
Fucoxanthin Holds Potential to Become a Drug Adjuvant in Breast Cancer Treatment: Evidence from 2D and 3D Cell Cultures

 ,
,  ,
, 
 and
and
Abstract
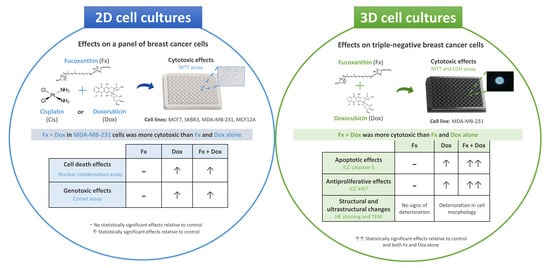
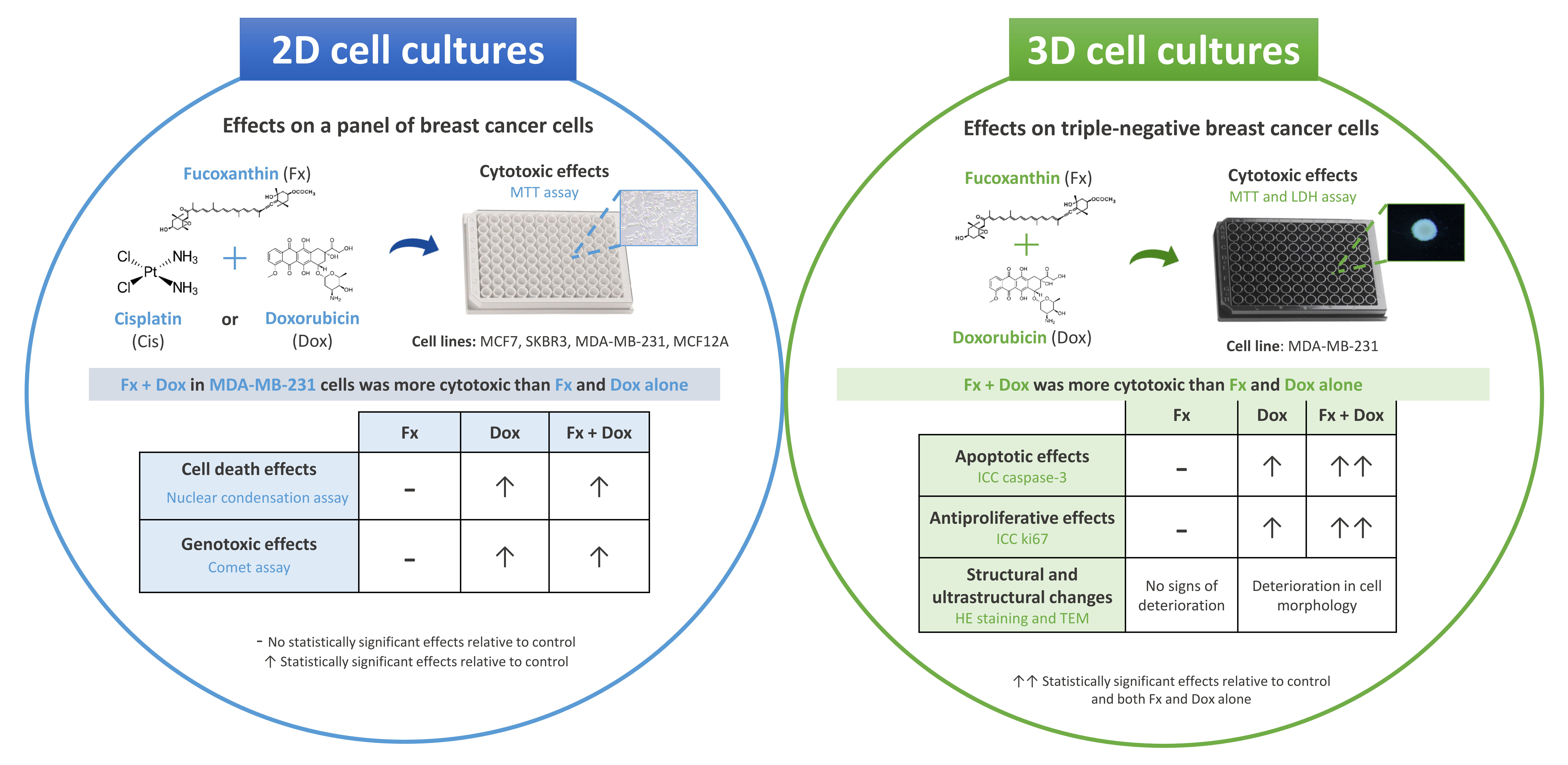
:
1. Introduction
2. Results
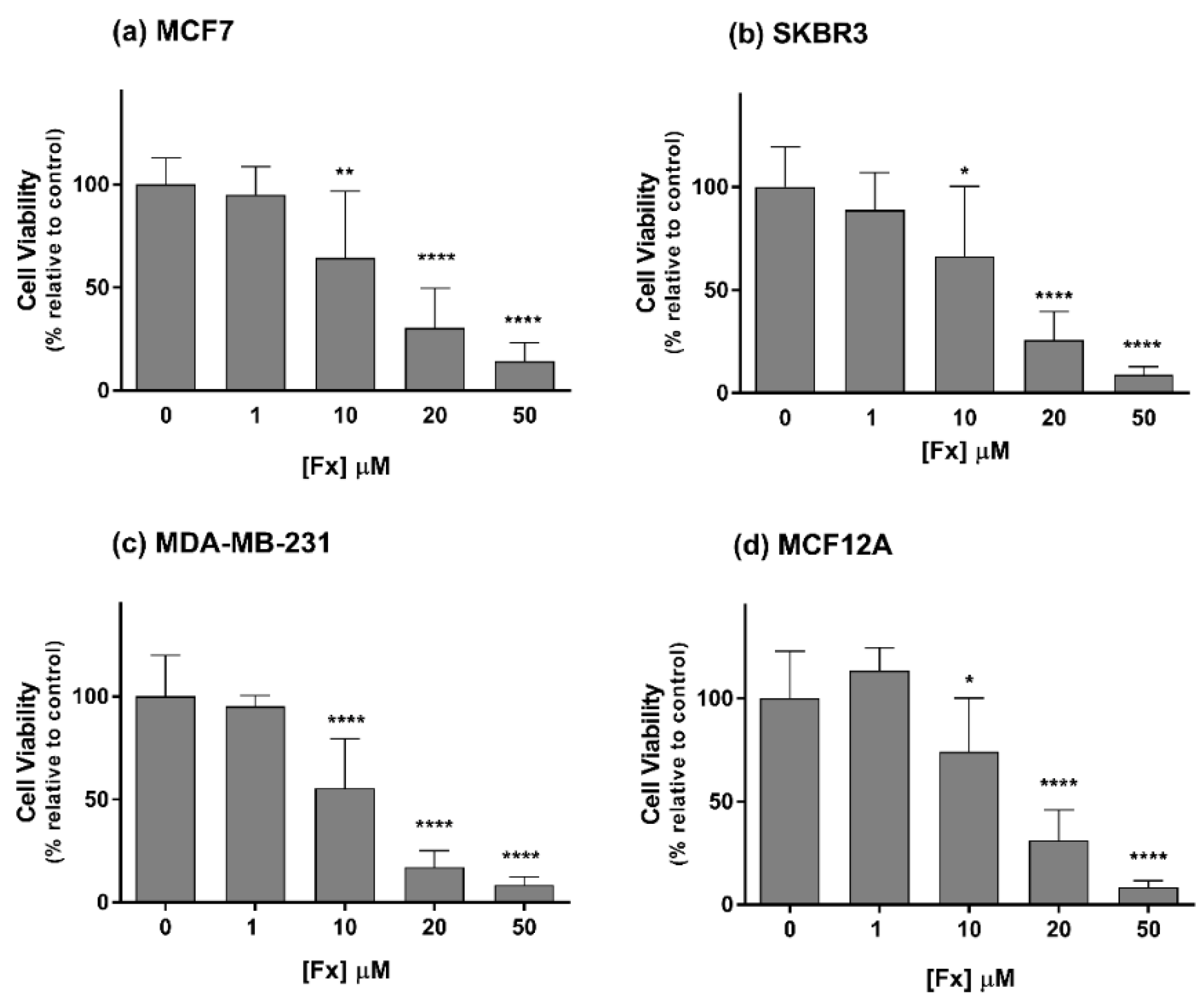
2.1. Phase 1—Cytotoxic Effects of Fx Alone in 2D Cell Cultures
2.2. Phase 2—Cytotoxic Effects of Fx Combined with Dox or Cis in 2D Cell Cultures
2.3. Phase 3—Effects of Fx Combined with Dox on Cell Death and DNA Damage in 2D Cell Cultures
2.4. Phase 4—Effects of Fx Combined with Dox in 3D Cell Cultures (Multicellular Aggregates-MCAs)
2.4.1. Cytotoxic Effects
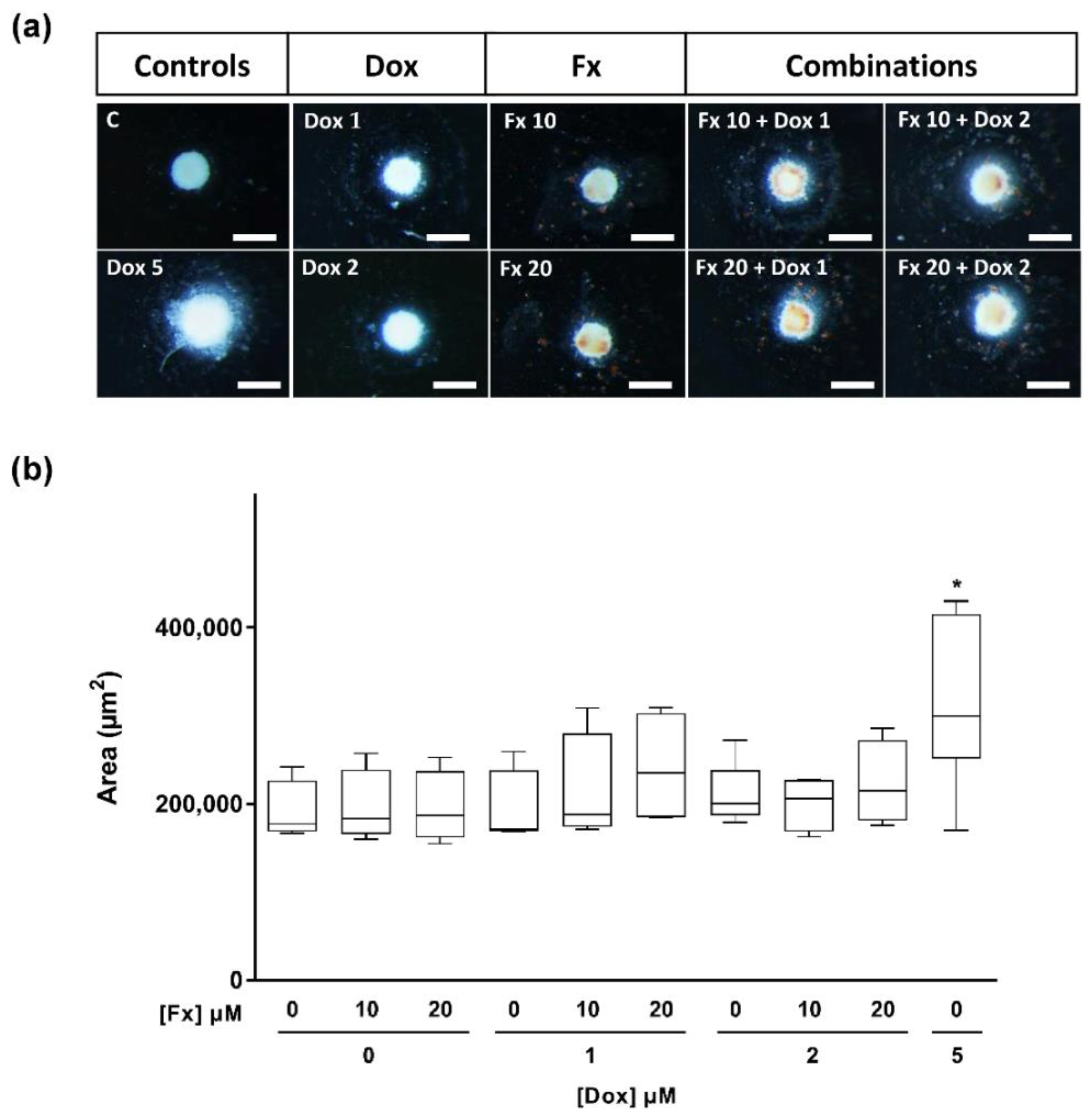
2.4.2. Stereomicroscopic Analysis and Area Measurements
2.4.3. Structural and Ultrastructural Analysis
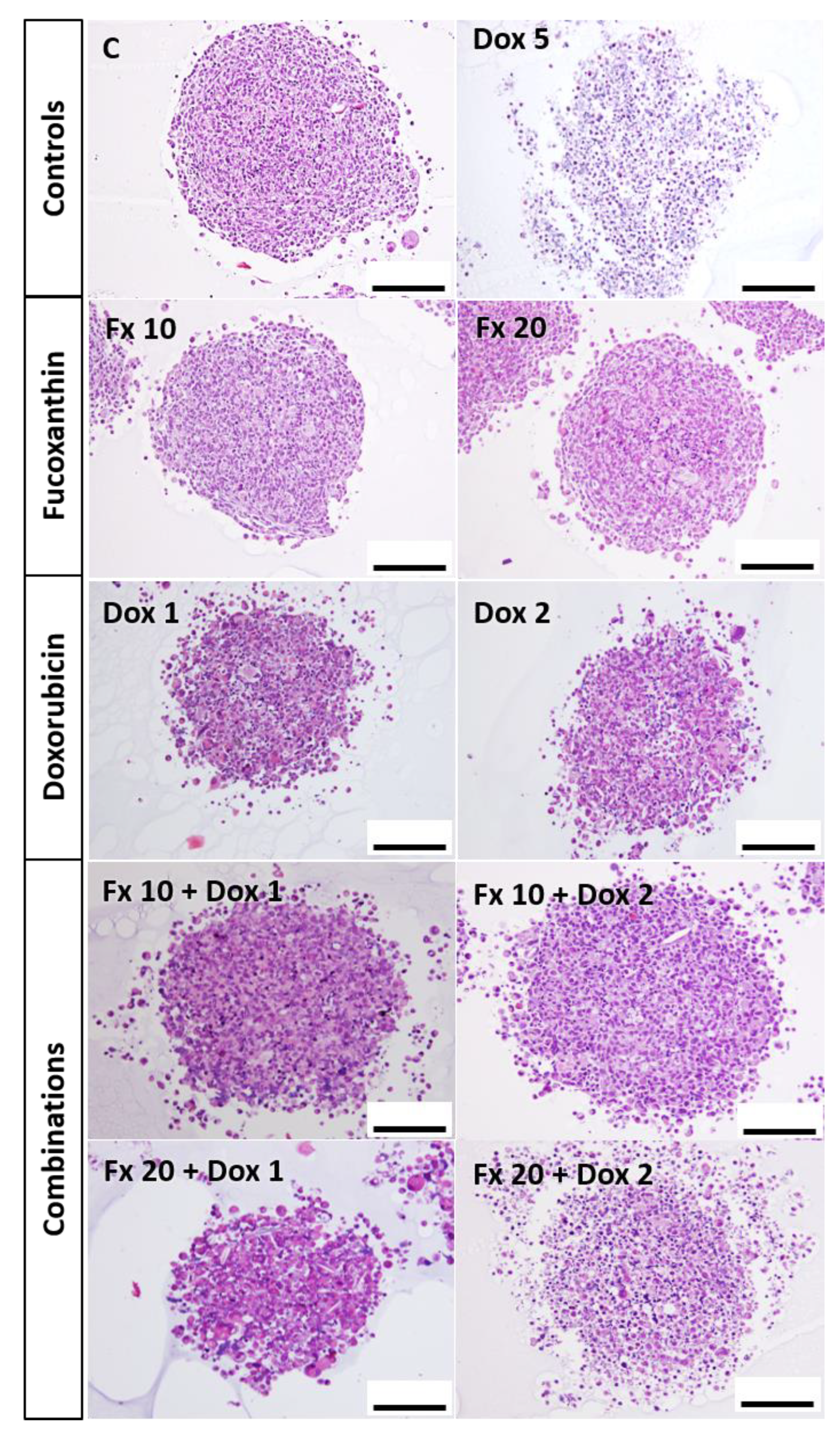
HE Staining
ICC
Transmission Electron Microscopy (TEM)
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Stock and Exposure Solutions
4.3. Cell Culture
4.3.1. 2D Cell Culture
4.3.2. 3D Cell Culture—Multicellular Aggregates (MCAs)
4.4. Study Design
4.5. Exposures (Single or Combination) in 2D Cell Cultures
4.5.1. MTT Assay
4.5.2. Nuclear Condensation Assay
4.5.3. Comet Assay
4.6. Exposures (Single or Combination) in 3D Cell Cultures
4.6.1. MTT Assay
4.6.2. Lactate Dehydrogenase (LDH) Assay
4.6.3. Stereomicroscopic Analysis and Area Measurements
4.6.4. Hematoxylin and Eosin (HE) Staining
4.6.5. Immunocytochemical Analysis
4.6.6. Transmission Electron Microscopy (TEM)
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Wright, G.D. Unlocking the potential of natural products in drug discovery. Microb. Biotechnol. 2019, 12, 55–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharifi-Rad, J.; Ozleyen, A.; Boyunegmez Tumer, T.; Oluwaseun Adetunji, C.; El Omari, N.; Balahbib, A.; Taheri, Y.; Bouyahya, A.; Martorell, M.; Martins, N.; et al. Natural products and synthetic analogs as a source of antitumor drugs. Biomolecules 2019, 9, 679. [Google Scholar] [CrossRef]
- Ruan, B.F.; Ge, W.W.; Lin, M.X.; Li, Q.S. A review of the components of seaweeds as potential candidates in cancer therapy. Curr. Med. Chem. Anticancer Agents 2018, 18, 354–366. [Google Scholar] [CrossRef]
- Schumacher, M.; Kelkel, M.; Dicato, M.; Diederich, M. Gold from the sea: Marine compounds as inhibitors of the hallmarks of cancer. Biotechnol. Adv. 2011, 29, 531–547. [Google Scholar] [CrossRef]
- Proksch, P.; Edrada-Ebel, R.; Ebel, R. Drugs from the sea—Opportunities and obstacles. Mar. Drugs 2003, 1, 5–17. [Google Scholar] [CrossRef] [Green Version]
- Khalifa, S.A.M.; Elias, N.; Farag, M.A.; Chen, L.; Saeed, A.; Hegazy, M.F.; Moustafa, M.S.; Abd El-Wahed, A.; Al-Mousawi, S.M.; Musharraf, S.G.; et al. Marine natural products: A source of novel anticancer drugs. Mar. Drugs 2019, 17, 491. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, P.C.; Wilke, D.V.; Branco, P.C.; Bauermeister, A.; Rezende-Teixeira, P.; Gaudêncio, S.P.; Costa-Lotufo, L.V. Enriching cancer pharmacology with drugs of marine origin. Br. J. Pharmacol. 2020, 177, 3–27. [Google Scholar] [CrossRef] [Green Version]
- Dyshlovoy, S.A.; Honecker, F. Marine compounds and cancer: The first two decades of XXI century. Mar. Drugs 2019, 18, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalid, S.; Abbas, M.; Saeed, F.; Bader-Ul-Ain, H.; Ansar Rasul Suleria, H. Therapeutic potential of seaweed bioactive compounds. In Seaweed Biomaterials; Maiti, S., Ed.; IntechOpen: London, UK, 2018; pp. 8–25. [Google Scholar] [CrossRef] [Green Version]
- Teas, J.; Vena, S.; Cone, D.L.; Irhimeh, M. The consumption of seaweed as a protective factor in the etiology of breast cancer: Proof of principle. J. Appl. Phycol. 2013, 25, 771–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.J.; Nam, S.-J.; Kong, G.; Kim, M.K. A case–control study on seaweed consumption and the risk of breast cancer. Br. J. Nutr. 2010, 103, 1345–1353. [Google Scholar] [CrossRef] [Green Version]
- Funahashi, H.; Imai, T.; Mase, T.; Sekiya, M.; Yokoi, K.; Hayashi, H.; Shibata, A.; Hayashi, T.; Nishikawa, M.; Suda, N.; et al. Seaweed prevents breast cancer? Jpn. J. Cancer Res. 2001, 92, 483–487. [Google Scholar] [CrossRef]
- Rwigemera, A.; Mamelona, J.; Martin, L.J. Inhibitory effects of fucoxanthinol on the viability of human breast cancer cell lines MCF-7 and MDA-MB-231 are correlated with modulation of the NF-kappaB pathway. Cell Biol. Toxicol. 2014, 30, 157–167. [Google Scholar] [CrossRef]
- Bae, M.; Kim, M.B.; Park, Y.K.; Lee, J.Y. Health benefits of fucoxanthin in the prevention of chronic diseases. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158618. [Google Scholar] [CrossRef]
- Martin, L.J. Fucoxanthin and its metabolite fucoxanthinol in cancer prevention and treatment. Mar. Drugs 2015, 13, 4784–4798. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.R.; Hosokawa, M.; Miyashita, K. Fucoxanthin: A marine carotenoid exerting anti-cancer effects by affecting multiple mechanisms. Mar. Drugs 2013, 11, 5130–5147. [Google Scholar] [CrossRef] [Green Version]
- Meresse, S.; Fodil, M.; Fleury, F.; Chenais, B. Fucoxanthin, a marine-derived carotenoid from brown seaweeds and microalgae: A promising bioactive compound for cancer therapy. Int. J. Mol. Sci. 2020, 21, 9273. [Google Scholar] [CrossRef]
- Garg, S.; Afzal, S.; Elwakeel, A.; Sharma, D.; Radhakrishnan, N.; Dhanjal, J.K.; Sundar, D.; Kaul, S.C.; Wadhwa, R. Marine carotenoid fucoxanthin possesses anti-metastasis activity: Molecular evidence. Mar. Drugs 2019, 17, 338. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Tang, Y.; Zhang, Y.; Zhang, S.; Qu, J.; Wang, X.; Kong, R.; Han, C.; Liu, Z. Fucoxanthin: A promising medicinal and nutritional ingredient. Evid. Based Complement. Alternat. Med. 2015, 2015, 723515. [Google Scholar] [CrossRef]
- Ishikawa, C.; Tafuku, S.; Kadekaru, T.; Sawada, S.; Tomita, M.; Okudaira, T.; Nakazato, T.; Toda, T.; Uchihara, J.N.; Taira, N.; et al. Anti-adult T-cell leukemia effects of brown algae fucoxanthin and its deacetylated product, fucoxanthinol. Int. J. Cancer 2008, 123, 2702–2712. [Google Scholar] [CrossRef]
- Satomi, Y. Antitumor and cancer-preventative function of fucoxanthin: A marine carotenoid. Anticancer Res. 2017, 37, 1557–1562. [Google Scholar] [CrossRef] [Green Version]
- Rwigemera, A.; Mamelona, J.; Martin, L.J. Comparative effects between fucoxanthinol and its precursor fucoxanthin on viability and apoptosis of breast cancer cell lines MCF-7 and MDA-MB-231. Anticancer Res. 2015, 35, 207–219. [Google Scholar] [PubMed]
- Wang, J.; Jiang, Y.-F. Natural compounds as anticancer agents: Experimental evidence. World J. Exp. Med. 2012, 2, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ma, Y.; Yang, J.; Jin, L.; Gao, Z.; Xue, L.; Hou, L.; Sui, L.; Liu, J.; Zou, X. Fucoxanthin inhibits tumour-related lymphangiogenesis and growth of breast cancer. J. Cell Mol. Med. 2019, 23, 2219–2229. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- World Health Organization. Breast Cancer—WHO|World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/breast-cancer (accessed on 1 May 2021).
- Fragomeni, S.M.; Sciallis, A.; Jeruss, J.S. Molecular Subtypes and Local-Regional Control of Breast Cancer. Surg. Oncol. Clin. N. Am. 2018, 27, 95–120. [Google Scholar] [CrossRef]
- Goldhirsch, A.; Winer, E.P.; Coates, A.S.; Gelber, R.D.; Piccart-Gebhart, M.; Thürlimann, B.; Senn, H.J. Personalizing the treatment of women with early breast cancer: Highlights of the St Gallen International Expert Consensus on the primary therapy of early breast cancer 2013. Ann. Oncol. 2013, 24, 2206–2223. [Google Scholar] [CrossRef]
- Yersal, O.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J. Clin. Oncol. 2014, 5, 412–424. [Google Scholar] [CrossRef]
- Dai, J.; Jian, J.; Bosland, M.; Frenkel, K.; Bernhardt, G.; Huang, X. Roles of hormone replacement therapy and iron in proliferation of breast epithelial cells with different estrogen and progesterone receptor status. Breast 2008, 17, 172–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trendowski, M. Recent advances in the development of antineoplastic agents derived from natural products. Drugs 2015, 75, 1993–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights into molecular classifications of triple-negative breast cancer: Improving patient selection for treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef] [Green Version]
- Yin, L.; Duan, J.J.; Bian, X.W.; Yu, S.C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef] [PubMed]
- Fisusi, F.A.; Akala, E.O. Drug combinations in breast cancer therapy. Pharm. Nanotechnol. 2019, 7, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Masui, K.; Gini, B.; Wykosky, J.; Zanca, C.; Mischel, P.S.; Furnari, F.B.; Cavenee, W.K. A tale of two approaches: Complementary mechanisms of cytotoxic and targeted therapy resistance may inform next-generation cancer treatments. Carcinogenesis 2013, 34, 725–738. [Google Scholar] [CrossRef]
- Amable, L. Cisplatin resistance and opportunities for precision medicine. Pharmacol. Res. 2016, 106, 27–36. [Google Scholar] [CrossRef]
- Pádua, D.; Rocha, E.; Gargiulo, D.; Ramos, A.A. Bioactive compounds from brown seaweeds: Phloroglucinol, fucoxanthin and fucoidan as promising therapeutic agents against breast cancer. PhytoChem. Lett. 2015, 14, 91–98. [Google Scholar] [CrossRef]
- Yun, C.W.; Kim, H.J.; Lee, S.H. Therapeutic application of diverse marine-derived natural products in cancer therapy. Anticancer Res. 2019, 39, 5261–5284. [Google Scholar] [CrossRef] [Green Version]
- Lopes-Costa, E.; Abreu, M.; Gargiulo, D.; Rocha, E.; Ramos, A.A. Anticancer effects of seaweed compounds fucoxanthin and phloroglucinol, alone and in combination with 5-fluorouracil in colon cells. J. Toxicol. Environ. Health A 2017, 80, 776–787. [Google Scholar] [CrossRef]
- Almeida, T.P.; Ferreira, J.; Vettorazzi, A.; Azqueta, A.; Rocha, E.; Ramos, A.A. Cytotoxic activity of fucoxanthin, alone and in combination with the cancer drugs imatinib and doxorubicin, in CML cell lines. Environ. Toxicol. Pharmacol. 2018, 59, 24–33. [Google Scholar] [CrossRef]
- Vijay, K.; Sowmya, P.R.; Arathi, B.P.; Shilpa, S.; Shwetha, H.J.; Raju, M.; Baskaran, V.; Lakshminarayana, R. Low-dose doxorubicin with carotenoids selectively alters redox status and upregulates oxidative stress-mediated apoptosis in breast cancer cells. Food Chem. Toxicol. 2018, 118, 675–690. [Google Scholar] [CrossRef]
- Wang, Z.; Li, H.; Dong, M.; Zhu, P.; Cai, Y. The anticancer effects and mechanisms of fucoxanthin combined with other drugs. J. Cancer Res. Clin. Oncol. 2019, 145, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Cherry, P.; O’Hara, C.; Magee, P.J.; McSorley, E.M.; Allsopp, P.J. Risks and benefits of consuming edible seaweeds. Nutr. Rev. 2019, 77, 307–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.B.; Stein, R.; O’Hare, M.J. Three-dimensional in vitro tissue culture models of breast cancer—A review. Breast Cancer Res. Treat. 2004, 85, 281–291. [Google Scholar] [CrossRef]
- Verjans, E.T.; Doijen, J.; Luyten, W.; Landuyt, B.; Schoofs, L. Three-dimensional cell culture models for anticancer drug screening: Worth the effort? J. Cell Physiol. 2018, 233, 2993–3003. [Google Scholar] [CrossRef] [PubMed]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, Ł.; Lamperska, K. 2D and 3D cell cultures—A comparison of different types of cancer cell cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef]
- Santo, V.E.; Rebelo, S.P.; Estrada, M.F.; Alves, P.M.; Boghaert, E.; Brito, C. Drug screening in 3D in vitro tumor models: Overcoming current pitfalls of efficacy read-outs. Biotechnol. J. 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Imamura, Y.; Mukohara, T.; Shimono, Y.; Funakoshi, Y.; Chayahara, N.; Toyoda, M.; Kiyota, N.; Takao, S.; Kono, S.; Nakatsura, T.; et al. Comparison of 2D- and 3D-culture models as drug-testing platforms in breast cancer. Oncol. Rep. 2015, 33, 1837–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konishi, I.; Hosokawa, M.; Sashima, T.; Kobayashi, H.; Miyashita, K. Halocynthiaxanthin and fucoxanthinol isolated from Halocynthia roretzi induce apoptosis in human leukemia, breast and colon cancer cells. Comp. BioChem. Physiol. C Toxicol. Pharmacol. 2006, 142, 53–59. [Google Scholar] [CrossRef]
- Yamamoto, K.; Ishikawa, C.; Katano, H.; Yasumoto, T.; Mori, N. Fucoxanthin and its deacetylated product, fucoxanthinol, induce apoptosis of primary effusion lymphomas. Cancer Lett. 2011, 300, 225–234. [Google Scholar] [CrossRef]
- De la Mare, J.A.; Sterrenberg, J.N.; Sukhthankar, M.G.; Chiwakata, M.T.; Beukes, D.R.; Blatch, G.L.; Edkins, A.L. Assessment of potential anti-cancer stem cell activity of marine algal compounds using an in vitro mammosphere assay. Cancer Cell Int. 2013, 13, 39. [Google Scholar] [CrossRef] [Green Version]
- Møller, P. The alkaline comet assay: Towards validation in biomonitoring of DNA damaging exposures. Basic. Clin. Pharmacol. Toxicol. 2006, 98, 336–345. [Google Scholar] [CrossRef]
- Leon-Galicia, I.; Diaz-Chavez, J.; Albino-Sanchez, M.E.; Garcia-Villa, E.; Bermudez-Cruz, R.; Garcia-Mena, J.; Herrera, L.A.; García-Carrancá, A.; Gariglio, P. Resveratrol decreases Rad51 expression and sensitizes cisplatin-resistant MCF-7 breast cancer cells. Oncol. Rep. 2018, 39, 3025–3033. [Google Scholar] [CrossRef]
- Lee, K.S.; Lee, M.G.; Kwon, Y.S.; Nam, K.S. Arctigenin enhances the cytotoxic effect of doxorubicin in MDA-MB-231 breast cancer cells. Int. J. Mol. Sci. 2020, 21, 2997. [Google Scholar] [CrossRef]
- Antberg, L.; Cifani, P.; Levander, F.; James, P. Pathway-centric analysis of the DNA damage response to chemotherapeutic agents in two breast cell lines. EuPA Open Proteom. 2015, 8, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Pilco-Ferreto, N.; Calaf, G.M. Influence of doxorubicin on apoptosis and oxidative stress in breast cancer cell lines. Int. J. Oncol. 2016, 49, 753–762. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Teves, S.S.; Kemp, C.J.; Henikoff, S. Doxorubicin, DNA torsion, and chromatin dynamics. Biochim. Biophys. Acta 2014, 1845, 84–89. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Lee, Y.S.; Kim, D.K. Doxorubicin exerts cytotoxic effects through cell cycle arrest and Fas-mediated cell death. Pharmacology 2009, 84, 300–309. [Google Scholar] [CrossRef]
- Lin, S.R.; Chang, C.H.; Hsu, C.F.; Tsai, M.J.; Cheng, H.; Leong, M.K.; Sung, P.J.; Chen, J.C.; Weng, C.F. Natural compounds as potential adjuvants to cancer therapy: Preclinical evidence. Br. J. Pharmacol. 2020, 177, 1409–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liston, D.R.; Davis, M. Clinically relevant concentrations of anticancer drugs: A guide for nonclinical studies. Clin. Cancer Res. 2017, 23, 3489–3498. [Google Scholar] [CrossRef] [Green Version]
- Menéndez-Menéndez, J.; Hermida-Prado, F.; Granda-Díaz, R.; González, A.; García-Pedrero, J.M.; Del-Río-Ibisate, N.; González-González, A.; Cos, S.; Alonso-González, C.; Martínez-Campa, C. Deciphering the molecular basis of melatonin protective effects on breast cells treated with doxorubicin: TWIST1 a transcription factor involved in EMT and metastasis, a novel target of melatonin. Cancers 2019, 11, 1011. [Google Scholar] [CrossRef] [Green Version]
- Prabhakaran, P.; Hassiotou, F.; Blancafort, P.; Filgueira, L. Cisplatin induces differentiation of breast cancer cells. Front. Oncol. 2013, 3, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosokawa, M.; Kudo, M.; Maeda, H.; Kohno, H.; Tanaka, T.; Miyashita, K. Fucoxanthin induces apoptosis and enhances the antiproliferative effect of the PPARgamma ligand, troglitazone, on colon cancer cells. Biochim. Biophys. Acta 2004, 1675, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zeng, Y.; Liu, Y.; Hu, X.; Li, S.; Wang, Y.; Li, L.; Lei, Z.; Zhang, Z. Fucoxanthin induces growth arrest and apoptosis in human bladder cancer T24 cells by up-regulation of p21 and down-regulation of mortalin. Acta Biochim. Biophys. Sin. 2014, 46, 877–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.L.; Fu, X.Y.; Cao, W.Q.; Xiang, W.Z.; Hou, Y.J.; Ma, J.K.; Wang, Y.; Fan, C.D. Induction of apoptosis in human glioma cells by fucoxanthin via triggering of ROS-mediated oxidative damage and regulation of MAPKs and PI3K-AKT pathways. J. Agric. Food Chem. 2019, 67, 2212–2219. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Song, M.-H.; Oh, J.-W.; Keum, Y.-S.; Saini, R.K. Pro-oxidant actions of carotenoids in triggering apoptosis of cancer cells: A review of emerging evidence. Antioxidants 2020, 9, 532. [Google Scholar] [CrossRef]
- Liu, C.L.; Chiu, Y.T.; Hu, M.L. Fucoxanthin enhances HO-1 and NQO1 expression in murine hepatic BNL CL.2 cells through activation of the Nrf2/ARE system partially by its pro-oxidant activity. J. Agric. Food Chem. 2011, 59, 11344–11351. [Google Scholar] [CrossRef]
- Zurina, I.M.; Gorkun, A.A.; Dzhussoeva, E.V.; Kolokoltsova, T.D.; Markov, D.D.; Kosheleva, N.V.; Morozov, S.G.; Saburina, I.N. Human melanocyte-derived spheroids: A precise test system for drug screening and a multicellular unit for tissue engineering. Front. Bioeng. Biotechnol. 2020, 8, 540. [Google Scholar] [CrossRef]
- Terasaki, M.; Maeda, H.; Miyashita, K.; Mutoh, M. Induction of anoikis in human colorectal cancer cells by fucoxanthinol. Nutr. Cancer 2017, 69, 1–10. [Google Scholar] [CrossRef]
- Terasaki, M.; Matsumoto, N.; Hashimoto, R.; Endo, T.; Maeda, H.; Hamada, J.; Osada, K.; Miyashita, K.; Mutoh, M. Fucoxanthin administration delays occurrence of tumors in xenograft mice by colonospheres, with an anti-tumor predictor of glycine. J. Clin. BioChem. Nutr. 2019, 64, 52–58. [Google Scholar] [CrossRef] [Green Version]
- Hongisto, V.; Jernström, S.; Fey, V.; Mpindi, J.P.; Kleivi Sahlberg, K.; Kallioniemi, O.; Perälä, M. High-throughput 3D screening reveals differences in drug sensitivities between culture models of JIMT1 breast cancer cells. PLoS ONE 2013, 8, e77232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovitt, C.J.; Shelper, T.B.; Avery, V.M. Doxorubicin resistance in breast cancer cells is mediated by extracellular matrix proteins. BMC Cancer 2018, 18, 41. [Google Scholar] [CrossRef] [Green Version]
- Prabst, K.; Engelhardt, H.; Ringgeler, S.; Hubner, H. Basic. colorimetric proliferation assays: MTT, WST, and Resazurin. Methods Mol. Biol. 2017, 1601, 1–17. [Google Scholar] [CrossRef]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of cell viability by the lactate dehydrogenase assay. Cold Spring Harb. Protoc. 2018, 2018. [Google Scholar] [CrossRef]
- Malhão, F.; Ramos, A.A.; Macedo, A.C.; Rocha, E. Cytotoxicity of seaweed compounds, alone or combined to reference drugs, against breast cell lines cultured in 2D and 3D. Toxics 2021, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Yu, P.; Tang, J. Characterization of triple-negative breast cancer MDA-MB-231 cell spheroid model. Oncol. Targets Ther. 2020, 13, 5395–5405. [Google Scholar] [CrossRef]
- Fischer, U.; Schulze-Osthoff, K. Apoptosis-based therapies and drug targets. Cell Death Differ. 2005, 12, 942–961. [Google Scholar] [CrossRef]
- Chen, D.L.; Engle, J.T.; Griffin, E.A.; Miller, J.P.; Chu, W.; Zhou, D.; Mach, R.H. Imaging caspase-3 activation as a marker of apoptosis-targeted treatment response in cancer. Mol. Imaging Biol. 2015, 17, 384–393. [Google Scholar] [CrossRef] [Green Version]
- Muftah, A.A.; Aleskandarany, M.A.; Al-Kaabi, M.M.; Sonbul, S.N.; Diez-Rodriguez, M.; Nolan, C.C.; Caldas, C.; Ellis, I.O.; Rakha, E.A.; Green, A.R. Ki67 expression in invasive breast cancer: The use of tissue microarrays compared with whole tissue sections. Breast Cancer Res. Treat. 2017, 164, 341–348. [Google Scholar] [CrossRef] [Green Version]
- Soliman, N.A.; Yussif, S.M. Ki-67 as a prognostic marker according to breast cancer molecular subtype. Cancer Biol. Med. 2016, 13, 496–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershey, B.J.; Vazzana, R.; Joppi, D.L.; Havas, K.M. Lipid droplets define a sub-population of breast cancer stem cells. J. Clin. Med. 2019, 9, 87. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Fujita, A.; Ohsaki, Y.; Suzuki, M.; Shinohara, Y.; Fujimoto, T. Quantitative electron microscopy shows uniform incorporation of triglycerides into existing lipid droplets. HistoChem. Cell Biol. 2009, 132, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Gammone, M.A.; D’Orazio, N. Anti-obesity activity of the marine carotenoid fucoxanthin. Mar. Drugs 2015, 13, 2196–2214. [Google Scholar] [CrossRef]
- Shyu, P., Jr.; Wong, X.; Fah, A.; Crasta, K.; Thibault, G. Dropping in on lipid droplets: Insights into cellular stress and cancer. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Bojko, A.; Staniak, K.; Czarnecka-Herok, J.; Sunderland, P.; Dudkowska, M.; Śliwińska, M.A.; Salmina, K.; Sikora, E. Improved autophagic flux in escapers from doxorubicin-induced senescence/polyploidy of breast cancer cells. Int. J. Mol. Sci. 2020, 21, 6084. [Google Scholar] [CrossRef] [PubMed]
- Holliday, D.L.; Speirs, V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011, 13, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subik, K.; Lee, J.-F.; Baxter, L.; Strzepek, T.; Costello, D.; Crowley, P.; Xing, L.; Hung, M.-C.; Bonfiglio, T.; Hicks, D.G.; et al. The expression patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by immunohistochemical analysis in breast cancer cell lines. Breast Cancer 2010, 4, 35–41. [Google Scholar] [CrossRef]
- Malhão, F.; Ramos, A.A.; Buttachon, S.; Dethoup, T.; Kijjoa, A.; Rocha, E. Cytotoxic and antiproliferative effects of preussin, a hydroxypyrrolidine derivative from the marine sponge-associated fungus Aspergillus candidus KUFA 0062, in a panel of breast cancer cell lines and using 2D and 3D cultures. Mar. Drugs 2019, 17, 448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccinini, F. AnaSP: A software suite for automatic image analysis of multicellular spheroids. Comput. Methods Programs Biomed. 2015, 119, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Sales Gil, R.; Vagnarelli, P. Ki-67: More hidden behind a ‘classic proliferation marker’. Trends BioChem. Sci. 2018, 43, 747–748. [Google Scholar] [CrossRef]
- Bressenot, A.; Marchal, S.; Bezdetnaya, L.; Garrier, J.; Guillemin, F.; Plenat, F. Assessment of apoptosis by immunohistochemistry to active caspase-3, active caspase-7, or cleaved PARP in monolayer cells and spheroid and subcutaneous xenografts of human carcinoma. J. HistoChem. CytoChem. 2009, 57, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Hammer, O.; Harper, D.; Ryan, P. PAST: Paleontological Statistics Software Package for education and data analysis. Palaeontol. Electron. 2001, 4, 1–9. [Google Scholar]
- Gaetano, J. Holm-Bonferroni Sequential Correction: An Excel Calculator (v. 1.3) Microsoft Excel Workbook. Available online: https://www.researchgate.net/publication/322569220_Holm-Bonferroni_sequential_correction_An_Excel_calculator_13 (accessed on 1 June 2021).
- Holm, S. A simple sequentially rejective multiple test procedure. Scand. Stat. Theory Appl. 1979, 6, 65–70. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Fucoxanthin (Fx) | Cisplatin (Cis) | Doxorubicin (Dox) | |
|---|---|---|---|---|
| Cell Line | ||||
| MCF7 | 1; 10 µM | 10; 20 µM | 0.01; 0.1 µM | |
| SKBR3 | ||||
| MDA-MB-231 | 0.1; 1 µM | |||
| MCF12A | 1; 10 µM | 0.01; 0.1 µM | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malhão, F.; Macedo, A.C.; Costa, C.; Rocha, E.; Ramos, A.A. Fucoxanthin Holds Potential to Become a Drug Adjuvant in Breast Cancer Treatment: Evidence from 2D and 3D Cell Cultures. Molecules 2021, 26, 4288. https://doi.org/10.3390/molecules26144288
Malhão F, Macedo AC, Costa C, Rocha E, Ramos AA. Fucoxanthin Holds Potential to Become a Drug Adjuvant in Breast Cancer Treatment: Evidence from 2D and 3D Cell Cultures. Molecules. 2021; 26(14):4288. https://doi.org/10.3390/molecules26144288
Chicago/Turabian StyleMalhão, Fernanda, Ana Catarina Macedo, Carla Costa, Eduardo Rocha, and Alice Abreu Ramos. 2021. "Fucoxanthin Holds Potential to Become a Drug Adjuvant in Breast Cancer Treatment: Evidence from 2D and 3D Cell Cultures" Molecules 26, no. 14: 4288. https://doi.org/10.3390/molecules26144288