
Bioassay-Guided Isolation of Two Eudesmane Sesquiterpenes from Lindera strychnifolia Using Centrifugal Partition Chromatography

 ,
,
Abstract
:1. Introduction
2. Results
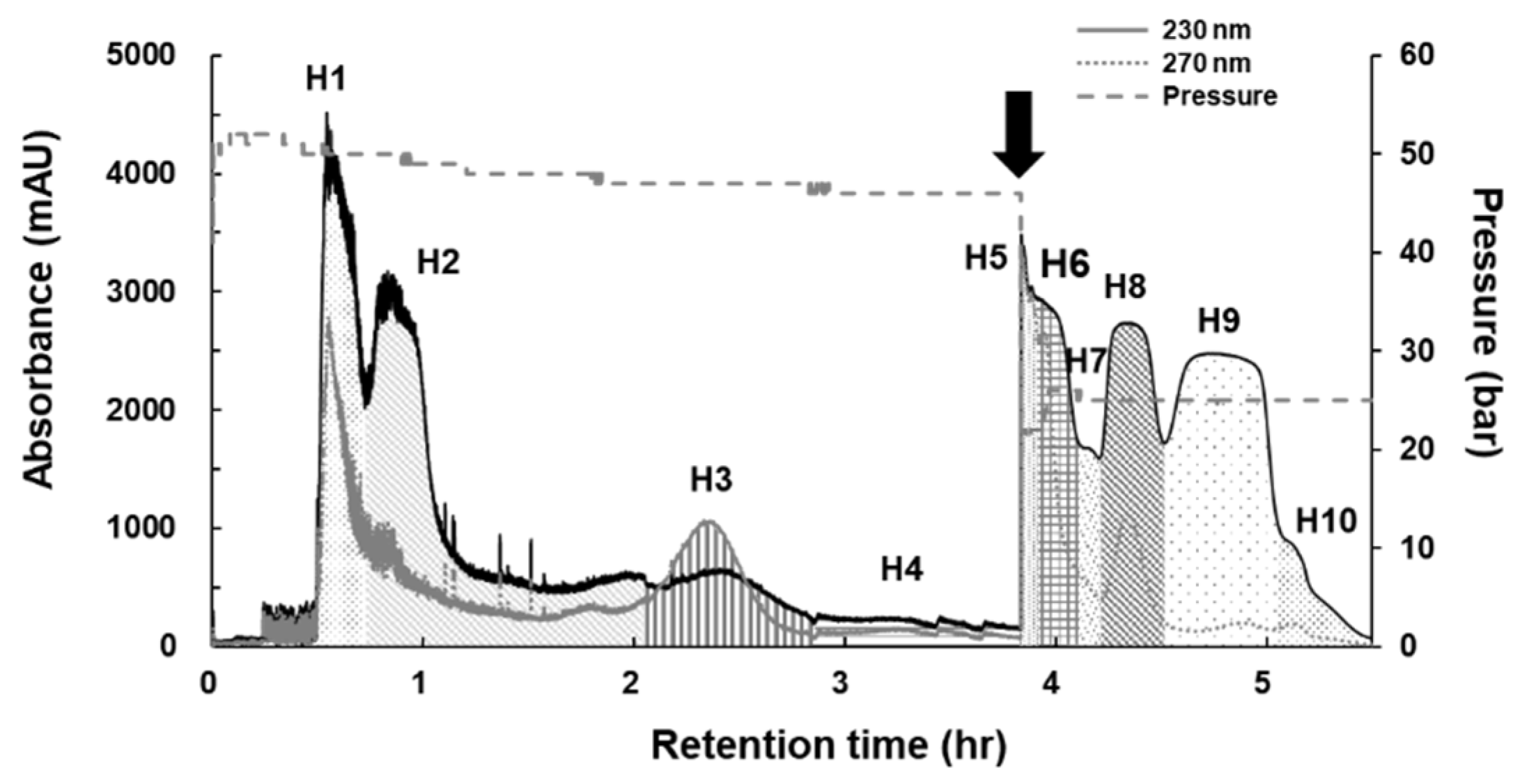
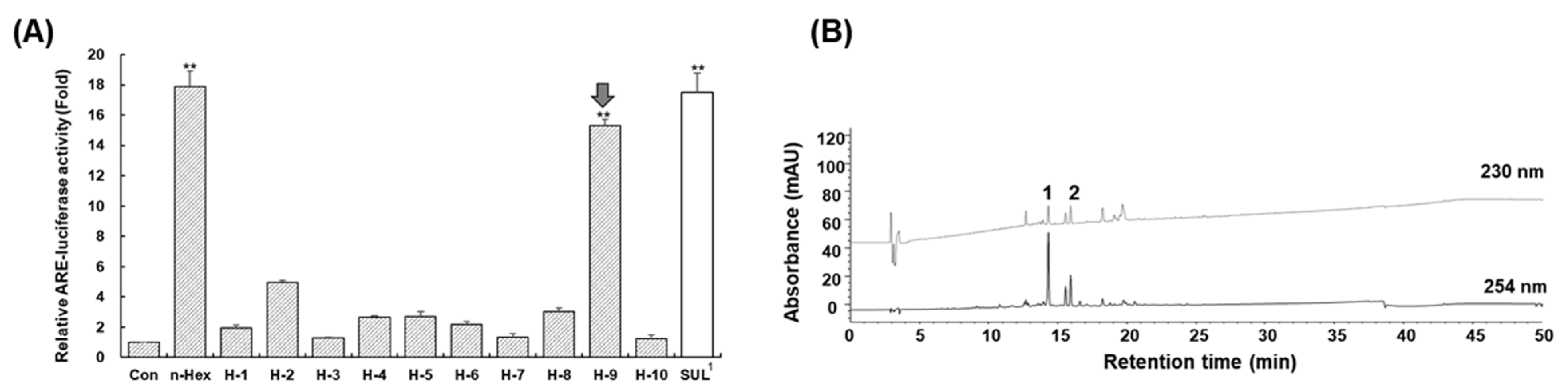
2.1. Isolation of Active Compounds Using Dual-Mode CPC and HPLC
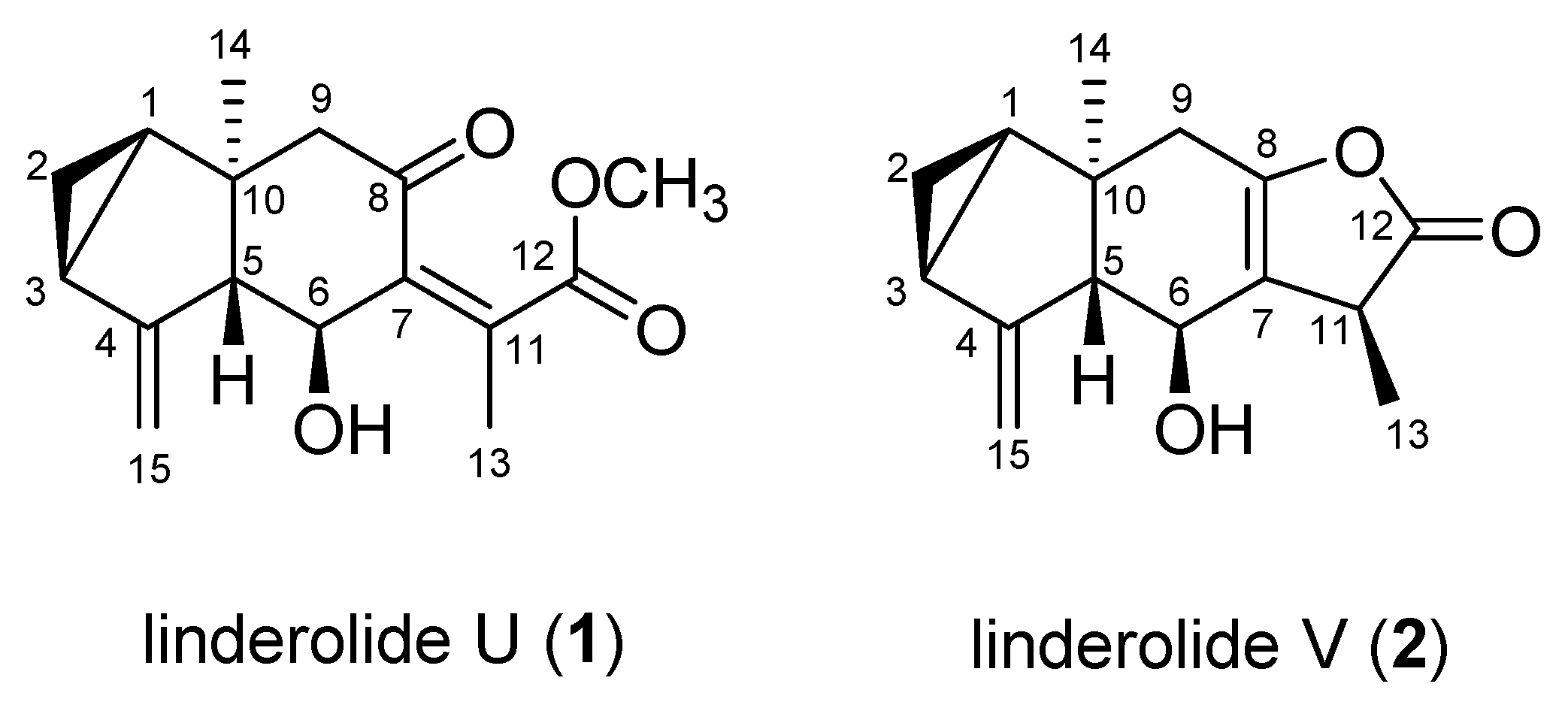
2.2. Structural Elucidation of Compounds 1 and 2
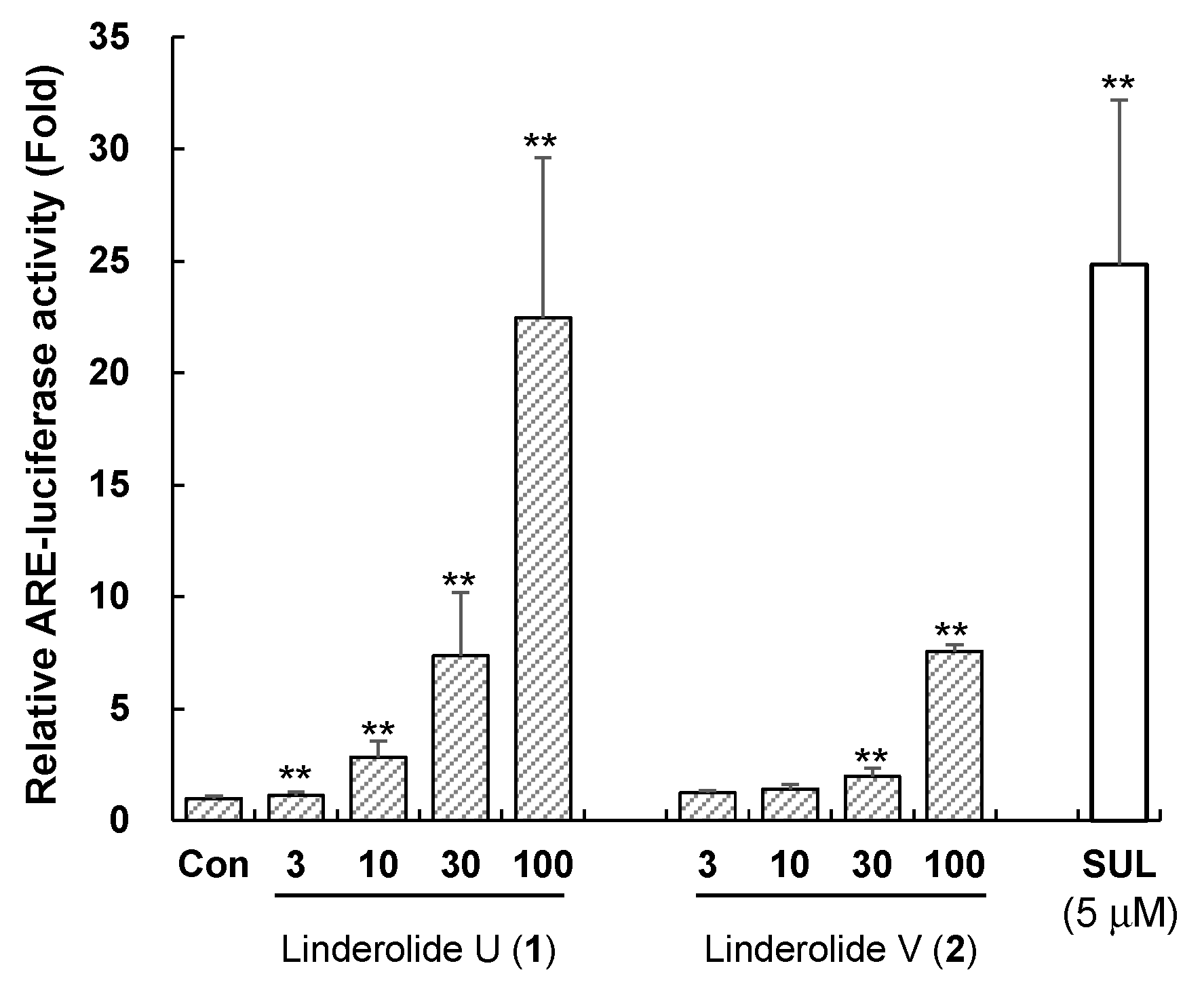
2.3. Induction of Antioxidant Response Element by Compounds 1 and 2
3. Materials and Methods
3.1. Apparatus
3.2. Chemicals and Reagents
3.3. Preparation of Crude Samples
3.4. HPLC Analysis
3.5. Selection of Solvent System
3.6. CPC Operation for Active n-Hexane Extract
3.7. Isolation and Structural Elucidation of Active Compounds 1 and 2
3.8. Preparation of the (R)- and (S)-MTPA Ester Derivatives
3.9. Assay of ARE-Inducing Activities
3.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nothias, L.F.; Nothias-Esposito, M.; da Silva, R.; Wang, M.; Protsyuk, I.; Zhang, Z.; Sarvepalli, A.; Leyssen, P.; Touboul, D.; Costa, J.; et al. Bioactivity-Based Molecular Networking for the Discovery of Drug Leads in Natural Product Bioassay-Guided Fractionation. J. Nat. Prod. 2018, 81, 758–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Jung, E.J.; Lee, Y.J.; Gao, E.M.; Syed, A.S.; Kim, C.Y. Bioassay-Guided Separation of Centipeda minima Using Comprehensive Linear Gradient Centrifugal Partition Chromatography. Molecules 2020, 25, 3077. [Google Scholar] [CrossRef]
- Wang, D.; Khan, M.S.; Cui, L.; Song, X.Y.; Zhu, H.; Ma, T.Y.; Sun, R. Preparative separation of mangiferin glycosides by high speed counter current chromatography and comparison of their antioxidant and antitumor activities. RSC Adv. 2019, 9, 4892–4899. [Google Scholar] [CrossRef] [Green Version]
- Santos, J.H.; Almeida, M.R.; Martins, C.I.; Dias, A.C.; Freire, M.G.; Coutinho, J.A.; Ventura, S.P. Separation of phenolic compounds by centrifugal partition chromatography. Continuous separation of cytochrome-c PEGylated conjugates by fast centrifugal partition chromatography. Green Chem. 2018, 20, 1906–1916. [Google Scholar] [CrossRef]
- da Silva, L.A.L.; Sandjo, L.P.; Fratoni, E.; Moon, Y.J.K.; Dalmarco, E.M.; Biavatti, M.W. A single-step isolation by centrifugal partition chromatography of the potential anti-inflammatory glaucolide B from Lepidaploa chamissonis. J. Chromatogr. A 2019, 1605, 460362. [Google Scholar] [CrossRef]
- Abedini, A.; Chollet, S.; Angelis, A.; Borie, N.; Nuzillard, J.M.; Skaltsounis, A.L.; Reynaud, R.; Gangloff, S.C.; Renault, J.-H.; Hubert, J. Bioactivity-guided identification of antimicrobial metabolites in Alnus glutinosa bark and optimization of oregonin purification by Centrifugal Partition Chromatography. J. Chromatogr. B 2016, 1029, 121–127. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, C.; Zhang, Z.; Wang, J.; Wu, G.; Li, S.J. Comprehensive separation and identification of chemical constituents from Apocynum venetum leaves by high-performance counter-current chromatography and high performance liquid chromatography coupled with mass spectrometry. Chromatogr. B 2010, 878, 3149–3155. [Google Scholar] [CrossRef]
- Wang, Z.; Hwang, S.H.; Lim, S.S. Comprehensive profiling of minor tyrosinase inhibitors from Gastrodia elata using an off-line hyphenation of ultrafiltration, high-speed countercurrent chromatography, and high-performance liquid chromatography. J. Chromatogr. A 2017, 1529, 63–71. [Google Scholar] [CrossRef]
- Karkoula, E.; Angelis, A.; Koulakiotis, N.S.; Gikas, E.; Halabalaki, M.; Tsarbopoulos, A.; Skaltsounis, A.L. Rapid isolation and characterization of crocins, picrocrocin, and crocetin from saffron using centrifugal partition chromatography and LC-MS. J. Sep. Sci. 2018, 41, 4105–4114. [Google Scholar] [CrossRef]
- Kim, M.I.; Kim, J.H.; Syed, A.S.; Kim, Y.M.; Choe, K.K.; Kim, C.Y. Application of Centrifugal Partition Chromatography for Bioactivity-Guided Purification of Antioxidant-Response-Element-Inducing Constituents from Atractylodis Rhizoma Alba. Molecules 2018, 23, 2274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mroczek, T.; Dymek, A.; Widelski, J.; Wojtanowski, K.K. The Bioassay-Guided Fractionation and Identification of Potent Acetylcholinesterase Inhibitors from Narcissus c.v. ‘Hawera’ Using Optimized Vacuum Liquid Chromatography, High Resolution Mass Spectrometry and Bioautography. Metabolites 2020, 10, 395. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Sun, X.; Zhao, L.; Wang, X.; Wang, X. An efficient method to obtain anti-inflammatory phenolic derivatives from Scindapsus officinalis (Roxb.) Schott. by a high speed counter-current chromatography coupled with a recycling mode. RSC Adv. 2020, 10, 11132–11138. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Xuan, B.; Peng, B.; Li, C.; Chai, X.; Tu, P. The genus Lindera: A source of structurally diverse molecules having pharmacological significance. Phytochem. Rev. 2016, 15, 869–906. [Google Scholar] [CrossRef]
- Liu, Q.; Ahn, J.H.; Kim, S.B.; Lee, C.; Hwang, B.Y.; Lee, M.K. Sesquiterpene lactones from the roots of Lindera strychnifolia. Phytochemistry 2013, 87, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Sumioka, H.; Harinantenaina, L.; Matsunami, K.; Otsuka, H.; Kawahata, M.; Yamaguchi, K. Linderolides A–F, eudesmane-type sesquiterpene lactones and linderoline, a germacrane-type sesquiterpene from the roots of Lindera strychnifolia and their inhibitory activity on NO production in RAW 264.7 cells in vitro. Phytochemistry 2011, 72, 2165–2171. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Jo, Y.H.; Kim, S.B.; Jin, Q.; Hwang, B.Y.; Lee, M.K. Sesquiterpenes from the roots of Lindera strychnifolia with inhibitory effects on nitric oxide production in RAW 264.7 cells. Bioorg. Med. Chem. Lett. 2016, 26, 4950–4954. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Jeong, G.S.; Kang, D.G.; Lee, H.S.; Kim, Y.C. Cytoprotective effects of lindenenyl acetate isolated from Lindera strychnifolia on mouse hippocampal HT22 cells. Eur. J. Pharmacol. 2009, 614, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Han, F.; Luan, S.S.; Ai, R.; Zhang, P.; Li, H.; Chen, L.X. Triterpenoids from Ganoderma lucidum and Their Potential Anti-inflammatory Effects. J. Agric. Food Chem. 2019, 67, 5147–5158. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.J.; Kwon, E.B.; Li, W. Linderolide U, a new sesquiterpene from Lindera aggregata root. Nat. Prod. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, J.; Tahara, S.; Mizutani, J.; Furusaki, A.; Hashiba, N.; Matsumoto, T. Shizukanolides, Two Sesquiterpenoids from Chloranthus japonicus (Chloranthaceae). Agric. Biol. Chem. 1979, 43, 885–887. [Google Scholar] [CrossRef]

 C) and NOESY (H
C) and NOESY (H  H) correlations of compound 2.
H) correlations of compound 2.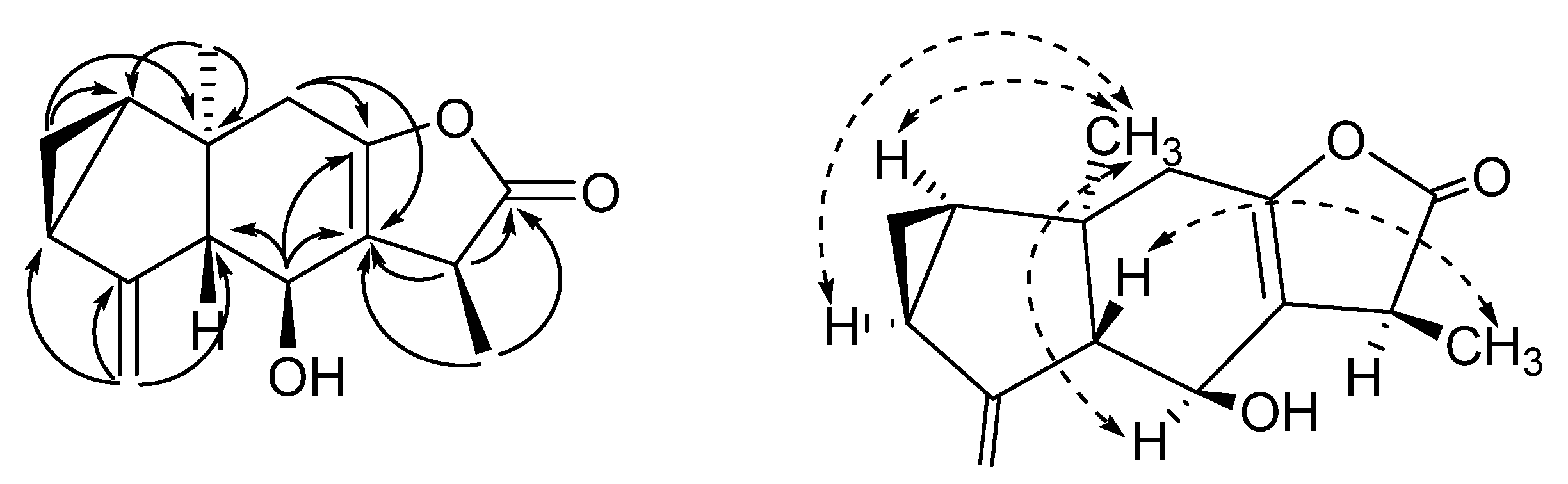

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent System (v/v/v) n-Hexane:Methanol:Water | Partition Coefficient (K 1) | ||||
|---|---|---|---|---|---|
| Peak a | Peak b | Peak c | Peak d | Peak e | |
| 10:9.5:0.5 | 5.47 | 4.22 | 3.90 | 2.86 | 0.67 |
| 10:9:1 | 5.02 | 3.51 | 3.67 | 2.15 | 0.45 |
| 10:8.5:1.5 | 4.28 | 2.65 | 2.78 | 1.84 | 0.29 |
| 10:8:2 | 2.29 | 1.54 | 1.46 | 1.06 | 0.17 |
| Fractions | H1 | H2 | H3 | H4 | H5 | H6 | H7 | H8 | H9 | H10 | Sum |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Weight (mg) | 940.8 | 964.1 | 89.1 | 114.0 | 550.4 | 417.5 | 45.2 | 339.8 | 1226.5 | 88.9 | 4776.3 |
| Weight ratio (%) | 19.7 | 20.2 | 1.9 | 2.4 | 11.5 | 8.7 | 0.9 | 7.1 | 25.7 | 1.9 | 100 |
| Treatment (µg/mL) | 5.91 | 6.06 | 0.56 | 0.72 | 3.46 | 2.62 | 0.28 | 2.13 | 7.70 | 1.86 | 30 |
| No. | Linderolide U (1) | Linderolide V (2) | ||||
|---|---|---|---|---|---|---|
| 1H (J in Hz) | 13C | 1H (J in Hz) | 13C | |||
| 1 | 1.45 (ddd, J = 8.0, 7.1, 3.6) | 29.1 | CH | 1.52 (dt, J = 7.5, 3.9) | 28.4 | CH |
| 2 | 0.90 (ddd, J = 8.9, 8.0, 5.2), 0.81 (dt, J = 5.2, 3.6) | 17.6 | CH2 | 0.88 (ddd, J = 10.7, 7.9, 4.2) | 18.5 | CH2 |
| 3 | 2.04 (m) | 24.4 | CH | 1.98 (t, J = 9.4) | 23.7 | CH |
| 4 | 152.0 | C | 151.3 | C | ||
| 5 | 2.79 (ddd, J = 11.0, 3.2, 2.2) | 67.2 | CH | 2.69 (dt, J = 9.6, 2.6) | 67.4 | CH |
| 6 | 4.42 (dd, J = 11.0, 0.9) | 65.9 | CH | 4.10 (d, J = 9.6) | 64.1 | CH |
| 7 | 139.9 | C | 120.9 | C | ||
| 8 | 202.3 | C | 151.2 | C | ||
| 9 | 2.58 (dd, J = 4.3, 0.9) | 54.4 | CH2 | 2.59 (dt, J = 16.3, 3.5), 2.36 (d, J = 16.3) | 38.1 | CH2 |
| 10 | 37.7 | C | 40.6 | C | ||
| 11 | 146.0 | C | 3.37 (m) | 39.8 | CH | |
| 12 | 173.5 | C | 181.7 | C | ||
| 13 | 2.10 (d, J = 0.9) | 17.9 | CH3 | 1.33 (d, J = 7.6) | 14.3 | CH3 |
| 14 | 0.74 (s) | 20.2 | CH3 | 0.71 (s) | 17.3 | CH3 |
| 15 | 5.24 (d, J = 1.4), 5.10 (dt, J = 2.7, 1.4) | 108.6 | CH2 | 5.20 (brs), 5.06 (d, J = 1.2) | 108.6 | CH2 |
| OCH3 | 3.72 (s, 3H) | 52.8 | CH3 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.H.; Jeon, J.-S.; Kim, J.H.; Jung, E.J.; Lee, Y.J.; Gao, E.M.; Syed, A.S.; Son, R.H.; Kim, C.Y. Bioassay-Guided Isolation of Two Eudesmane Sesquiterpenes from Lindera strychnifolia Using Centrifugal Partition Chromatography. Molecules 2021, 26, 5269. https://doi.org/10.3390/molecules26175269
Kim JH, Jeon J-S, Kim JH, Jung EJ, Lee YJ, Gao EM, Syed AS, Son RH, Kim CY. Bioassay-Guided Isolation of Two Eudesmane Sesquiterpenes from Lindera strychnifolia Using Centrifugal Partition Chromatography. Molecules. 2021; 26(17):5269. https://doi.org/10.3390/molecules26175269
Chicago/Turabian StyleKim, Ji Hoon, Je-Seung Jeon, Jung Hoon Kim, Eun Ju Jung, Yun Jung Lee, En Mei Gao, Ahmed Shah Syed, Rak Ho Son, and Chul Young Kim. 2021. "Bioassay-Guided Isolation of Two Eudesmane Sesquiterpenes from Lindera strychnifolia Using Centrifugal Partition Chromatography" Molecules 26, no. 17: 5269. https://doi.org/10.3390/molecules26175269