
Donor Atom Preference of Organoruthenium and Organorhodium Cations on the Interaction with Novel Ambidentate (N,N) and (O,O) Chelating Ligands in Aqueous Solution


Abstract
:1. Introduction
2. Experimental
2.1. Materials and Methods
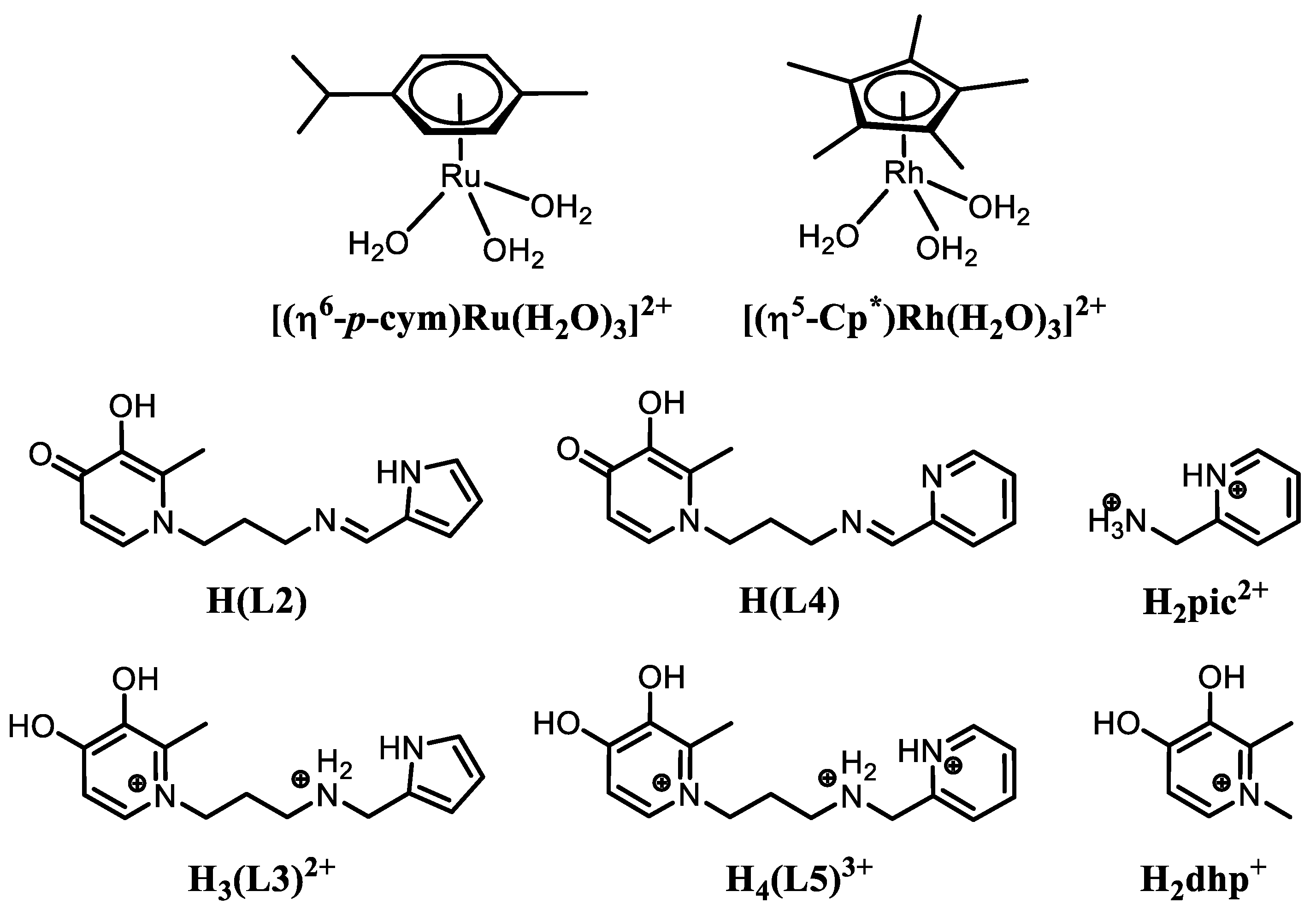
2.2. Synthesis of the Ligands
2.3. Solution Studies
2.4. X-ray Diffraction Analysis
3. Results and Discussion
3.1. Synthesis, Characterization and Acid-Base Properties of the Ligands
3.2. Complexation Properties of H(L3)
3.3. Complexation Properties of H(L5)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Amable, L. Cisplatin resistance and opportunities for precision medicine. Pharmacol. Res. 2016, 106, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Kenny, R.G.; Marmion, C.J. Toward Multi-Targeted Platinum and Ruthenium Drugs-A New Paradigm in Cancer Drug Treatment Regimens? Chem. Rev. 2019, 119, 1058–1137. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.J.; Lippard, S.J. Synthetic methods for the preparation of platinum anticancer complexes. Chem. Rev. 2014, 114, 4470–4495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869. [Google Scholar] [CrossRef] [Green Version]
- Shen, D.-W.; Pouliot, L.M.; Hall, M.D.; Gottesman, M.M. Cisplatin resistance: A cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol. Rev. 2012, 64, 706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Failes, T.W.; Cullinane, C.; Diakos, C.I.; Yamamoto, N.; Lyons, J.G.; Hambley, T.W. Studies of a cobalt(III) complex of the MMP inhibitor marimastat: A potential hypoxia-activated prodrug. Chem. Eur. J. 2007, 13, 2974–2982. [Google Scholar] [CrossRef]
- Failes, T.W.; Hambley, T.W. Models of hypoxia activated prodrugs: Co(III) complexes of hydroxamic acids. Dalton Trans. 2006, 1895–1901. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.J.; Hocking, R.; Clegg, J.K.; Turner, P.; Neville, S.M.; Hambley, T.W. Cobalt complexes with tripodal ligands: Implications for the design of drug chaperones. Dalton Trans. 2012, 41, 11293–11304. [Google Scholar]
- Buglyó, P.; Kacsir, I.; Kozsup, M.; Nagy, I.; Nagy, S.; Bényei, A.C.; Kovats, É.; Farkas, E. Tuning the redox potentials of ternary cobalt(III) complexes containing various hydroxamates. Inorg. Chim. Acta 2018, 472, 234–242. [Google Scholar] [CrossRef] [Green Version]
- Kozsup, M.; Zhou, X.; Farkas, E.; Bényei, A.C.; Bonnet, S.; Patonay, T.; Kónya, K.; Buglyó, P. Synthesis, characterization and cytotoxicity studies of Co(III)-flavonolato complexes. J. Inorg. Biochem. 2021, 217, 11138. [Google Scholar] [CrossRef]
- Ozsváth, A.; Farkas, E.; Diószegi, R.; Buglyó, P. Versatility and trends in the interaction between Pd(ii) and peptide hydroxamic acids. New J. Chem. 2019, 43, 8239–8249. [Google Scholar] [CrossRef]
- Ozsváth, A.; Diószegi, R.; Bényei, A.C.; Buglyó, P. Pd(ii)-Complexes of a novel pyridinone based tripeptide conjugate: Solution and solid state studies. Dalton Trans. 2020, 49, 9254–9267. [Google Scholar] [CrossRef]
- Parajdi-Losonczi, P.L.; Bényei, A.C.; Kováts, É.; Timári, I.; Muchova, T.R.; Kasparkova, J.; Buglyó, P.J. [(η(6)-p-cymene)Ru(H2O)3](2+) binding capability of aminohydroxamates—A solution and solid state study. Inorg. Biochem. 2016, 160, 236–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parajdi-Losonczi, P.L.; Buglyó, P.; Skakalova, H.; Kasparkova, J.; Lihi, N.; Farkas, E. Half-sandwich type rhodium(iii)–aminohydroxamate complexes: The role of the position of the amino group in metal ion binding. New J. Chem. 2018, 42, 7659–7670. [Google Scholar] [CrossRef]
- Ozsváth, A.; Bíró, L.; Nagy, E.M.; Buglyó, P.; Sanna, D.; Farkas, E. Trends and Exceptions in the Interaction of Hydroxamic Acid Derivatives of Common Di- and Tripeptides with Some 3d and 4d Metal Ions in Aqueous Solution. Molecules 2019, 24, 3941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bihari, Z.; Ugone, V.; Garribba, E.; Lihi, N.; Buglyó, P. Complex formation between [(eta(6)-p-cym)Ru(H2O)(3)](2+) and oligopeptides containing three histidyl moieties. J. Organomet. Chem. 2016, 823, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Balogh, B.D.; Bihari, Z.; Buglyó, P.; Csire, G.; Kerekes, Z.; Lukács, M.; Sóvágó, I.; Várnagy, K. Metal binding selectivity of an N-terminally free multihistidine peptide HAVAHHH-NH2. New J. Chem. 2019, 43, 907–916. [Google Scholar] [CrossRef]
- Nagy, I.; Farkas, E.; Kasparkova, J.; Kostrhunova, H.; Brabec, V.; Buglyó, P. Synthesis and characterization of (Ru(II), Co(III)) heterobimetallic complexes formed with a 1,10-phenanthroline based hydroxamic acid conjugate. J. Organomet. Chem. 2020, 916, 121265. [Google Scholar] [CrossRef]
- Mészáros, J.P.; Poljarevic, J.M.; Szatmári, I.; Csuvik, O.; Fülöp, F.; Szoboszlai, N.; Spengler, G.; Enyedy, É.A. An 8-hydroxyquinoline-proline hybrid with multidrug resistance reversal activity and the solution chemistry of its half-sandwich organometallic Ru and Rh complexes. Dalton Trans. 2020, 49, 7977–7992. [Google Scholar] [CrossRef]
- Mészáros, J.P.; Németi, G.; Poljarevic, J.M.; Holczbauer, T.; May, N.V.; Enyedy, É.A. Effect of the additional carboxyl group in half-sandwich organometallic 2,4-dipicolinate complexes on solution speciation and structure. Eur. J. Inorg. Chem. 2021. [Google Scholar] [CrossRef]
- Santos, M.A.; Grazina, R.; Neto, A.Q.; Cantinho, G.; Gano, L.; Patricio, L. Synthesis, chelating properties towards gallium and biological evaluation of two N-substituted 3-hydroxy-4-pyridinones. J. Inorg. Biochem. 2000, 78, 303–311. [Google Scholar] [CrossRef]
- Gran, G. Determination of the Equivalent Point in Potentiometric Titrations. Acta Chem. Scand. 1950, 4, 559–577. [Google Scholar] [CrossRef] [Green Version]
- Irving, H.M.; Miles, M.G.; Pettit, L.D. A study of some problems in determining the stoichiometric proton dissociation constants of complexes by potentiometric titrations using a glass electrode. Anal. Chim. Acta 1967, 38, 475–488. [Google Scholar] [CrossRef]
- Zékány, L.; Nagypál, I. Computational Methods for the Determination of Stability Constants Leggett; Plenum: New York, NY, USA, 1985; pp. 291–299. [Google Scholar]
- Gans, P.; Sabatini, A.; Vacca, A. SUPERQUAD: An improved general program for computation of formation constants from potentiometric data. J. Chem. Soc. Dalton Trans. 1985, 1195–1200. [Google Scholar] [CrossRef]
- Bíró, L.; Godó, A.J.; Bihari, Z.; Garribba, E.; Buglyó, P. Tuning the Hydrolytic Properties of Half-Sandwich Type Organometallic Cations in Aqueous Solution. Eur. J. Inorg. Chem. 2013, 3090–3100. [Google Scholar] [CrossRef]
- Dömötör, O.; Aicher, s.; Schmidlehner, M.; Novak, M.S.; Roller, A.; Jakupec, M.A.; Kandioller, W.; Hartinger, C.G.; Keppler, B.K.; Enyedy, É.A. Antitumor pentamethylcyclopentadienyl rhodium complexes of maltol and allomaltol: Synthesis, solution speciation and bioactivity. J. Inorg. Biochem. 2014, 134, 57–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ősz, K.; Lente, G.; Kállay, C. New protonation microequilibrium treatment in the case of some amino acid and peptide derivatives containing a bis(imidazolyl)methyl group. J. Phys. Chem. B 2005, 109, 1039–1047. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westrip, S.P. publCIF: Software for editing, validating and formatting crystallographic information files. J. Appl. Crystallogr. 2010, 43, 920–925. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Linnell, R. Notes—Dissociation Constants of 2-Substituted Pyridines. J. Org. Chem. 1960, 25, 290. [Google Scholar] [CrossRef]
- Enyedy, É.A.; Dömötör, O.; Hackl, C.M.; Roller, A.; Novak, M.S.; Jakupec, M.A.; Keppler, B.K.; Kandioller, W. Solution equilibria and antitumor activities of pentamethylcyclopentadienyl rhodium complexes of picolinic acid and deferiprone. J. Coord. Chem. 2015, 68, 1583–1601. [Google Scholar] [CrossRef] [Green Version]
- Bíró, L.; Farkas, E.; Buglyó, P. Complex formation between Ru(η(6)-p-cym)(H2O)3]2+ and (O,O) donor ligands with biological relevance in aqueous solution. Dalton Trans. 2010, 39, 10272–10278. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, J.P.; Dömötör, O.; Hackl, C.M.; Roller, A.; Keppler, B.K.; Kandioller, W.; Enyedy, É.A. Structural and solution equilibrium studies on half-sandwich organorhodium complexes of (N,N) donor bidentate ligands. New J. Chem. 2018, 42, 11174–11184. [Google Scholar] [CrossRef] [Green Version]
- Bíró, L.; Buglyó, P.; Farkas, E. Factors determining the metal ion binding ability and selectivity of hydroxamate based compounds. Curr. Med. Chem. 2021. accepted. [Google Scholar] [CrossRef]
- Buglyó, P.; Parajdi-Losonczi, P.L.; Bényei, A.C.; Lihi, N.; Bíró, L.; Farkas, E. Versatility of Coordination Modes in Complexes of Monohydroxamic Acids with Half-Sandwich Type Ruthenium, Rhodium, Osmium and Iridium Cations. ChemistrySelect 2017, 2, 8127–8136. [Google Scholar] [CrossRef]
- Gerhartz, W.; Campbell, F.T.; Pfefferkorn, R.; Rounsaville, J.F. Ullmann’s Encyclopedia of Industrial Chemistry, 5th ed.; VCH Publishers: Deerfield Beach, FL, USA, 1985; Volume A1, pp. A22, (93), 453. [Google Scholar]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| L3 | L5 | dhp | pic | ||||
|---|---|---|---|---|---|---|---|
| pKaromatic N | – | 1.3(1) | N/A c | – | 2.29 f | ||
| pKOH | 3.19(1) | 3.46(2) c | 3.20(3) | 3.50(4) c | 3.64 d | – | |
| pKaliphatic N | 8.57(1) | 8.53(7) c | 7.72(2) | 7.73(8) c | – | 8.69 f | |
| pKOH | 9.83(1) | 9.61(5) c | 9.78(1) | 9.60(9) c | 9.77 d | – | |
| Rh2+ g | Ru2+ g | Rh2+ g | Rh2+ g | Ru2+ g | Rh2+ g | ||
| logβ[MH2L] | – | – | 23.7(3) c | – | – | – | |
| logβ[MHL] | 17.09(3) | 20.30(6) | 20.57(2) | – | – | – | |
| logβ[ML] | 8.10(11) | 11.92(4) | 11.03(4) | 8.93 d | 11.86 e | 13.59 f | |
| logβ[MH–1L] | −3.1(3) | 1.86(6) | 0.00(5) | −2.97 d | 1.83 e | – | |
| logβ[M2L] | 14.98(4) | – | 19.71(3) | – | – | – | |
| logβ[M2H–1L] | 6.62(5) | – | 9.30(6) | – | – | – | |
| logβ[M2H–2L] | – | – | −2.04(10) | – | – | – | |
| Fitting parameter (mL) b | 0.0118 | 0.0115 | 0.0109 | – | – | – | |
| # of fitted data | 455 | 303 | 289 | – | – | – | |
| pK[MH2L] | – | – | 3.13 | – | – | – | |
| pK[MHL] | 8.99 | 8.38 | 9.54 | – | – | – | |
| pK[ML] | 11.20 | 10.06 | 11.03 | 11.90 d | 10.03 e | 8.48 f | |
| pK[M2L] | 8.36 | – | 10.41 | – | – | – | |
| pK[M2H–1L] | – | – | 11.34 | – | – | – | |
| pM’1:10 h | 7.08 | 9.76 | 11.26 | 7.53 | 10.04 | 13.23 | |
| pM’1:1 i | 6.27 | 6.73 | 8.48 | 6.39 | 6.83 | 9.15 | |
| m/z | |||||
|---|---|---|---|---|---|
| Ligand | M2+ | Species | pH (at 1:1/2:1 Ratios) | Obtained | Calculated |
| H(L3) | [(η5-Cp*)Rh]2+ | [M(L3)]+ | 2.3; 5.1; 7.0; 9.1/2.3; 6.0; 8.6; 10.5 | 498.1619 | 498.1622 |
| [M(L1)]+ a | 2.3; 5.1; 7.0; 9.1/2.3; 6.0; 8.6; 10.5 | 419.1200 | 419.1200 | ||
| [M2H–1(L3)]2+ | 5.1; 7.0; 9.1/2.3; 6.0; 8.6; 10.5 | 367.5881 | 367.5884 | ||
| [M2H–1(L3)+Cl]+ | 7.0; 9.1/6.0; 8.6; 10.5 | 770.1460 | 770.1461 | ||
| [H2(L1)]+ a | 2.3; 5.1; 7.0; 9.1/2.3 | 183.1130 | 183.1128 | ||
| [M2Cl2]+ | 2.3/2.3, 6.0 | 272.9912 | 272.9912 | ||
| [M2(η-O)(η-OH)]+ | /10.5 | 509.0430 | 509.0430 | ||
| [(η6-p-cym)Ru]2+ | [M(L3)]+ | 2.3; 7.1/2.4; 4.0; | 496.1537 | 496.1539 | |
| [M(L1)]+ | 2.3; 7.1; 10.4/2.4; 4.0; 9.5; | 417.1115 | 417.1116 | ||
| [H2(L1)]+ a | 2.3; 7.1/2.4 | 183.1131 | 183.1128 | ||
| [M2(η-OH)3]+ | 10.4/9.5; 10.3 | 522.0374 | 522.0376 | ||
| [M2(η-O)(η-OH)]+ | 10.4/9.5; 10.3 | 504.0268 | 504.0270 | ||
| H(L5) | [(η5-Cp*)Rh]2+ | [M(L5)]+ | 2.2; 4.2; 8.5; 10.4/2.3; 3.7; 9.7; 10.3 | 510.1618 | 510.1622 |
| [MH(L5)+Cl]+ | 2.2; 4.2; 8.5; 10.4/2.3 | 546.1386 | 546.1389 | ||
| [MH(L5)]2+ | 2.2/2.3 | 255.5845 | 255.5848 | ||
| [M2Cl2]+ | 2.2; 4.2/2.3; 3.7; 9.7; 10.3 | 272.9912 | 272.9912 | ||
| [M2(L5)+2 Cl]+ | 4.2/2.3; 3.7; 9.7; 10.3 | 818.1220 | 818.1228 | ||
| [M2H–1(L5)]2+ | 4.2/2.3; 9.7; 10.3 | 373.5881 | 373.5884 | ||
| [(η6-p-cym)Ru]2+ | [M(L5)]+ | 2.3; 7.9; 9.0/2.1; 4.3; 7.9; 10.0 | 508.1533 | 508.1539 | |
| [MH(L5)+Cl]+ | 2.3; 7.9; 9.0/2.1; 7.9 | 544.1298 | 544.1303 | ||
| [M2H–1(L5)]2+ | /2.1; 4.3; 7.9 | 371.0797 | 371.0806 | ||
| [MH(L5)]2+ | 2.3; 7.9/2.1; 4.3; 7.9 | 254.5795 | 254.5806 | ||
| [M(L5)+Cl]+ | 2.3 | 270.9814 | 270.9821 | ||
| [M2(η-OH)3]+ | 7.9; 9.0; 10.5/7.9; 10.0 | 522.0374 | 522.0376 | ||
| [M2(η-O)(η-OH)]+ | 7.9; 9.0; 10.5/7.9; 10.0 | 504.0268 | 504.0270 | ||
| [M2(L5)+2Cl]+ | /2.1; 4.3; | 814.1059 | 814.1058 | ||
| [M2(L5)+Cl]2+ | /2.1; 4.3; 7.9 | 389.0681 | 389.0687 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagy, S.; Ozsváth, A.; Bényei, A.C.; Farkas, E.; Buglyó, P. Donor Atom Preference of Organoruthenium and Organorhodium Cations on the Interaction with Novel Ambidentate (N,N) and (O,O) Chelating Ligands in Aqueous Solution. Molecules 2021, 26, 3586. https://doi.org/10.3390/molecules26123586
Nagy S, Ozsváth A, Bényei AC, Farkas E, Buglyó P. Donor Atom Preference of Organoruthenium and Organorhodium Cations on the Interaction with Novel Ambidentate (N,N) and (O,O) Chelating Ligands in Aqueous Solution. Molecules. 2021; 26(12):3586. https://doi.org/10.3390/molecules26123586
Chicago/Turabian StyleNagy, Sándor, András Ozsváth, Attila Cs. Bényei, Etelka Farkas, and Péter Buglyó. 2021. "Donor Atom Preference of Organoruthenium and Organorhodium Cations on the Interaction with Novel Ambidentate (N,N) and (O,O) Chelating Ligands in Aqueous Solution" Molecules 26, no. 12: 3586. https://doi.org/10.3390/molecules26123586