
Parallel Interconnected Kinetic Asymmetric Transformation (PIKAT) with an Immobilized ω-Transaminase in Neat Organic Solvent


Abstract
:
1. Introduction
2. Results
2.1. Biocatalyst Preparation
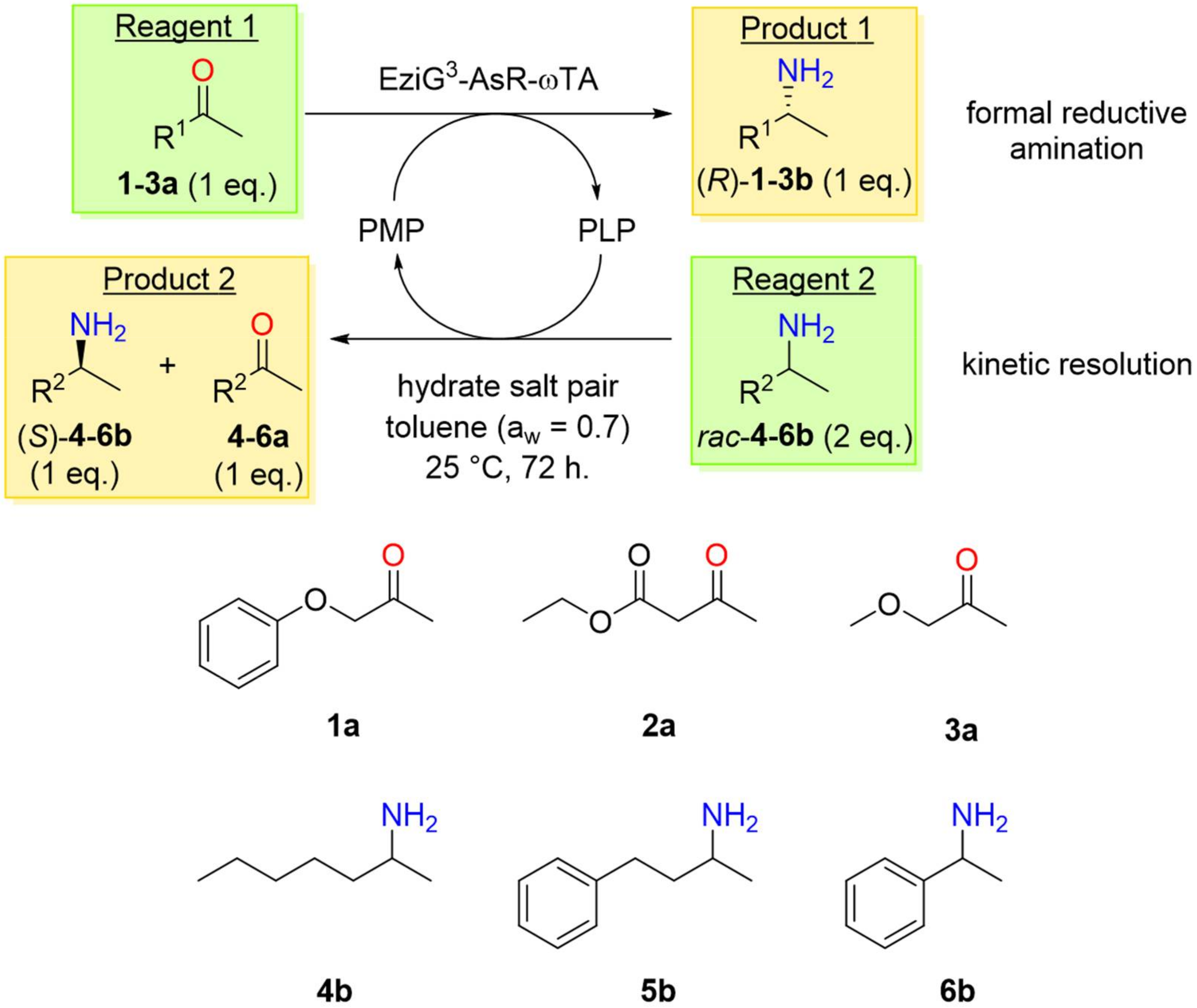
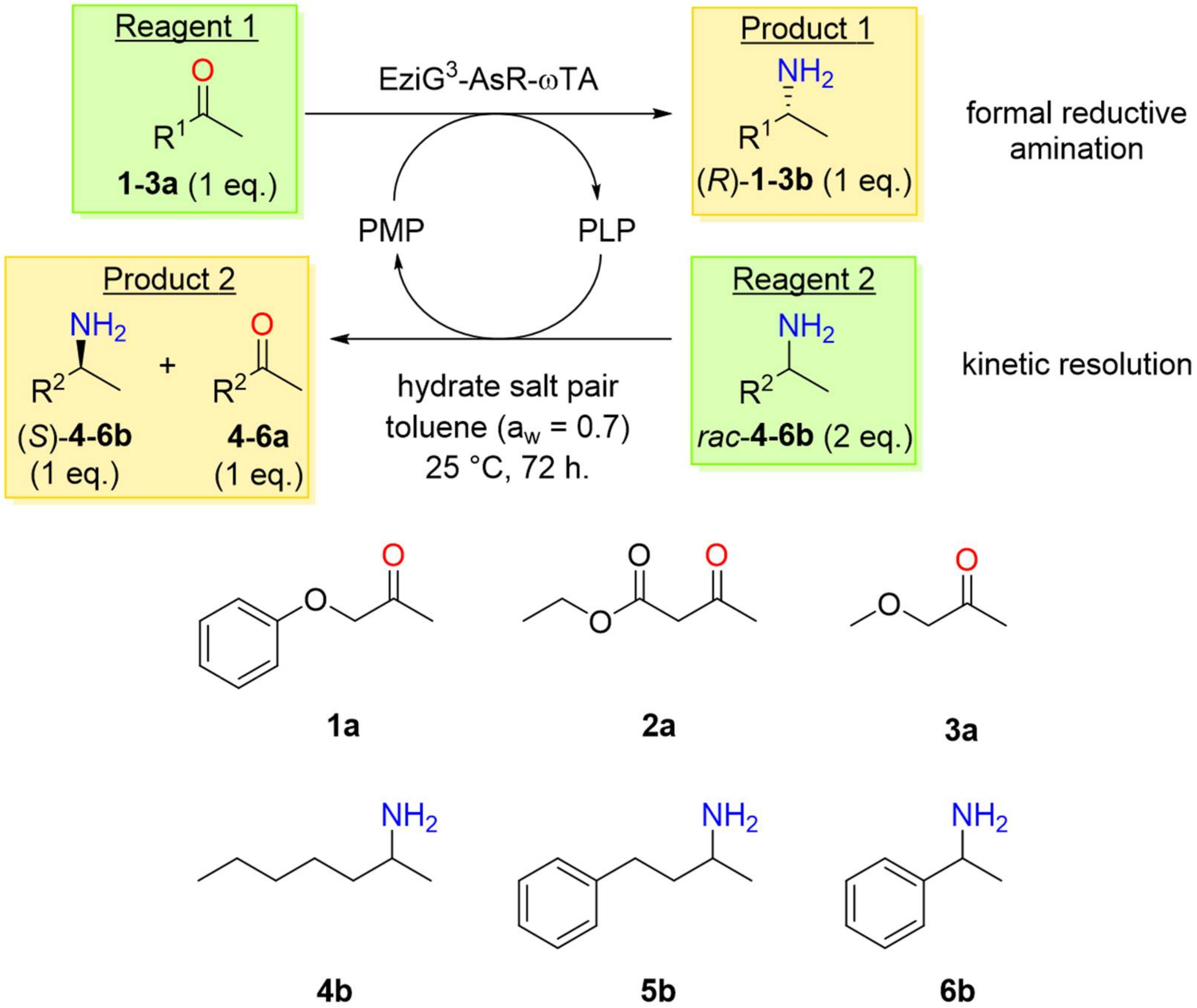
2.2. PIKAT Substrate Selection
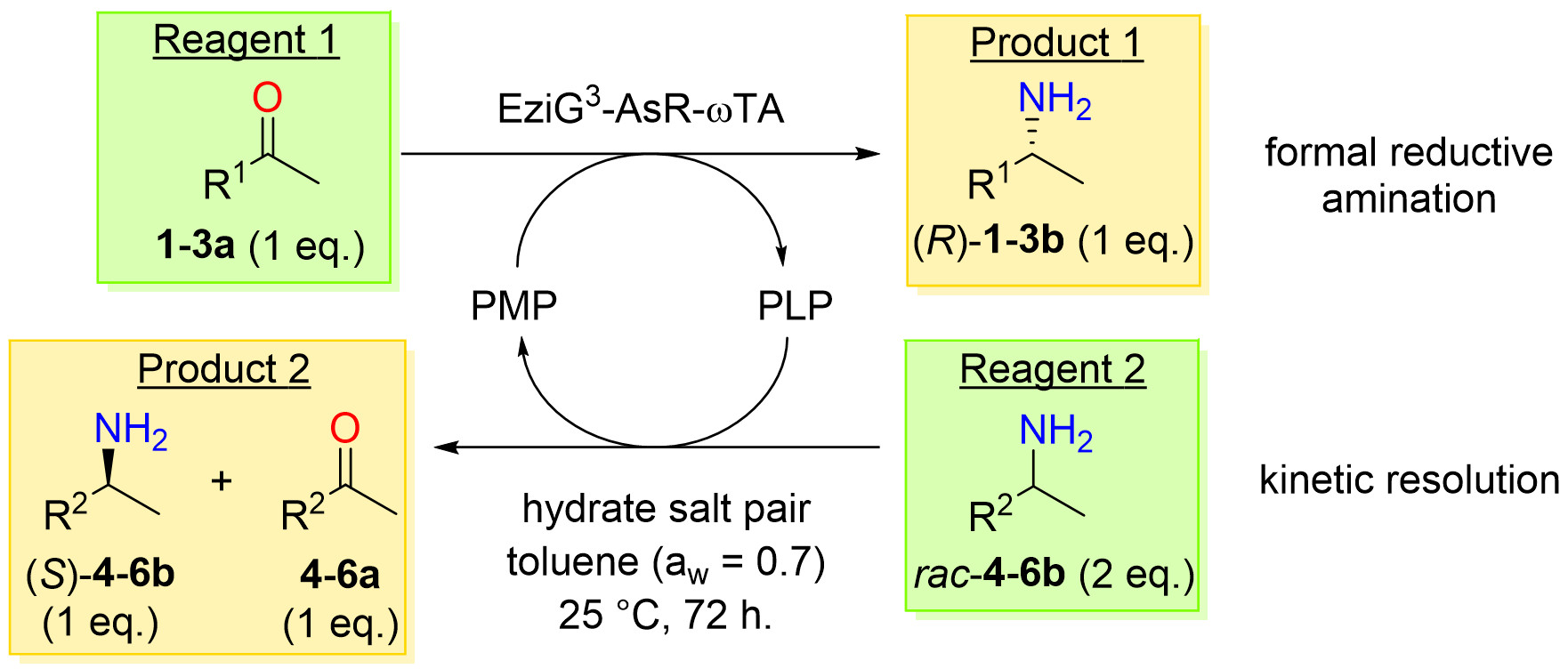
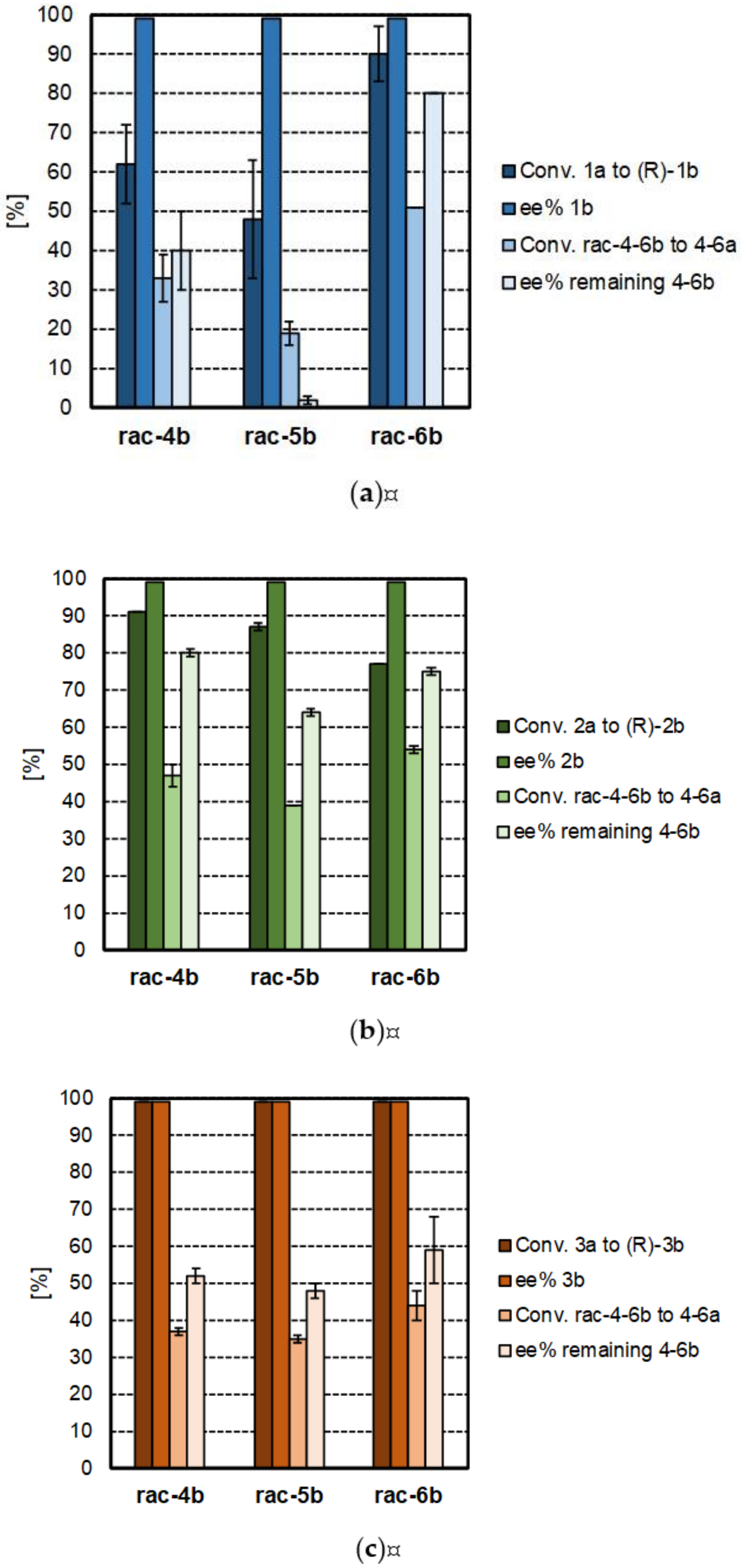
2.3. PIKAT Screening of Prochiral Ketones and Racemic Amines
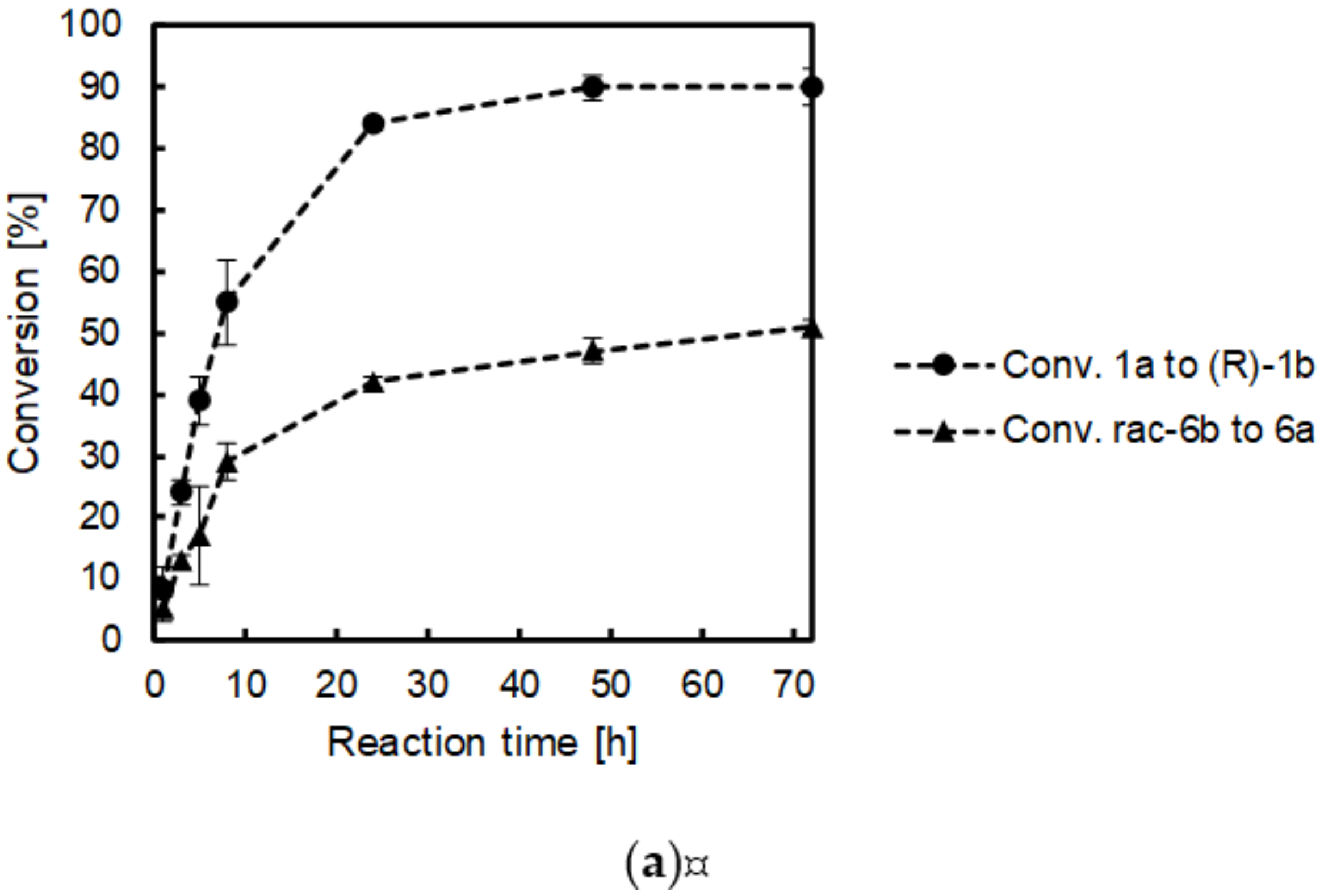
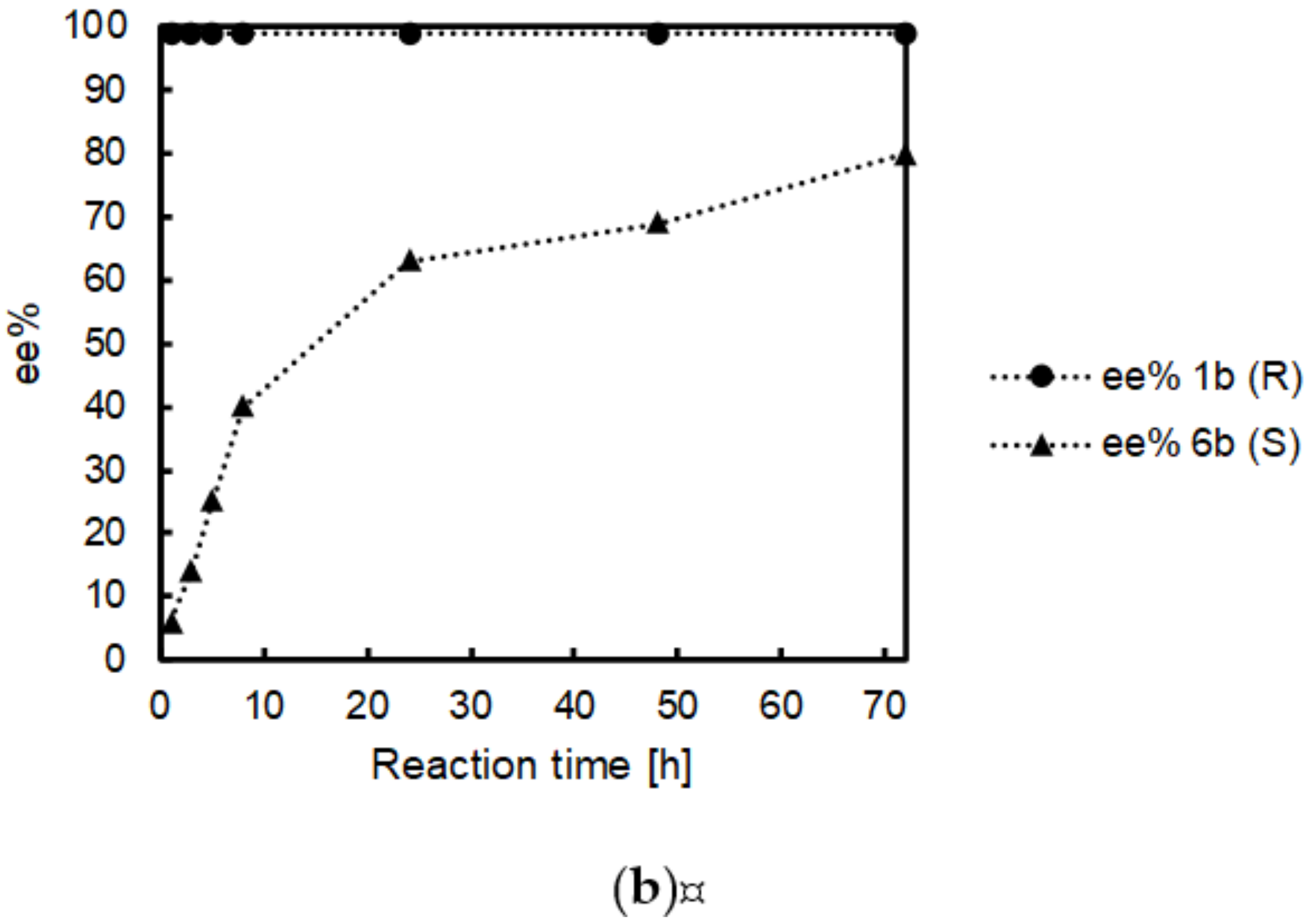
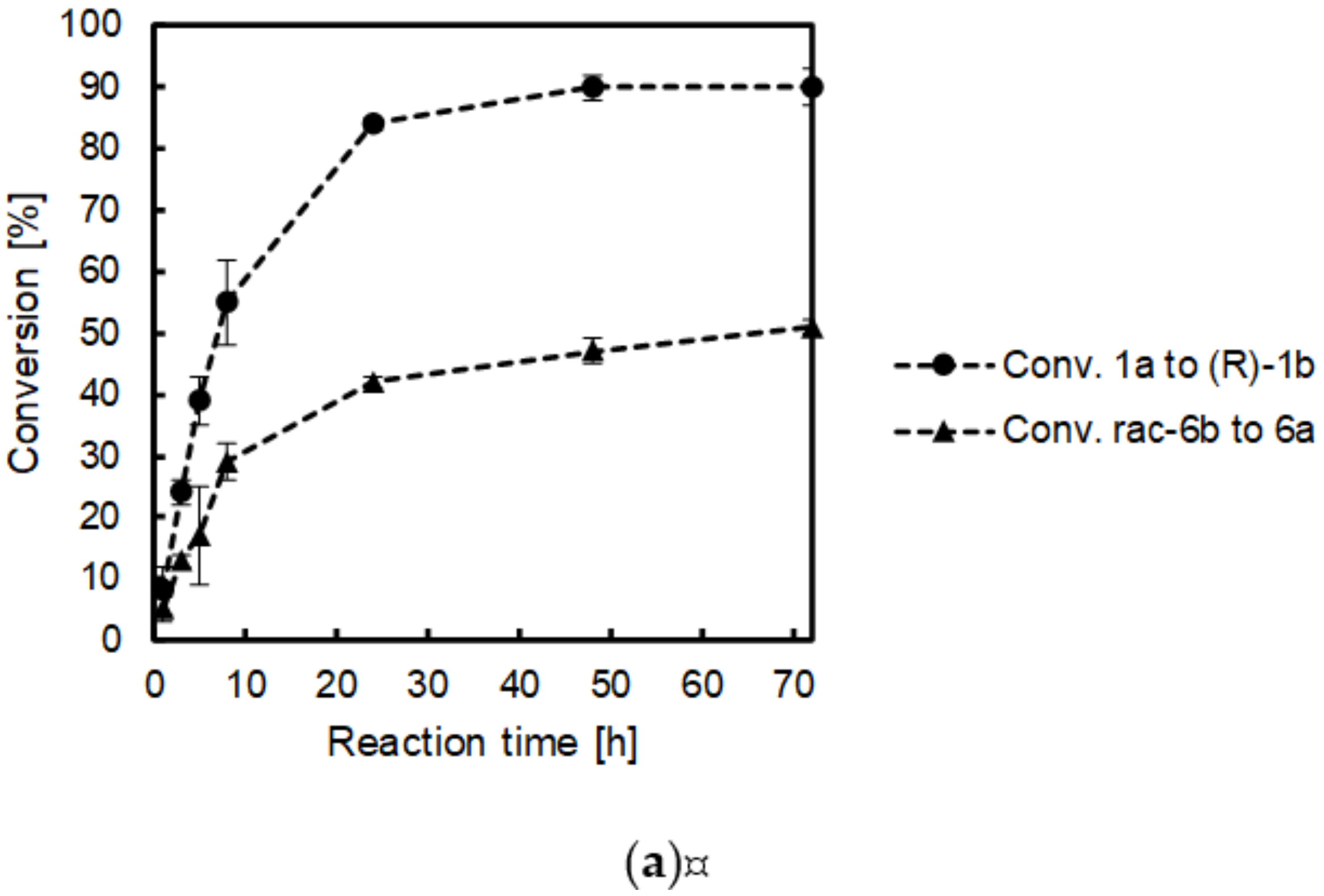
2.4. Time Studies
2.5. Further Studies
3. Discussion
4. Materials and Methods
4.1. Chemicals and Carrier Materials
4.2. Expression and Purification of ωTAs
4.3. Bradford Assay
4.4. Immobilization of AsR-ωTAs on EziG3 Carrier Materials
4.5. PIKAT in Organic Solvents with Immobilized ωTAs
4.6. Analytical Equipment and Determination
- DB1701-30 m-method-A: constant pressure 6.9 psi, T injector 250 °C, split ratio 50:1, T initial 80 °C, hold 6.5 min; gradient 10 °C/min up to 160 °C, hold 5 min; gradient 20 °C/min up to 200 °C, hold 2 min; gradient 20 °C/min up to 280 °C, hold 1 min.
- DB1701-30 m-method-B: constant pressure 6.9 psi, T injector 250 °C, split ratio 50:1, T initial 60 °C, hold 6.5 min; gradient 20 °C/min up to 100 °C, hold 1 min; gradient 20 °C/min up to 280 °C, hold 1 min.
- HP-5-method-A: constant pressure 4 psi, T injector 250 °C, split ratio 30:1, T initial 60 °C; gradient 5 °C/min up to 150 °C, hold 1 min; gradient 10 °C/min up to 250 °C, hold 1 min.
- HP-5-method-B: constant pressure 4 psi, T injector 250 °C, split ratio 30:1, T initial 40 °C, hold 2 min; gradient 5 °C/min up to 80 °C, hold 2 min; gradient 20 °C/min up to 250 °C.
- CP-DEX-method-A: constant flow 1.4 mL/min, T injector 200 °C, split ratio 40:1, T initial 100 °C, hold 2 min; gradient 1 °C/min up to 130 °C, hold 5 min; gradient 10 °C/min up to 170 °C, hold 10 min.; gradient 10 °C/min up to 180 °C, hold 1 min.
- CP-DEX-method-B: constant flow 1.4 mL/min, T injector 200 °C, split ratio 40:1, T initial 100 °C, hold 2 min; gradient 1 °C/min up to 118 °C, hold 5 min; gradient 10 °C/min up to 170 °C, hold 10 min.; gradient 10 °C/min up to 180 °C, hold 1 min.
- CP-DEX-method-C: constant flow 1.5 mL/min, T injector 200 °C, split ratio 20:1, T initial 60 °C, hold 2 min; gradient 5 °C/min up to 100 °C, hold 2 min; gradient 10 °C/min up to 180 °C, hold 1 min.
- HydroDex-β-TBDAc-method: constant flow 1 mL/min, T injector 220 °C, split ratio 20:1, T initial 100 °C, hold 2 min; gradient 1 °C/min up to 150 °C, hold 8 min; gradient 10 °C/min up to 170 °C, hold 1 min.
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formal Reductive Amination Step | Kinetic Resolution Step | |||||
|---|---|---|---|---|---|---|
| Entry | Ketone Substrate | Amine Donor | Conversion 1–3a to 1–3b [%] | ee 1–3b [%] | Conversion 4–6b to 4–6a [%] | ee 4–6b [%] |
| 1 | 1a | 4b | 62 ± 10 | >99 (R) | 33 ± 6 | 40 ± 10 (S) |
| 2 | 5b | 48 ± 15 | >99 (R) | 19 ± 3 | 2 ± 1 (S) | |
| 3 | 6b | 90 ± 7 | >99 (R) | 51 ± 1 | 80 ± 0 (S) | |
| 4 | 2a | 4b | 91 ± 0 | >99 (R) | 47 ± 3 | 80 ± 1 (S) |
| 5 | 5b | 87 ± 1 | >99 (R) | 39 ± 0 | 64 ± 1 (S) | |
| 6 | 6b | 77 ± 0 | >99 (R) | 54 ± 1 | 75 ± 1 (S) | |
| 7 | 3a | 4b | >99 | >99 (R) | 37 ± 1 | 52 ± 2 (S) |
| 8 | 5b | >99 | >99 (R) | 35 ± 1 | 48 ± 2 (S) | |
| 9 | 6b | >99 | >99(R) | 44 ± 4 | 59 ± 9 (S) | |
| Formal Reductive Amination Step | Kinetic Resolution Step | ||||
|---|---|---|---|---|---|
| Entry | Reaction Time [h] | Conv. 1a to 1b [%] | ee 1b [%] | Conv. 6b to 6a [%] | ee * 6b [%] |
| 1 | 1 | 8 ± 4 | >99 (R) | 5 ± 2 | 6 (S) |
| 2 | 2 | 15 ± 8 | >99 (R) | 9 ± 4 | 9 (S) |
| 3 | 3 | 24 ± 2 | >99 (R) | 13 ± 1 | 14 (S) |
| 4 | 5 | 39 ± 4 | >99 (R) | 17 ± 8 | 25 (S) |
| 5 | 8 | 55 ± 7 | >99 (R) | 29 ± 3 | 40 (S) |
| 6 | 24 | 84 ± 1 | >99 (R) | 42 ± 1 | 63 (S) |
| 7 | 48 | 90 ± 3 | >99 (R) | 47 ± 2 | 69 (S) |
| 8 | 72 | 90 ± 7 | >99 (R) | 51 ± 1 | 80 (S) |
| Formal Reductive Amination Step | Kinetic Resolution Step | |||||
|---|---|---|---|---|---|---|
| Entry | Ketone 1a Conc. [mM] | Conv. 1a to 1b [%] | Productivity 1a to 1b [mM] | ee 1b [%] | Conv. 6b to 6a [%] | ee 6b [%] |
| 1 | 50 | 79 ± 1 | 40 | >99 (R) | 49 ± 1 | 77 ± 2 (S) |
| 2 | 60 | 66 ± 3 | 40 | >99 (R) | 49 ± 3 | 78 ± 2 (S) |
| 3 | 70 | 51 ± 2 | 36 | >99 (R) | 44 ± 2 | 63 ± 2 (S) |
| 4 | 80 | 49 ± 4 | 39 | >99 (R) | 47 ± 4 | 70 ± 10 (S) |
| 5 | 90 | 34 ± 7 | 31 | >99 (R) | 38 ± 7 | 48 ± 15 (S) |
| 6 | 100 | 20 ± 6 | 20 | >99 (R) | 28 ± 6 | 27 ± 11 (S) |
| Compound | Retention Time | GC Method |
|---|---|---|
| 1a | 13.6 | HP-5-method-A |
| 1b | 13.7 | HP-5-method-A |
| (R)-1b | 43.7 | CP-DEX-method-A |
| (S)-1b | 43.3 | CP-DEX-method-A |
| 2a | 6.5 | HP-5-method-A |
| 2b | 6.9 | HP-5-method-A |
| (R)-2b | 14.6 | HydroDex-β-TBDAc-method |
| (S)-2b | 14.4 | HydroDex-β-TBDAc-method |
| 3a | 2.8 | DB1701-30m-method-B |
| 3b | 2.7 | DB1701-30m-method-B |
| (R)-3b | 15.1 | CP-DEX-method-C |
| (S)-3b | 14.4 | CP-DEX-method-C |
| 4a | 3.7 | DB1701-30m-method-A |
| 4b | 4.5 | DB1701-30m-method-A |
| (R)-4b | 13.3 | CP-DEX-method-A |
| (S)-4b | 12.6 | CP-DEX-method-A |
| 5a | 14.2 | DB1701-30m-method-A |
| 5b | 13.5 | DB1701-30m-method-A |
| (R)-5b | 41.0 | CP-DEX-method-A |
| (S)-5b | 40.9 | CP-DEX-method-A |
| 6a | 10.3 | DB1701-30m-method-A |
| 6b | 9.0 | DB1701-30m-method-A |
| (R)-6b | 27.9 | CP-DEX-method-A |
| (S)-6b | 27.5 | CP-DEX-method-A |
| naphthalene (internal standard) | 12.1 | DB1701-30m-method-A |
| 12.8 | DB1701-30m-method-B | |
| 12.4 | HP-5-method-A |
References
- Guo, F.; Berglund, P. Transaminase biocatalysis: Optimization and application. Green Chem. 2017, 19, 333–360. [Google Scholar] [CrossRef] [Green Version]
- Slabu, I.; Galman, J.L.; Lloyd, R.C.; Turner, N.J. Discovery, engineering, and synthetic application of transaminase biocatalysts. ACS Catal. 2017, 7, 8263–8284. [Google Scholar] [CrossRef]
- Kelly, S.A.; Pohle, S.; Wharry, S.; Mix, S.; Allen, C.C.R.; Moody, T.S.; Gilmore, B.F. Application of ω-transaminases in the pharmaceutical industry. Chem. Rev. 2018, 118, 349–367. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.D.; Grogan, G.; Bommarius, A.; Yun, H. Recent advances in ω-transaminase-mediated biocatalysis for the enantioselective synthesis of chiral amines. Catalysts 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Gomm, A.; O’Reilly, E. Transaminases for chiral amine synthesis. Curr. Opin. Chem. Biol. 2018, 43, 106–112. [Google Scholar] [CrossRef]
- Fuchs, M.; Farnberger, J.E.; Kroutil, W. The industrial age of biocatalytic transamination. Eur. J. Org. Chem. 2015, 32, 6965–6982. [Google Scholar] [CrossRef] [Green Version]
- Mathew, S.; Yun, H. ω-Transaminases for the production of optically pure amines and unnatural amino acids. ACS Catal. 2012, 2, 993–1001. [Google Scholar] [CrossRef]
- Koszelewski, D.; Tauber, K.; Faber, K.; Kroutil, W. ω-Transaminases for the synthesis of non-racemic alpha-chiral primary amines. Trends Biotechnol. 2010, 28, 324–332. [Google Scholar] [CrossRef]
- Savile, C.K.; Janey, J.M.; Mundorff, E.C.; Moore, J.C.; Tam, S.; Jarvis, W.R.; Colbeck, J.C.; Krebber, A.; Fleitz, F.J.; Brands, J.; et al. Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture. Science 2010, 329, 305–309. [Google Scholar] [CrossRef] [Green Version]
- Pavlidis, I.V.; Weiß, M.S.; Genz, M.; Spurr, P.; Hanlon, S.P.; Wirz, B.; Iding, H.; Bornscheuer, U.T. Identification of (S)-selective transaminases for the asymmetric synthesis of bulky chiral amines. Nat. Chem. 2016, 8, 1076–1082. [Google Scholar] [CrossRef]
- Wang, B.; Land, H.; Berglund, P. An efficient single-enzymatic cascade for asymmetric synthesis of chiral amines catalyzed by ω-transaminase. Chem. Commun. 2013, 49, 161–163. [Google Scholar] [CrossRef] [PubMed]
- Green, A.P.; Turner, N.J.; O’Reilly, E. Chiral amine synthesis using omega-transaminases: An amine donor that displaces equilibria and enables high-throughput screening. Angew. Chem. Int. Ed. 2014, 53, 10714–10717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Montero, L.; Gotor, V.; Gotor-Fernández, V.; Lavandera, I. But-2-ene-1,4-diamine and But-2-ene-1,4-diol as donors for thermodynamically favored transaminase- and alcohol dehydrogenase-catalyzed processes. Adv. Synth. Catal. 2016, 358, 1618–1624. [Google Scholar] [CrossRef] [Green Version]
- Gomm, A.; Lewis, W.; Green, A.P.; O’Reilly, E. A New generation of smart amine donors for transaminase-mediated biotransformations. Chem. Eur. J. 2016, 22, 12692–12695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payer, S.E.; Schrittwieser, J.H.; Kroutil, W. Vicinal diamines as smart cosubstrates in the transaminase-catalyzed asymmetric amination of ketones. Eur. J. Org. Chem. 2017, 2017, 2553–2559. [Google Scholar] [CrossRef]
- Mutti, F.G.; Sattler, J.; Tauber, K.; Kroutil, W. Creating a biocatalyst for the production of an optically pure sterically hindered amine. Chem. Cat. Chem. 2011, 3, 109–111. [Google Scholar] [CrossRef]
- Contente, M.L.; Paradisi, F. Self-sustaining closed-loop multienzyme-mediated conversion of amines into alcohols in continuous reactions. Nat. Catal. 2018, 1, 452–459. [Google Scholar] [CrossRef]
- Bajić, M.; Plazl, I.; Stloukal, R.; Žnidaršič-Plazl, P. Development of a miniaturized packed bed reactor with ω-transaminase immobilized in LentiKats®. Process Biochem. 2017, 52, 63–72. [Google Scholar] [CrossRef]
- Benítez-Mateos, A.I.; Contente, M.L.; Velasco-Lozano, S.; Paradisi, F.; López-Gallego, F. Self-sufficient flow-biocatalysis by coimmobilization of pyridoxal 5′-phosphate and ω-transaminases onto porous carriers. ACS Sustain. Chem. Eng. 2018, 6, 13151–13159. [Google Scholar] [CrossRef]
- Planchestainer, M.; Contente, M.L.; Cassidy, J.; Molinari, F.; Tamborini, L.; Paradisi, F. Continuous flow biocatalysis: Production and in-line purification of amines by immobilised transaminase from Halomonas elongata. Green Chem. 2017, 19, 372–375. [Google Scholar] [CrossRef]
- Contente, M.L.; Dall’Oglio, F.; Tamborini, L.; Molinari, F.; Paradisi, F. Highly efficient oxidation of amines to aldehydes with flow-based biocatalysis. Chem. Cat. Chem. 2017, 9, 3843–3848. [Google Scholar] [CrossRef]
- Abaházi, E.; Sátorhelyi, P.; Erdélyi, B.; Vértessy, B.G.; Land, H.; Paizs, C.; Berglund, P.; Poppe, L. Covalently immobilized Trp60Cys mutant of ω-transaminase from Chromobacterium violaceum for kinetic resolution of racemic amines in batch and continuous-flow modes. Biochem. Eng. J. 2018, 132, 270–278. [Google Scholar] [CrossRef]
- Nagy-Győr, L.; Abaházi, E.; Bódai, V.; Sátorhelyi, P.; Erdélyi, B.; Balogh-Weiser, D.; Paizs, C.; Hornyánszky, G.; Poppe, L. Co-immobilized whole cells with ω-transaminase and ketoreductase activities for continuous-flow cascade reactions. Chem. Bio. Chem. 2018, 19, 1845–1848. [Google Scholar] [CrossRef] [PubMed]
- Gruber, P.; Carvalho, F.; Marques, M.P.C.; O’Sullivan, B.; Subrizi, F.; Dobrijevic, D.; Ward, J.; Hailes, H.C.; Fernandes, P.; Wohlgemuth, R.; et al. Enzymatic synthesis of chiral amino-alcohols by coupling transketolase and transaminase-catalyzed reactions in a cascading continuous-flow microreactor system. Biotechnol. Bioeng. 2018, 115, 586–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tufvesson, P.; Jensen, J.S.; Kroutil, W.; Woodley, J.M. Experimental determination of thermodynamic equilibrium in biocatalytic transamination. Biotechnol. Bioeng. 2012, 109, 2159–2162. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.-S.; Kim, B.-G. Asymmetric synthesis of chiral amines with ω-transaminase. Biotechnol. Bioeng. 1999, 65, 206–211. [Google Scholar] [CrossRef]
- Truppo, M.D.; Rozzell, J.D.; Turner, N.J. Efficient production of enantiomerically pure chiral amines at concentrations of 50 g/L using transaminases. Org. Process Res. Dev. 2010, 14, 234–237. [Google Scholar] [CrossRef]
- Shin, J.-S.; Kim, B.-G. Comparison of the ω-transaminases from different microorganisms and application to production of chiral amines. Biosci. Biotechnol. Biochem. 2001, 65, 1782–1788. [Google Scholar] [CrossRef] [Green Version]
- Rios-Solis, L.; Bayir, N.; Halim, M.; Du, C.; Ward, J.M.; Baganz, F.; Lye, G.J. Non-linear kinetic modelling of reversible bioconversions: Application to the transaminase catalyzed synthesis of chiral amino-alcohols. Biochem. Eng. J. 2013, 73, 38–48. [Google Scholar] [CrossRef]
- Al-Haque, N.; Santacoloma, P.A.; Neto, W.; Tufvesson, P.; Gani, R.; Woodley, J.M. A robust methodology for kinetic model parameter estimation for biocatalytic reactions. Biotechnol. Prog. 2012, 28, 1186–1196. [Google Scholar] [CrossRef]
- Cassimjee, K.E.; Humble, M.S.; Miceli, V.; Colomina, C.G.; Berglund, P. Active site quantification of an ω-transaminase by performing a half transamination reaction. ACS Catal. 2011, 1, 1051–1055. [Google Scholar] [CrossRef]
- Park, E.; Kim, M.; Shin, J.-S. One-pot conversion of l-threonine into l-homoalanine: Biocatalytic production of an unnatural amino acid from a natural one. Adv. Synth. Catal. 2010, 352, 3391–3398. [Google Scholar] [CrossRef]
- Mutti, F.G.; Kroutil, W. Asymmetric bio-amination of ketones in organic solvents. Adv. Synth. Catal. 2012, 354, 3409–3413. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Simon, R.C.; Riethorst, W.; Zepeck, F.; Kroutil, W. Synthesis of (R)- or (S)-valinol using ω-transaminases in aqueous and organic media. Bioorg. Med. Chem. 2014, 22, 5558–5562. [Google Scholar] [CrossRef]
- Böhmer, W.; Volkov, A.; Engelmark Cassimjee, K.; Mutti, F.G. Continuous flow bioamination of ketones in organic solvents at controlled water activity using immobilized ω-transaminases. Adv. Synth. Catal. 2020, 362, 1858–1867. [Google Scholar] [CrossRef] [Green Version]
- Britton, J.; Majumdar, S.; Weiss, G.A. Continuous flow biocatalysis. Chem. Soc. Rev. 2018, 47, 5891–5918. [Google Scholar] [CrossRef]
- Barbosa, O.; Ortiz, C.; Berenguer-Murcia, A.; Torres, R.; Rodrigues, R.C.; Fernandez-Lafuente, R. Strategies for the one-step immobilization-purification of enzymes as industrial biocatalysts. Biotechnol. Adv. 2015, 33, 435–456. [Google Scholar] [CrossRef] [Green Version]
- Cantone, S.; Ferrario, V.; Corici, L.; Ebert, C.; Fattor, D.; Spizzo, P.; Gardossi, L. Efficient immobilisation of industrial biocatalysts: Criteria and constraints for the selection of organic polymeric carriers and immobilisation methods. Chem. Soc. Rev. 2013, 42, 6262–6276. [Google Scholar] [CrossRef] [Green Version]
- DiCosimo, R.; McAuliffe, J.; Poulose, A.J.; Bohlmann, G. Industrial use of immobilized enzymes. Chem. Soc. Rev. 2013, 42, 6437–6474. [Google Scholar] [CrossRef]
- Santos, J.C.S.D.; Barbosa, O.; Ortiz, C.; Berenguer-Murcia, A.; Rodrigues, R.C.; Fernandez-Lafuente, R. Importance of the support properties for immobilization or purification of enzymes. Chem. Cat. Chem. 2015, 7, 2413–2432. [Google Scholar] [CrossRef] [Green Version]
- Sheldon, R.A.; van Pelt, S. Enzyme immobilisation in biocatalysis: Why, what and how. Chem. Soc. Rev. 2013, 42, 6223–6235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alami, A.T.; Richina, F.; Correro, M.R.; Dudal, Y.; Shahgaldian, P. Surface immobilization and shielding of a transaminase enzyme for the stereoselective synthesis of pharmaceutically relevant building blocks. Chimia 2018, 72, 345–346. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Gao, J.; Zhou, L.; Huang, Z.; He, Y.; Zhu, M.; Jiang, Y. Fabrication of Ni2+-nitrilotriacetic acid functionalized magnetic mesoporous silica nanoflowers for one pot purification and immobilization of His-tagged ω-transaminase. Biochem. Eng. J. 2017, 128, 116–125. [Google Scholar] [CrossRef]
- Cardenas-Fernandez, M.; Neto, W.; Lopez, C.; Alvaro, G.; Tufvesson, P.; Woodley, J.M. Immobilization of Escherichia coli containing ω-transaminase activity in LentiKats(R). Biotechnol. Prog. 2012, 28, 693–698. [Google Scholar] [CrossRef]
- Engelmark Cassimjee, K.; Kadow, M.; Wikmark, Y.; Svedendahl Humble, M.; Rothstein, M.L.; Rothstein, D.M.; Backvall, J.E. A general protein purification and immobilization method on controlled porosity glass: Biocatalytic applications. Chem. Commun. 2014, 50, 9134–9137. [Google Scholar] [CrossRef] [Green Version]
- De Souza, S.P.; Junior, I.I.; Silva, G.M.A.; Miranda, L.S.M.; Santiago, M.F.; Leung-Yuk Lam, F.; Dawood, A.; Bornscheuer, U.T.; de Souza, R.O.M.A. Cellulose as an efficient matrix for lipase and transaminase immobilization. RSC Adv. 2016, 6, 6665–6671. [Google Scholar] [CrossRef]
- Jia, H.; Huang, F.; Gao, Z.; Zhong, C.; Zhou, H.; Jiang, M.; Wei, P. Immobilization of omega-transaminase by magnetic PVA-Fe3O4 nanoparticles. Biotechnol. Rep. 2016, 10, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Mallin, H.; Hohne, M.; Bornscheuer, U.T. Immobilization of (R)- and (S)-amine transaminases on chitosan support and their application for amine synthesis using isopropylamine as donor. J. Biotechnol. 2014, 191, 32–37. [Google Scholar] [CrossRef]
- Sun, J.; Cui, W.H.; Du, K.; Gao, Q.; Du, M.; Ji, P.; Feng, W. Immobilization of R-ω-transaminase on MnO2 nanorods for catalyzing the conversion of (R)-1-phenylethylamine. J. Biotechnol. 2017, 245, 14–20. [Google Scholar] [CrossRef]
- Neto, W.; Schürmann, M.; Panella, L.; Vogel, A.; Woodley, J.M. Immobilisation of ω-transaminase for industrial application: Screening and characterisation of commercial ready to use enzyme carriers. J. Mol. Catal. B Enzym. 2015, 117, 54–61. [Google Scholar] [CrossRef]
- Böhmer, W.; Knaus, T.; Volkov, A.; Slot, T.K.; Shiju, N.R.; Engelmark Cassimjee, K.; Mutti, F.G. Highly efficient production of chiral amines in batch and continuous flow by immobilized omega-transaminases on controlled porosity glass metal-ion affinity carrier. J. Biotechnol. 2019, 291, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Schrittwieser, J.H.; Velikogne, S.; Hall, M.; Kroutil, W. Artificial biocatalytic linear cascades for preparation of organic molecules. Chem. Rev. 2018, 118, 270–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrittwieser, J.H.; Sattler, J.; Resch, V.; Mutti, F.G.; Kroutil, W. Recent biocatalytic oxidation-reduction cascades. Curr. Opin. Chem. Biol. 2011, 15, 249–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tassano, E.; Hall, M. Enzymatic self-sufficient hydride transfer processes. Chem. Soc. Rev. 2019, 48, 5596–5615. [Google Scholar] [CrossRef]
- Knaus, T.; Mutti, F.G. Biocatalytic hydrogen-borrowing cascades. Chim. Oggi-Chem. Today 2017, 35, 34–37. [Google Scholar]
- Ricca, E.; Brucher, B.; Schrittwieser, J.H. Multi-enzymatic cascade reactions: Overview and perspectives. Adv. Synth. Catal. 2011, 353, 2239–2262. [Google Scholar] [CrossRef]
- Bortolini, O.; Fantin, G.; Fogagnolo, M.; Giovannini, P.P.; Guerrini, A.; Medici, A. An easy approach to the synthesis of optically active vic-diols: A new single-enzyme system. J. Org. Chem. 1997, 62, 1854–1856. [Google Scholar] [CrossRef]
- Bisogno, F.R.; Lavandera, I.; Kroutil, W.; Gotor, V. Tandem concurrent processes: One-pot single-catalyst biohydrogen transfer for the simultaneous preparation of enantiopure secondary alcohols. J. Org. Chem. 2009, 74, 1730–1732. [Google Scholar] [CrossRef] [Green Version]
- Bisogno, F.R.; Rioz-Martínez, A.; Rodríguez, C.; Lavandera, I.; de Gonzalo, G.; Torres Pazmiño, D.E.; Fraaije, M.W.; Gotor, V. Oxidoreductases working together: Concurrent obtaining of valuable derivatives by employing the pikat method. Chem. Cat. Chem. 2010, 2, 946–949. [Google Scholar] [CrossRef] [Green Version]
- Rioz-Martinez, A.; Bisogno, F.R.; Rodriguez, C.; de Gonzalo, G.; Lavandera, I.; Torres Pazmino, D.E.; Fraaije, M.W.; Gotor, V. Biocatalysed concurrent production of enantioenriched compounds through parallel interconnected kinetic asymmetric transformations. Org. Biomol. Chem. 2010, 8, 1431–1437. [Google Scholar] [CrossRef] [Green Version]
- Malik, M.S.; Park, E.-S.; Shin, J.-S. ω-Transaminase-catalyzed kinetic resolution of chiral amines using l-threonine as an amino acceptor precursor. Green Chem. 2012, 14, 2137–2140. [Google Scholar] [CrossRef]
- Engelmark Cassimjee, K.; Federsel, H.J. EziG: A universal platform for enzyme immobilisation. In Biocatalysis: An Industrial Perspective; De Gonzalo, G., Dominguez De Maria, P., Eds.; Royal Society of Chemistry: London, UK, 2018; pp. 345–362. [Google Scholar]
- Iwasaki, A.; Matsumoto, K.; Hasegawa, J.; Yasohara, Y. A novel transaminase, (R)-amine:pyruvate aminotransferase, from Arthrobacter sp. KNK168 (FERM BP-5228): Purification, characterization, and gene cloning. Appl. Microbiol. Biotechnol. 2012, 93, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Mutti, F.G.; Fuchs, C.S.; Pressnitz, D.; Sattler, J.H.; Kroutil, W. Stereoselectivity of four (R)-selective transaminases for the asymmetric amination of ketones. Adv. Synth. Catal. 2011, 353, 3227–3233. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Böhmer, W.; Koenekoop, L.; Simon, T.; Mutti, F.G. Parallel Interconnected Kinetic Asymmetric Transformation (PIKAT) with an Immobilized ω-Transaminase in Neat Organic Solvent. Molecules 2020, 25, 2140. https://doi.org/10.3390/molecules25092140
Böhmer W, Koenekoop L, Simon T, Mutti FG. Parallel Interconnected Kinetic Asymmetric Transformation (PIKAT) with an Immobilized ω-Transaminase in Neat Organic Solvent. Molecules. 2020; 25(9):2140. https://doi.org/10.3390/molecules25092140
Chicago/Turabian StyleBöhmer, Wesley, Lucien Koenekoop, Timothée Simon, and Francesco G. Mutti. 2020. "Parallel Interconnected Kinetic Asymmetric Transformation (PIKAT) with an Immobilized ω-Transaminase in Neat Organic Solvent" Molecules 25, no. 9: 2140. https://doi.org/10.3390/molecules25092140