- Shimasaki, Y.; Koshino, S.; Hayashi, Y. Formal Synthesis of Ezetimibe Using a Proline-mediated,Asymmetric Three-component Mannich Reaction. Chem. Lett. 2016, 45, 30–32. [Google Scholar] [CrossRef]
- Sun, S.; Mao, Y.; Lou, H.; Liu, L. Copper (II)/Amine Synergistically Catalyzed Enantioselective Alkylation of Cyclic N-Acyl Hemiaminals with Aldehydes. Chem. Commun. 2015, 51, 10691–10694. [Google Scholar] [CrossRef]
- Berti, F.; Malossi, F.; Marchetti, F.; Pineschi, M. A Highly Enantioselective Mannich Reaction of Aldehydes with Cyclic N-Acyliminium Ions by Synergistic Catalysis. Chem. Commun. 2015, 51, 13694–13697. [Google Scholar] [CrossRef]
- Volla, C.M.R.; Fava, E.; Atodiresei, I.; Rueping, M. Dual Metal and Lewis Base Catalysis Approach for Asymmetric Synthesis of Dihydroquinolines and the α-Arylation of Aldehydes via N-Acyliminium Ions. Chem. Commun. 2015, 51, 15788–15791. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.-Y.; Cai, C.; Yang, X.; Lv, Z.-C.; Schneider, U. Catalytic Asymmetric Reactions with N.O-Aminals. ACS Catal. 2016, 6, 5747–5763. [Google Scholar] [CrossRef]
- Mengozzi, L.; Gualandi, A.; Cozzi, P.G. Organocatalytic Stereoselective Addition of Aldehydes to Acylquinolinium Ions. Eur. J. Org. Chem. 2016, 3200–3207. [Google Scholar] [CrossRef]
- Gualandi, A.; Mengozzi, L.; Manoni, A.E.; Cozzi, P.G. Stereoselective Organocatalytic Addition of Nucleophiles to Isoquinolinium and 3,5-Dihydroisoquinolinium Ions: A Simple Approach for the Synthesis of Isoquinoline Alkaloids. Cataly. Lett. 2015, 145, 398–419. [Google Scholar] [CrossRef]
- Mengozzi, L.; Gualandi, A.; Cozzi, P.G. A Highly Enantioselective Acyl-Mannich Reaction of Isoquinolines with Aldehydes Promoted by Proline Derivatives: An Approach to 13-Alkyl-tetrahydroprotoberberine Alkaloids. Chem. Sci. 2014, 5, 3915–3921. [Google Scholar] [CrossRef]
- Sun, S.; Li, C.; Floreancig, P.E.; Lou, H.; Liu, L. Highly Enantioselective Catalytic Cross-Dehydrogenative Coupling of N-Carbamoyl Tetrahydroisoquinolines and Terminal Alkynes. Org. Lett. 2015, 17, 1684–1687. [Google Scholar] [CrossRef]
- Berti, F.; Favero, L.; Pineschi, M. Asymmetric Synthesis of Methylphenidate and Quinolizidonones by Addition of Aldheydes to Piperidine-Based Conjugated Acyliminium Ions. Synthesis 2016, 48, 2645–2652. [Google Scholar]
- Vargiu, M.; Favero, L.; Menichetti, A.; Di Bussolo, V.; Marchetti, F.; Pescitelli, G.; Di Pietro, S.; Pineschi, M. Direct Enantioselective Vinylogous Mannich-type Reaction of Acyclic Enals: New Experimental Insights into E/Z-dilemma. Chirality 2019, 31, 522–533. [Google Scholar] [CrossRef]
- Dubs, C.; Hamashima, Y.; Sasamoto, N.; Seidel, T.M.; Suzuki, S.; Hashizume, D.; Sodeoka, M. Mechanistic Studies on the Catalytic Asymmetric Mannich-Type Reaction with Dihydroisoquinolines and Development of Oxidative Mannich-Type Reactions Starting from Tetrahydroisoquinolines. J. Org. Chem. 2008, 73, 5859–5871. [Google Scholar] [CrossRef]
- Palanichamy, K.; Kaliappan, K.P. Synthesis of Saturated Six-Membered Ring Lactones. In Topics in Heterocyclic Chemistry; Cossy, J., Ed.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 97–140. [Google Scholar]
- Zhu, R.; Buchwald, S.L. Versatile Enantioselective Synthesis of Functionalized Lactones via Copper-Catalyzed Radical Oxyfunctionalization of Alkenes. J. Am. Chem. Soc. 2015, 137, 8069–8077. [Google Scholar] [CrossRef] [Green Version]
- Bryans, J.S.; Wustrow, D.J. 3-Substituted GABA Analogs with Central Nervous System Activity: A Review. Med. Res. Rev. 1999, 19, 149–177. [Google Scholar] [CrossRef]
- Dworkin, R.H.; Kirkpatrick, P. Pregabalin. Nat. Rev. Drug Discov. 2005, 4, 455–456. [Google Scholar] [CrossRef]
- Barbosa, L.C.A.; Teixera, R.R.; Amarante, G.W. Synthetic Strategies for the Preparation of Butenolides and Their Transformation into Other Derivatives. Curr. Org. Synth. 2015, 12, 746–771. [Google Scholar] [CrossRef]
- Ogura, A.; Yamada, K.; Yokoshima, S.; Fukuyama, T. Total Synthesis of (-)-Anisatin. Org. Lett. 2012, 14, 1632–1635. [Google Scholar]
- Kim, S.-G. Organocatalytic Asymmetric 1,4-Addition of Organoboronic Acids to γ-Hydroxy α,β-Unsaturated Aldehyde: Facile Synthesis of Chiral β-Substituted γ-Lactones. Tetrahedron Lett. 2008, 49, 6148–6151. [Google Scholar] [CrossRef]
- Smith, A.J.; Abbott, L.K.; Martin, S.F. Enantioselective Conjugate Addition Employing 2-Heteroaryl Titanates and Zinc Reagents. Org. Lett. 2009, 11, 4200–4203. [Google Scholar] [CrossRef]
- Oliveira, C.C.; Angnes, R.A.; Correia, C.R.D. Intermolecular Enantioselective Heck-Matsuda Arylation of Acyclic Olefins: Application to the Synthesis of β-Aryl-γ-lactones and β-Aryl Aldehydes. J. Org. Chem. 2013, 78, 4373–4385. [Google Scholar] [CrossRef]
- Aycock, R.A.; Wang, H.; Jui, N.T. A Mild Catalytic System for Radical Conjugate Addition of Nitrogen Heterocycles. Chem. Sci. 2017, 8, 3121–3125. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian’16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. Design of Density Functionals That Are Broadly Accurate for Thermochemistry, Thermochemical Kinetics, and Nonbonded Interactions. J. Phys. Chem. A 2005, 109, 5656–5667. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. Software Update: The ORCA Program System, version 4.0. WIREs Comput. Mol. Sci 2018, 8, 1–6. [Google Scholar] [CrossRef]
- Grimme, S. Improved Second-order Moeller-Plesset Perturbation Theory by Separate Scaling of Parallel- and Antiparallel-spin Correlation Energies. Chem. Phys. 2003, 118, 9095–9102. [Google Scholar]
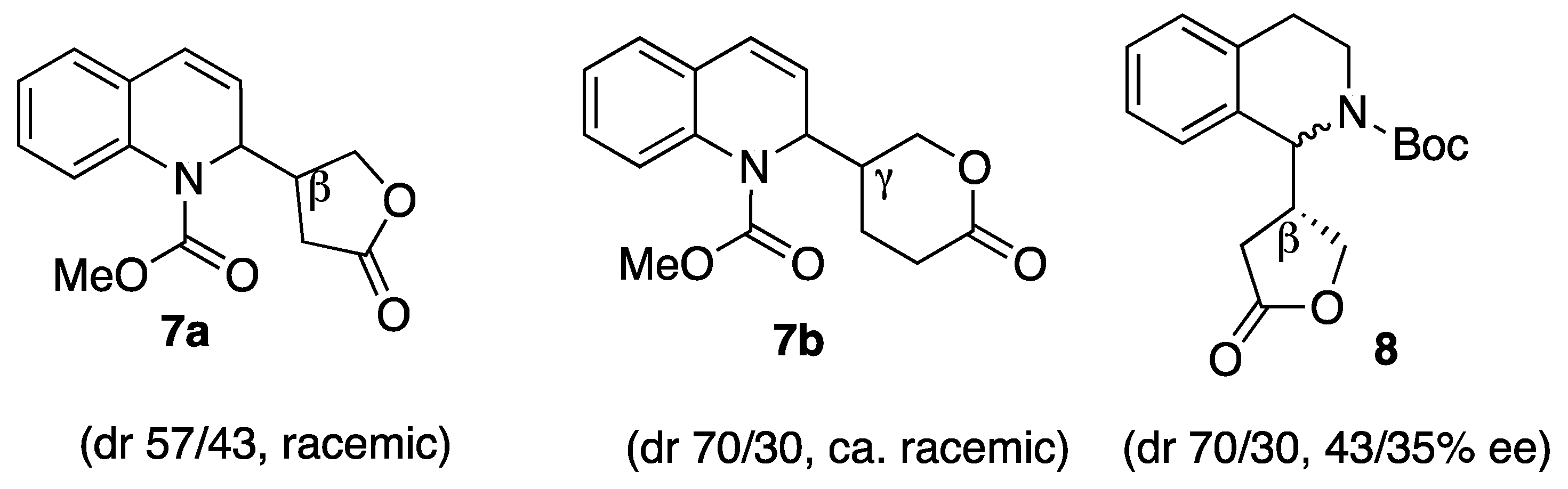
Sample Availability: Samples of compounds 7a,b and 8 are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
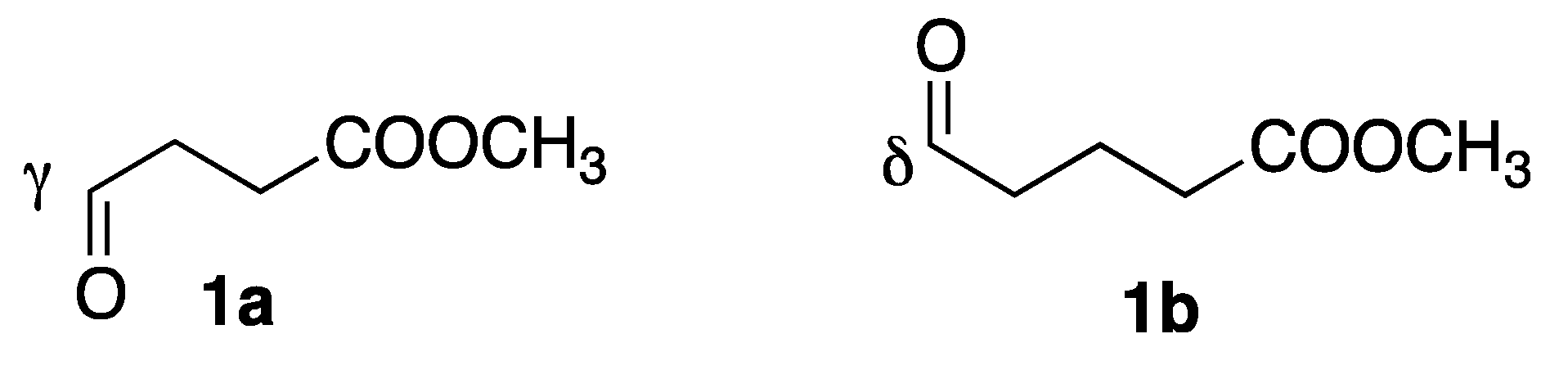
| Entry | Sub | L | Oxoester | T (h) | 1,2/1,4 b (3/4) | Dr b (Syn/Anti) | Ee (Syn/Anti) | Yield (%) c |
|---|---|---|---|---|---|---|---|---|
| 1 | 2a | L1 | 1b | 1 | 83/17 | 42/58 | 10/32 | 26(3ab) |
| 2 | 2a | L2 | 1b | 1 | 89/11 | 60/40 | 43/10 | 44(3ab) |
| 3 d | 2a | L2 | 1b | 4 | 89/11 | 60/40 | 58/32 | 60(3ab) |
| 4 d | 2a | L1 | 1b | 4 | 95/5 | 50/50 | 5/12 | 30(3ab) |
| 5 | 2a | L1 | 1a | 4 | 86/14 | 57/43 | 16/24 | 50(3aa) |
| 6 | 2a | L2 | 1a | 3 | 88/12 | 60/40 | 28/20 | 84(3aa) |
| 7 | 2b | L1 | 1b | 1 | 85/15 | 50/50 | nd | 14(3bb) |
| 8 | 2b | L2 | 1b | 1 | 89/11 | 70/30 | nd | 32(3bb) |
| 9 | 2b | L1 | 1a | 1 | 80/20 | 56/44 | nd | 25(3ba) |
| 10 | 2b | L2 | 1a | 1 | 83/17 | 68/32 | nd | 33(3ba) |
| 11 | 2c | L2 | 1a | 2 | 80/20 | 62/38 | nd | 66(3cb) f |
| Entry | Sub | L | Oxoester | T (h) | Dr b | ee | Yield (%) c (Product) |
|---|---|---|---|---|---|---|---|
| 1 | 5a | L1/L2 | 1a | 3 | Na | Low conversion | |
| 2 | 5b | L1 | 1b | 3 | 65/35 | 70/nd | 60 (6b) |
| 3 | 5b | L2 | 1b | 3 | 50/50 | 50/nd | 32 (6b) |
| 4 | 5b | L1 | 1a | 2 | 68/32 | 70/2 | 52 (6a) |
| 5 | 5b | L2 | 1a | 15 | 45/55 | 68/30 | 34 (6a) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menichetti, A.; Di Pietro, S.; Di Bussolo, V.; Favero, L.; Pineschi, M. Experimental and Computational Studies Unraveling the Peculiarity of Enolizable Oxoesters in the Organocatalyzed Mannich-Type Addition to Cyclic N-Acyl Iminium Ions. Molecules 2020, 25, 1903. https://doi.org/10.3390/molecules25081903
Menichetti A, Di Pietro S, Di Bussolo V, Favero L, Pineschi M. Experimental and Computational Studies Unraveling the Peculiarity of Enolizable Oxoesters in the Organocatalyzed Mannich-Type Addition to Cyclic N-Acyl Iminium Ions. Molecules. 2020; 25(8):1903. https://doi.org/10.3390/molecules25081903
Chicago/Turabian StyleMenichetti, Andrea, Sebastiano Di Pietro, Valeria Di Bussolo, Lucilla Favero, and Mauro Pineschi. 2020. "Experimental and Computational Studies Unraveling the Peculiarity of Enolizable Oxoesters in the Organocatalyzed Mannich-Type Addition to Cyclic N-Acyl Iminium Ions" Molecules 25, no. 8: 1903. https://doi.org/10.3390/molecules25081903