
Immunoproteasome and Non-Covalent Inhibition: Exploration by Advanced Molecular Dynamics and Docking Methods

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
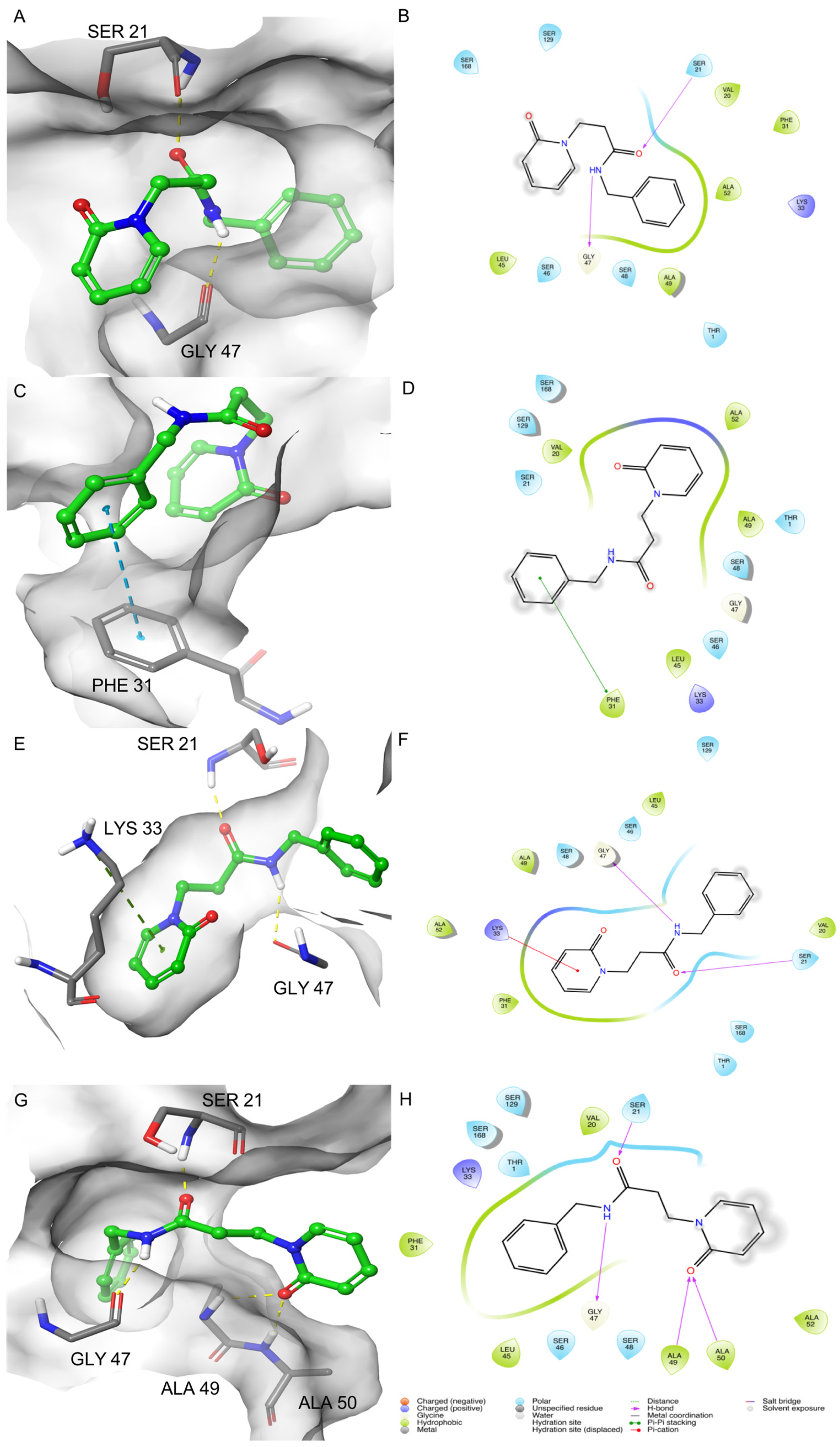
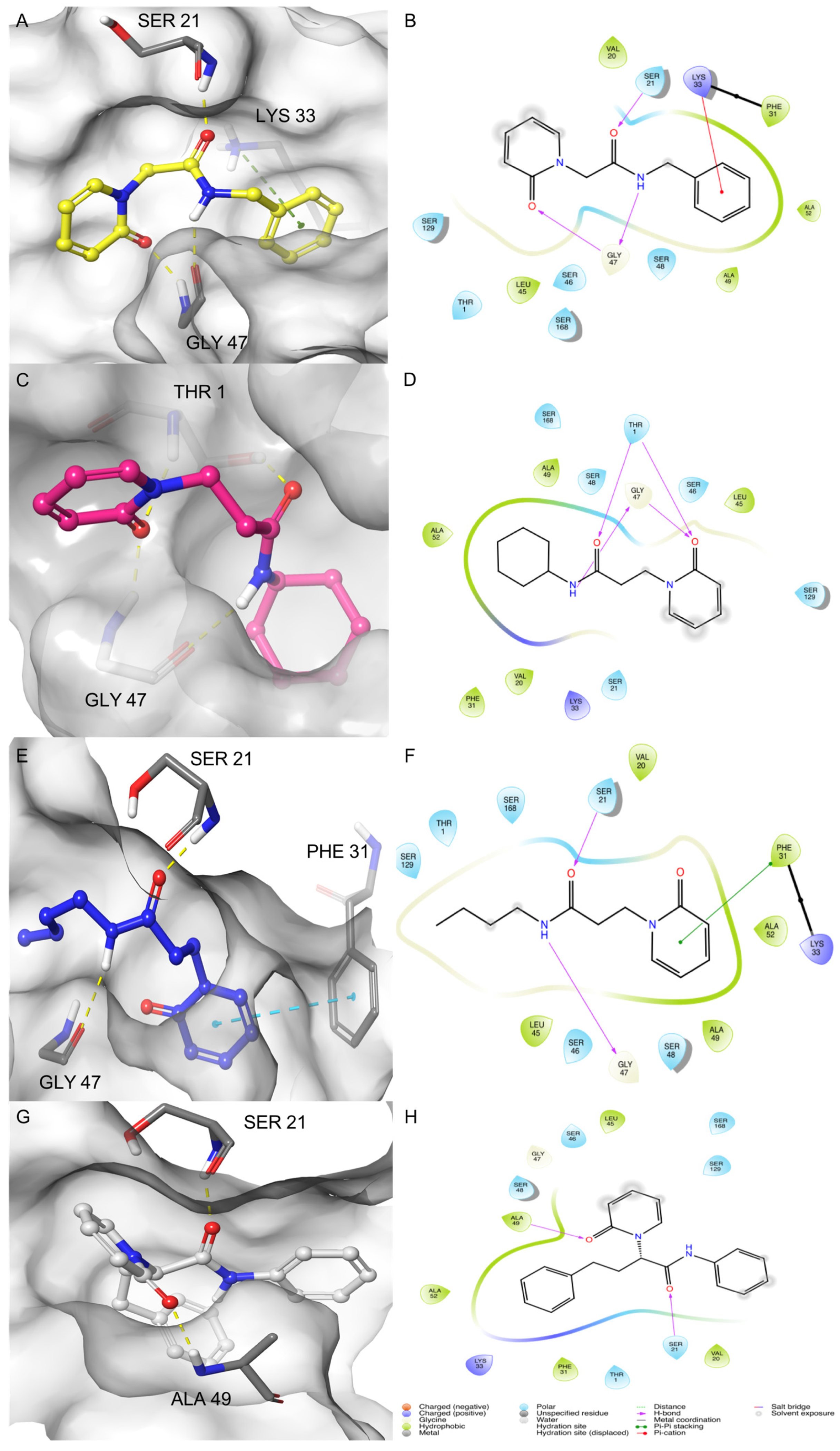
2.1. MD-Binding (MDB) Analysis
2.2. Induced Fit Docking (IFD)
2.3. Binding Pose MetaDynamics Analysis
3. Discussion
4. Materials and Methods
4.1. System and Ligand Preparation
4.2. MD-Binding Simulations
4.3. Plain MD Simulations
4.4. Binding Pose MetaDynamics (BPMD)
4.5. Induced-Fit Docking
4.6. MM-GBSA-Binding Free Energy Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- DeMartino, G.N.; Gillette, T.G. Proteasomes: Machines for all Reasons. Cell 2007, 129, 659–662. [Google Scholar] [CrossRef] [Green Version]
- Ferrington, D.A.; Gregerson, D.S. Immunoproteasome: Structure, Function, and Antigen Presentation. Prog. Mol. Biol. Transl. Sci. 2012, 109, 75–112. [Google Scholar] [CrossRef] [Green Version]
- Ettari, R.; Previti, S.; Bitto, A.; Grasso, S.; Zappalà, M. Immunoproteasome-Selective Inhibitors: A Promising Strategy to Treat Hematologic Malignancies, Autoimmune and Inflammatory Diseases. Curr. Med. Chem. 2016, 23, 1217–1238. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, D.J.; Orlowski, R.Z. The Immunoproteasome as a Target in Hematologic Malignancies. Semin. Hematol. 2012, 49, 258–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ettari, R.; Zappalà, M.; Grasso, S.; Musolino, C.; Innao, V.; Allegra, A. Immunoproteasome-Selective and Non-Selective Inhibitors: A Promising Approach for the Treatment of Multiple Myeloma. Pharmacol. Ther. 2018, 182, 176–192. [Google Scholar] [CrossRef]
- Xi, J.; Zhuang, R.; Kong, L.; He, R.; Zhu, H.; Zhang, J. Immunoproteasome-Selective Inhibitors: An Overview of Recent Developments as Potential Drugs for Hematologic Malignancies and Autoimmune Diseases. Eur. J. Med. Chem. 2019, 182, 111646. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Samanta, I.; Mondal, A.; Liu, W.R. Covalent Inhibition in Drug Discovery. ChemMedChem 2019, 14, 889–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allardyce, D.J.; Bell, C.M.; Loizidou, E.Z. Argyrin B, a Non-Competitive Inhibitor of the Human Immunoproteasome Exhibiting Preference for β1i. Chem. Biol. Drug Des. 2019, 94, 1556–1567. [Google Scholar] [CrossRef]
- Zhan, W.; Singh, P.K.; Ban, Y.; Qing, X.; Ah Kioon, M.D.; Fan, H.; Zhao, Q.; Wang, R.; Sukenick, G.; Salmon, J.; et al. Structure–Activity Relationships of Noncovalent Immunoproteasome β5i-Selective Dipeptides. J. Med. Chem. 2020, 63, 13103–13123. [Google Scholar] [CrossRef]
- Villoutreix, B.O.; Miteva, M.A.; Khatib, A.-M.; Cheng, Y.; Marechal, X.; Reboud-Ravaux, M.; Vidal, J. Blockade of the malignant phenotype by β-subunit selective noncovalent inhibition of immuno- and constitutive proteasomes. Oncotarget 2017, 8, 10437–10449. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.K.; Fan, H.; Jiang, X.; Shi, L.; Nathan, C.F.; Lin, G. Immunoproteasome β5i-Selective Dipeptidomimetic Inhibitors. Chem. Med. Chem. 2016, 11, 2127–2131. [Google Scholar] [CrossRef] [PubMed]
- Ettari, R.; Cerchia, C.; Maiorana, S.; Guccione, M.; Novellino, E.; Bitto, A.; Grasso, S.; Lavecchia, A.; Zappalà, M. Development of Novel Amides as Noncovalent Inhibitors of Immunoproteasomes. Chem. Med. Chem. 2019, 14, 842–852. [Google Scholar] [CrossRef] [PubMed]
- Lei, B.; Abdul Hameed, M.D.; Hamza, A.; Wehenkel, M.; Muzyka, J.L.; Yao, X.J.; Kim, K.B.; Zhan, C.G. Molecular Basis of the Selectivity of the Immunoproteasome Catalytic Subunit LMP2-Specific Inhibitor Revealed by Molecular Modeling and Dynamics Simulations. J. Phys. Chem. B 2010, 114, 12333–12339. [Google Scholar] [CrossRef] [Green Version]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4035–4406. [Google Scholar] [CrossRef] [PubMed]
- Culletta, G.; Almerico, A.M.; Tutone, M. Comparing molecular dynamics-derived pharmacophore models with docking: A study on CDK-2 inhibitors. Chem. Data Coll. 2020, 28, 100485. [Google Scholar] [CrossRef]
- Tutone, M.; Culletta, G.; Livecchi, L.; Almerico, A.M. A Definitive Pharmacophore Modelling Study on CDK2 ATP Pocket Binders: Tracing the Path of New Virtual High-Throughput Screenings. Curr. Drug Discov. Technol. 2020, 17, 740–747. [Google Scholar] [CrossRef]
- Spitaleri, A.; Decherchi, S.; Cavalli, A.; Rocchia, W. Fast Dynamic Docking Guided by Adaptive Electrostatic Bias: The MD-Binding Approach. J. Chem. Theory Comput. 2018, 14, 1727–1736. [Google Scholar] [CrossRef]
- Fusani, L.; Palmer, D.S.; Somers, D.O.; Wall, I.D. Exploring Ligand Stability in Protein Crystal Structures Using Binding Pose Metadynamics. J. Chem. Inf. Model. 2020, 60, 1528–1539. [Google Scholar] [CrossRef]
- Decherchi, S.; Bottegoni, G.; Spitaleri, A.; Rocchia, W.; Cavalli, A. BiKi Life Sciences: A New Suite for Molecular Dynamics and Related Methods in Drug Discovery. J. Chem. Inf. Model. 2018, 58, 219–224. [Google Scholar] [CrossRef]
- Huber, E.M.; Basler, M.; Schwab, R.; Heinemeyer, W.; Kirk, C.J.; Groettrup, M.; Groll, M. Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell 2012, 148, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Ladi, E.; Everett, C.; Stivala, C.E.; Daniels, B.E.; Durk, M.R.; Harris, S.F.; Huestis, M.P.; Purkey, H.E.; Staben, S.T.; Augustin, M.; et al. Design and Evaluation of Highly Selective Human Immunoproteasome Inhibitors Reveal a Compensatory Process that Preserves Immune Cell Viability. J. Med. Chem. 2019, 62, 7032–7041. [Google Scholar] [CrossRef]
- Lexa, K.W.; Carlson, H.A. Protein flexibility in docking and surface mapping. Q. Rev. Biophys. 2012, 45, 301–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decherchi, S.; Rocchia, W. A general and Robust Ray-Casting-Based Algorithm for Triangulating Surfaces at the Nanoscale. PLoS ONE 2013, 8, e59744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Culletta, G.; Gulotta, M.R.; Perricone, U.; Zappalà, M.; Almerico, A.M.; Tutone, M. Exploring the SARS-CoV-2 Proteome in the Search of Potential Inhibitors via Structure-Based Pharmacophore Modeling/Docking Approach. Computation 2020, 8, 77. [Google Scholar] [CrossRef]
- Selvaraj, C.; Panwar, U.; Dinesh, D.C.; Boura, E.; Singh, P.; Dubey, V.K.; Singh, S.K. Microsecond MD Simulation and Multiple-Conformation Virtual Screening to Identify Potential Anti-COVID-19 Inhibitors Against SARS-CoV-2 Main Protease. Front. Chem. 2021, 8, 595273. [Google Scholar] [CrossRef] [PubMed]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Prime; Schrödinger, LLC: New York, NY, USA, 2021.
- Epik; Schrödinger, LLC: New York, NY, USA, 2021.
- Marvin Sketch 19.25, ChemAxon. Available online: https://www.chemaxon.com (accessed on 2 July 2021).
- Motta, S.; Callea, L.; Giani Tagliabue, S.; Bonati, L. Exploring the PXR Ligand-Binding Mechanism with Advanced Molecular Dynamics Methods. Sci. Rep. 2018, 8, 16207. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Lindorff-Larsen, J.K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved Side-Chain Torsion Potentials for the Amber ff99SB Protein Force Field. Proteins Struct. Funct. Genet. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE SC|06 Conference (SC’06); Association for Computing Machinery: Tampa, FL, USA, 2006; ISBN 978-0-7695-2700-0. [Google Scholar]
- Robertson, M.J.; Tirado-Rives, J.; Jorgensen, W.L. Improved Peptide and Protein Torsional Energetics with the OPLS-AA Force Field. J. Chem. Theory Comput. 2015, 11, 3499–3509. [Google Scholar] [CrossRef]
- Dyer, K.M.; Perkyns, J.S.; Stell, G.; Pettitt, B.M. Site-renormalised Molecular Fluid Theory: On the Utility of a Two-site Model of Water. Mol. Phys. 2009, 107, 423–431. [Google Scholar] [CrossRef] [Green Version]
- Almerico, A.M.; Tutone, M.; Pantano, L.; Lauria, A. Molecular Dynamics Studies on Mdm2 Complexes: An Analysis of the Inhibitor Influence. Biochem. Biophys. Res. Commun. 2012, 424, 341–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tutone, M.; Pibiri, I.; Lentini, L.; Pace, A.; Almerico, A.M. Deciphering the Nonsense Readthrough Mechanism of Action of Ataluren: An in Silico Compared Study. ACS Med. Chem. Lett. 2019, 10, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Pibiri, I.; Lentini, L.; Melfi, R.; Tutone, M.; Baldassano, S.; Ricco Galluzzo, P.; Di Leonardo, A.; Pace, A. Rescuing the CFTR protein function: Introducing 1,3,4-oxadiazoles as translational readthrough inducing drugs. Eur. J. Med. Chem. 2018, 159, 126–142. [Google Scholar] [CrossRef] [PubMed]
- Almerico, A.M.; Tutone, M.; Lauria, A. Docking and multivariate methods to explore HIV-1 drug-resistance: A comparative analysis. J. Comput.-Aided Mol. Des. 2008, 22, 287–297. [Google Scholar] [CrossRef]
- Wieder, M.; Garon, A.; Perricone, U.; Boresch, S.; Seidel, T.; Almerico, A.M.; Langer, T. Common Hits Approach: Combining Pharmacophore Modeling and Molecular Dynamics Simulations. J. Chem. Inf. Model. 2017, 57, 365–385. [Google Scholar] [CrossRef]
- Perricone, U.; Wieder, M.; Seidel, T.; Langer, T.; Padova, A.; Almerico, A.M.; Tutone, M. A Molecular Dynamics-Shared Pharmacophore Approach to Boost Early-Enrichment Virtual Screening: A Case Study on Peroxisome Proliferator-Activated Receptor α. Chem. Med. Chem. 2017, 12, 1399–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Culletta, G.; Zappalà, M.; Ettari, R.; Almerico, A.M.; Tutone, M. Immunoproteasome and Non-Covalent Inhibition: Exploration by Advanced Molecular Dynamics and Docking Methods. Molecules 2021, 26, 4046. https://doi.org/10.3390/molecules26134046
Culletta G, Zappalà M, Ettari R, Almerico AM, Tutone M. Immunoproteasome and Non-Covalent Inhibition: Exploration by Advanced Molecular Dynamics and Docking Methods. Molecules. 2021; 26(13):4046. https://doi.org/10.3390/molecules26134046
Chicago/Turabian StyleCulletta, Giulia, Maria Zappalà, Roberta Ettari, Anna Maria Almerico, and Marco Tutone. 2021. "Immunoproteasome and Non-Covalent Inhibition: Exploration by Advanced Molecular Dynamics and Docking Methods" Molecules 26, no. 13: 4046. https://doi.org/10.3390/molecules26134046