
Modulation of the Activity and Regioselectivity of a Glycosidase: Development of a Convenient Tool for the Synthesis of Specific Disaccharides

 , and
, and
Abstract
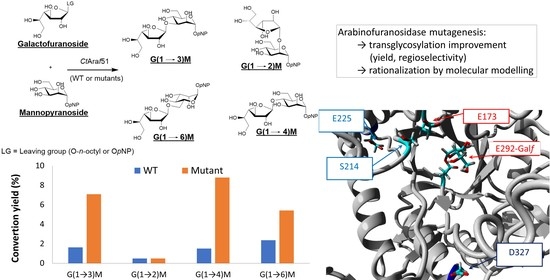
:
1. Introduction
2. Results and Discussion
2.1. Transglycosylation Profiles with p-Nitrophenyl α-d-Mannopyranoside (Manp-pNP) as Acceptor Using the Wild-Type (WT) CtAraf51 Enzyme
2.2. Mutagenesis Strategy
2.3. Transglycosylation Kinetics with the Selected Mutants
2.4. Molecular Modelling
2.4.1. Methodology and Molecular Dynamics (MD) on Glycosyl-Enzymes
2.4.2. Simulations of the Acceptor Approaching Glycosyl-Enzymes
3. Materials and Methods
3.1. Materials
3.2. Activity Units
3.3. Mutagenesis and Screening
3.3.1. Random Mutagenesis
3.3.2. Pre-Screen on Solid State Medium
3.3.3. Screening Methodology
3.4. General Procedure for Time Course Monitoring of Enzymatic Assays
3.5. Preparative Scale Synthesis of G(1→X)M Disaccharides
3.6. Molecular Modelling
3.6.1. Computational Methods and Details
3.6.2. Preparative Molecular Dynamics on Glycosyl-Enzyme
3.6.3. Local Dockings of Disaccharides and Elaboration of Manp-pNP Complexes
3.6.4. Molecular Dynamics of Glycosyl-Enzyme Complexes with Manp-pNP
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- WHO. Leishmaniasis. Available online: https://www.who.int/health-topics/leishmaniasis#tab=tab_3 (accessed on 27 June 2021).
- Novozhilova, N.M.; Bovin, N.V. Structure, Functions, and Biosynthesis of Glycoconjugates of Leishmania spp. Cell Surface. Biochem. Mosc. 2010, 75, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Peltier, P.; Euzen, R.; Daniellou, R.; Nugier-Chauvin, C.; Ferrières, V. Recent Knowledge and Innovations Related to Hexofuranosides: Structure, Synthesis and Applications. Carbohydr. Res. 2008, 343, 1897–1923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Brito, R.C.F.; Aguiar-Soares, R.D.O.; Cardoso, J.M.O.; Coura-Vital, W.; Roatt, B.M.; Reis, A.B. Recent Advances and New Strategies in Leishmaniasis Diagnosis. Appl. Microbiol. Biotechnol. 2020, 104, 8105–8116. [Google Scholar] [CrossRef] [PubMed]
- Ruda, K.; Lindberg, J.; Garegg, P.J.; Oscarson, S.; Konradsson, P. Synthesis of the Leishmania LPG Core Heptasaccharyl Myo-Inositol. J. Am. Chem. Soc. 2000, 122, 11067–11072. [Google Scholar] [CrossRef]
- Dureau, R.; Robert-Gangneux, F.; Gangneux, J.-P.; Nugier-Chauvin, C.; Legentil, L.; Daniellou, R.; Ferrières, V. Synthetic UDP-Furanoses Inhibit the Growth of the Parasite Leishmania. Carbohydr. Res. 2010, 345, 1299–1305. [Google Scholar] [CrossRef]
- Chlubnová, I.; Králová, B.; Dvořáková, H.; Spiwok, V.; Filipp, D.; Nugier-Chauvin, C.; Daniellou, R.; Ferrières, V. Biocatalyzed Synthesis of Difuranosides and Their Ability to Trigger Production of TNF-α. Bioorg. Med. Chem. Lett. 2016, 26, 1550–1553. [Google Scholar] [CrossRef]
- Chlubnová, I.; Filipp, D.; Spiwok, V.; Dvořáková, H.; Daniellou, R.; Nugier-Chauvin, C.; Králová, B.; Ferrières, V. Enzymatic Synthesis of Oligo-d-Galactofuranosides and l-Arabinofuranosides: From Molecular Dynamics to Immunological Assays. Org. Biomol. Chem. 2010, 8, 2092–2102. [Google Scholar] [CrossRef] [Green Version]
- Pavic, Q.; Pillot, A.; Tasseau, O.; Legentil, L.; Tranchimand, S. Improvement of the Versatility of an Arabinofuranosidase against Galactofuranose for the Synthesis of Galactofuranoconjugates. Org. Biomol. Chem. 2019, 17, 6799–6808. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.J.; Smith, N.L.; Turkenburg, J.P.; D’Souza, S.; Gilbert, H.J.; Davies, G.J. Structural Insight into the Ligand Specificity of a Thermostable Family 51 Arabinofuranosidase, Araf51, from Clostridium Thermocellum. Biochem. J. 2006, 395, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Teze, D.; Hendrickx, J.; Dion, M.; Tellier, C.; Woods, V.L.; Tran, V.; Sanejouand, Y.-H. Conserved Water Molecules in Family 1 Glycosidases: A DXMS and Molecular Dynamics Study. Biochemistry 2013, 52, 5900–5910. [Google Scholar] [CrossRef]
- Koné, F.M.T.; Le Béchec, M.; Sine, J.-P.; Dion, M.; Tellier, C. Digital Screening Methodology for the Directed Evolution of Transglycosidases. Protein Eng. Des. Sel. 2009, 22, 37–44. [Google Scholar] [CrossRef] [Green Version]
- McGregor, N.G.S.; Artola, M.; Nin-Hill, A.; Linzel, D.; Haon, M.; Reijngoud, J.; Ram, A.; Rosso, M.-N.; van der Marel, G.A.; Codée, J.D.C.; et al. Rational Design of Mechanism-Based Inhibitors and Activity-Based Probes for the Identification of Retaining α-l-Arabinofuranosidases. J. Am. Chem. Soc. 2020, 142, 4648–4662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Rijssel, E.R.; van Delft, P.; Lodder, G.; Overkleeft, H.S.; van der Marel, G.A.; Filippov, D.V.; Codée, J.D.C. Furanosyl Oxocarbenium Ion Stability and Stereoselectivity. Angew. Chem. Int. Ed. 2014, 53, 10381–10385. [Google Scholar] [CrossRef]
- Ferrières, V.; Bertho, J.-N.; Plusquellec, D. A Convenient Synthesis of Alkyl d-Glycofuranosiduronic Acids and Alkyl d-Glycofuranosides from Unprotected Carbohydrates. Carbohydr. Res. 1998, 311, 25–35. [Google Scholar] [CrossRef]
- Berlin, W.; Sauer, B. In Situ Color Detection of α-l-Arabinofuranosidase, a “No-Background” Reporter Gene, with 5-Bromo-3- Indolyl-α-l-Arabinofuranoside. Anal. Biochem. 1996, 243, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Vriend, G. YASARA View—Molecular Graphics for All Devices—from Smartphones to Workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, K.I.; Deepa, G.; Namboori, K. Computational Chemistry and Molecular Modeling: Principles and Applications; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Krieger, E.; Darden, T.; Nabuurs, S.B.; Finkelstein, A.; Vriend, G. Making Optimal Use of Empirical Energy Functions: Force-Field Parameterization in Crystal Space. Proteins Struct. Funct. Bioinform. 2004, 57, 678–683. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Di Pierro, M.; Elber, R.; Leimkuhler, B. A Stochastic Algorithm for the Isobaric–Isothermal Ensemble with Ewald Summations for All Long-Range Forces. J. Chem. Theory Comput. 2015, 11, 5624–5637. [Google Scholar] [CrossRef] [Green Version]
- Hooft, R.W.W.; Sander, C.; Vriend, G. Positioning Hydrogen Atoms by Optimizing Hydrogen-Bond Networks in Protein Structures. Proteins Struct. Funct. Bioinform. 1996, 26, 363–376. [Google Scholar] [CrossRef]
- Krieger, E.; Dunbrack, R.L.; Hooft, R.W.W.; Krieger, B. Assignment of Protonation States in Proteins and Ligands: Combining PKa Prediction with Hydrogen Bonding Network Optimization. In Computational Drug Discovery and Design; Baron, R., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2012; pp. 405–421. [Google Scholar]
- Krieger, E.; Vriend, G. New Ways to Boost Molecular Dynamics Simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, P.; Yang, Z.; Du, F.; Li, Z.; Wu, C.; Fang, A.; Xu, X.; Zhou, G. Molecular Dynamics Simulation Exploration of the Interaction between Curcumin and Myosin Combined with the Results of Spectroscopy Techniques. Food Hydrocoll. 2020, 101, 105455. [Google Scholar] [CrossRef]
- Martínez, L. Automatic Identification of Mobile and Rigid Substructures in Molecular Dynamics Simulations and Fractional Structural Fluctuation Analysis. PLoS ONE 2015, 10, e0119264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodsell, D.S.; Olson, A.J. Automated Docking of Substrates to Proteins by Simulated Annealing. Proteins Struct. Funct. Bioinform. 1990, 8, 195–202. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osanjo, G.; Dion, M.; Drone, J.; Solleux, C.; Tran, V.; Rabiller, C.; Tellier, C. Directed Evolution of the α-l-Fucosidase from Thermotoga Maritima into an α-l-Transfucosidase. Biochemistry 2007, 46, 1022–1033. [Google Scholar] [CrossRef]
- Kirschner, K.N.; Yongye, A.B.; Tschampel, S.M.; González-Outeiriño, J.; Daniels, C.R.; Foley, B.L.; Woods, R.J. GLYCAM06: A Generalizable Biomolecular Force Field. Carbohydrates. J. Comput. Chem. 2008, 29, 622–655. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Land, H.; Humble, M.S. YASARA: A Tool to Obtain Structural Guidance in Biocatalytic Investigations. In Protein Engineering: Methods and Protocols; Bornscheuer, U.T., Höhne, M., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2018; pp. 43–67. [Google Scholar]
- Spiwok, V. CH/π Interactions in Carbohydrate Recognition. Molecules 2017, 22, 1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Liu, Q.; Qu, G.; Feng, Y.; Reetz, M.T. Utility of B-Factors in Protein Science: Interpreting Rigidity, Flexibility, and Internal Motion and Engineering Thermostability. Chem. Rev. 2019, 119, 1626–1665. [Google Scholar] [CrossRef]
- Hayes, M.R.; Pietruszka, J. Synthesis of Glycosides by Glycosynthases. Molecules 2017, 22, 1434. [Google Scholar] [CrossRef]
- Williams, S.J.; Withers, S.G. Glycosyl Fluorides in Enzymatic Reactions. Carbohydr. Res. 2000, 327, 27–46. [Google Scholar] [CrossRef]
- Strazzulli, A.; Cobucci-Ponzano, B.; Carillo, S.; Bedini, E.; Corsaro, M.M.; Pocsfalvi, G.; Withers, S.G.; Rossi, M.; Moracci, M. Introducing Transgalactosylation Activity into a Family 42 β-Galactosidase. Glycobiology 2017, 27, 425–437. [Google Scholar] [CrossRef] [Green Version]
- Bissaro, B.; Monsan, P.; Fauré, R.; O’Donohue, M.J. Glycosynthesis in a Waterworld: New Insight into the Molecular Basis of Transglycosylation in Retaining Glycoside Hydrolases. Biochem. J. 2015, 467, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.-Y.; Drone, J.; Hoffmann, L.; Tran, V.; Tellier, C.; Rabiller, C.; Dion, M. Converting a β-Glycosidase into a β-Transglycosidase by Directed Evolution. J. Biol. Chem. 2005, 280, 37088–37097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, B.; Arnaud, P.; Tellier, C.; Sanejouand, Y.-H. Toward the Design of Efficient Transglycosidases: The Case of the GH1 of Thermus Thermophilus. Protein Eng. Des. Sel. 2019, 32, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Teze, D.; Zhao, J.; Wiemann, M.; Kazi, Z.G.A.; Lupo, R.; Zeuner, B.; Vuillemin, M.; Rønne, M.E.; Carlström, G.; Duus, J.Ø.; et al. Rational Enzyme Design without Structural Knowledge: A Sequence-Based Approach for Efficient Generation of Transglycosylases. Chem. Eur. J. 2021, 27, 1–13. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutant Name | Mutations |
|---|---|
| MYC44 | V58M, F255L, E452K |
| MYC69 = MYC70 = MYC98 | E80G, S214T, E225D, L451M, K503N |
| MYC80 | D327N |
| Distances aX (Å) | |||
|---|---|---|---|
| Initial (1→X) Orientation | X = 3 | X = 6 | X = 4 |
| CtAraf 51-wild type | 5.60 | 3.33 | 6.74 [6.10 (X = 3)] |
| CtAraf 51-S214T mutant | 4.16 | 6.78 [3.72 (X = 3); 3.48 (X = 4)] | 3.88 [3.37 (X = 3)] |
| CtAraf 51-S214T-E225D (MYC98) | 4.60 | 7.57 | 3.55 [3.64 (X = 3)] |
| CtAraf 51-E225D (MYC98-T214S) | 5.37 | 3.51 | 8.12 [5.37 (X = 3)] |
| CtAraf 51-D327N (MYC80) mutant | 3.95 | 4.10 [3.30 (X = 4)] | 3.36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabezas-Pérusse, Y.; Daligault, F.; Ferrières, V.; Tasseau, O.; Tranchimand, S. Modulation of the Activity and Regioselectivity of a Glycosidase: Development of a Convenient Tool for the Synthesis of Specific Disaccharides. Molecules 2021, 26, 5445. https://doi.org/10.3390/molecules26185445
Cabezas-Pérusse Y, Daligault F, Ferrières V, Tasseau O, Tranchimand S. Modulation of the Activity and Regioselectivity of a Glycosidase: Development of a Convenient Tool for the Synthesis of Specific Disaccharides. Molecules. 2021; 26(18):5445. https://doi.org/10.3390/molecules26185445
Chicago/Turabian StyleCabezas-Pérusse, Yari, Franck Daligault, Vincent Ferrières, Olivier Tasseau, and Sylvain Tranchimand. 2021. "Modulation of the Activity and Regioselectivity of a Glycosidase: Development of a Convenient Tool for the Synthesis of Specific Disaccharides" Molecules 26, no. 18: 5445. https://doi.org/10.3390/molecules26185445